

Abstract
The calcification of cartilage is an essential step in the process of normal bone growth through endochondral ossification. Chondrocyte apoptosis is generally observed prior to the transition of calcified cartilage to bone. There are, however, contradictory reports in the literature as to whether chondrocyte apoptosis is a precursor to cartilage calcification, a co-event, or occurs after calcification. The purpose of this study was to test the hypothesis that chondrocyte apoptosis is not a requirement for initial calcification using a cell culture system that mimics endochondral ossification. Mesenchymal stem cells harvested from Stages 21-23 chick limb buds were plated as micro-mass cultures in the presence of 4 mM inorganic phosphate (mineralizing conditions). The cultures were treated with either an apoptosis inhibitor or stimulator and compared to un-treated controls before the start of calcification on day 7. Inhibition of apoptosis with the caspase inhibitor Z-Val-Ala-Asp (O-Me)-fluoromethylketone (Z-VAD-fmk) caused no decreases in calcification as indicated by radioactive calcium uptake or Fourier transform infrared (FT-IR) analysis of mineral properties. When apoptosis was inhibited, the cultures showed more robust histological features (including more intense staining for proteoglycans, and more intact cells within the nodules as well as along the periphery of the cells as compared to untreated controls), more proliferation as noted by bromo-deoxyuridine (BrdU) labeling, decreases in terminal deoxynucleotidyl transferase (Tdt)-mediated dUTP nick-end labeling (TUNEL) staining, and fewer apoptotic bodies in electron microscopy. Stimulation of apoptosis with 40-120 nM staurosporine prior to the onset of calcification resulted in inhibition of calcium accretion, with the extent of total calcium uptake significantly decreased, the amount of matrix deposition impaired, and the formation of abnormal mineral crystals. These results indicate that chondrocyte apoptosis is not a pre-requisite for calcification in this culture system.
Keywords: micro-mass, apoptosis, calcification, staurosporine, caspase-inhibition, hydroxyapatite
Physiologic calcification is an essential step in endochondral ossification and the process of normal bone growth [Frost and Jee, 1994; Galotto et al., 1994; Gerber et al., 1999]. Aberrant calcification leads to the undesirable and irreversible sequella of osteoarthritis [Gordon et al., 1984], failed cartilage repair [Bab et al., 1982; Homminga et al., 1991; Hunziker, 1999; Chiang et al., 2005], and chondrodysplasias [Knopov et al., 1995; Garofalo et al., 1999]. It is recognized that terminally differentiated hypertrophic chondrocytes undergo programmed cell death (apoptosis) during endochondral ossification [Gibson et al., 1995; Roach, 1997; Vu et al., 1998; Adams and Shapiro, 2002]. It is not clear, however, whether apoptosis precedes calcification or occurs as a consequence thereof.
The purpose of this study was to look at the controversy concerning the sequence of events in cartilage calcification, namely chondrocyte apoptosis, and its involvement in calcification. There are contradictory reports in the literature that in the growth plate and related cartilaginous tissues calcification precedes [Kirsch et al., 1997; Johnson et al., 2001; Adams and Shapiro, 2002; Kirsch et al., 2003; Magne et al., 2003], is concurrent with [Mansfield et al., 2003], has no association with [Felisbino and Carvalho, 2001], or occurs after [Gibson, 1998], chondrocyte apoptosis. The concept that apoptosis is essential for initiation of calcification is contradicted by the observation that agents that inhibit apoptosis in other cell systems are often those which induce chondrocyte hypertrophy [Galotto et al., 1994; Gibson, 1998]. Thus, there is a conflict between the observation that the proliferating chondrocytes initiate an apoptotic process which concludes with the maturation of the hypertrophic chondrocyte, vascular invasion, and bone formation, and the anti-apoptotic nature of many of the agents used to promote the maturation of the hypertrophic chondrocyte into a cell that produces a calcified cartilage matrix [Desai and Gruber, 1999].
The elucidation of the properties of cartilage mineral and the events leading to cartilage calcification could lead to the development of novel therapeutic strategies to prevent abnormal calcifications and to better treat congenital limb deformities and other conditions associated with deficient or defective cartilage calcification such as the chondrodystrophies [e.g., Knopov et al., 1995; Garofalo et al., 1999; Waterham et al., 2003], as well as to the fundamental understanding of the calcification process. The system we have chosen for our studies, micro-mass cultures of differentiating chick limb-bud mesenchymal cells is a well-established model of growth plate development, which our group and others have used extensively to elucidate events in the endochondral ossification process [Boskey et al., 1992a, 1996, 2000, 2002; Mello and Tuan, 1999; Daumer et al., 2004]. Using this system, results obtained to date reveal that conclusions derived from this avian model are generally applicable to calcification mechanisms in mammals [Mello and Tuan, 1999].
Apoptosis can be induced through both intrinsic and extrinsic pathways, each involving the activation of the cellular caspase cascade [Adams and Shapiro, 2002; Mirkes, 2002; Kajta, 2004; Reed, 2004; Kim et al., 2005]. The caspase cascade has been shown to be well conserved in avian models [Sanders and Parker, 2001]. To test the hypothesis that chondrocyte apoptosis is not essential for cartilage calcification, we modulated this cascade in the differentiating chick limb-bud mesenchymal cell micro-mass culture system by adding agents that increased (Staurosporine treatment) or inhibited (Z-Val-Ala-Asp (O-Me)-fluoromethylketone, (Z-VAD-fmk) treatment) apoptosis prior to the start of calcification (day 7). Several previous studies have demonstrated that ZVAD-fmk can prevent staurosporine-induced apoptosis in chondrocytes [Nuttall et al., 2000] as well as a variety of other cells [e.g., Marcelli et al., 2000; Gylys et al., 2002] showing staurosporine acts through a caspase-2/-7 pathway. Rates of calcification, the nature and amount of the mineral deposited, and the light and electron microscopic appearance of the cells and matrix in mineralizing cultures in which apoptosis was inhibited or stimulated, were compared to untreated mineralizing cultures.
MATERIALS AND METHODS
Materials
Fertilized White Leghorn eggs were obtained from CBT Farms (Chestertown, MD). Except where noted, tissue culture reagents were purchased from Invitrogen (Carlsbad, CA). DMEM was from Gemini Bio-Products, Inc. (Calabasas, CA). Cell culture plastic ware was purchased from Falcon (Becton-Dickinson, Franklin Lakes, NJ). Glutamine, antibiotics, and trypsin/EDTA were obtained from Cellgro MediaTech (Herndon, VA). Antibiotic (50 units/ ml penicillin) and antimycotic (25 mg/ml streptomycin) were also from Cellgro. Fetal bovine serum (FBS) was from Gibco (Grand Island, NY). Chemicals, including calcium chloride, sodium acid phosphate, glutamine, sodium ascorbate, and spectral grade potassium bromide (KBr) were from Fischer Scientific (Springfield, NJ). 45Ca was from Perkin Elmer Life Sciences/New England Nuclear (Billerica MA). Aquasol was from New England Nuclear. Z-VAD-fmk was acquired from (Calbiochem, Inc., San Diego, CA) and was dissolved as concentrated solutions in dimethyl sulfoxide (CalBiochem, Inc.). The final media contained 0.1% DMSO. Staurosporine was from Roche Diagnostics (Roche Molecular Systems, Inc. Branchburg, NJ) and was dissolved directly in media. Hoechst dye, papain, and calf thymus DNA were from Sigma (St Louis, MO).
Reagents for histochemistry and apoptosis assays included: Annexin-V Binding Buffer, Annexin-V-Allophycocyanin, and Propidium Iodide were from (Becton, Dickson and Co. Biosciences, Inc., San Diego, CA); Epon Polybed 812 from (Polysciences, Inc., Warrington, PA). Fluorescein-Frag EL DNA detection kit was from (Calbiochem, Inc.). Serum-free protein solution was from Dako Cytomation (Carpinteria, CA). Paraformaldehyde, glutaraldehyde, and cacodylate buffer were from Electron Microscopy Sciences (Fort Washington, PA). The bromo-deoxyuridine (BrdU) monoclonal antibody G3G4 was acquired from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). All other chemicals were from standard laboratory suppliers and were of the highest purity available.
Cell Cultures
Fertilized eggs were incubated for 41/2 days in a Napco circulated air incubator. After incubation, Stages 21-23 limb buds [Hamburger and Hamilton, 1951] were removed from the chick embryos under a microscope and trypsinized in 1X Trypsin-EDTA solution in a water bath at 37°C for 10 min to liberate the mesenchymal stem cells. The suspension was filtered through a double layer of 20 μm Nitex membrane to remove any contaminants. Cells were then counted on a hemocytometer, checked for viability (trypan blue dye exclusion), and then pelleted at 1,000 rpm for 8 min at 4°C.
Cells were resuspended in Dulbecco's Modified Eagle Medium (DMEM) containing 1.3 mM CaCl2,25 mg/ml freshly prepared sodium ascorbate, 0.3 mg/ml L-glutamine, antibiotics, and 10% FBS and plated as a micro-mass spot at concentrations of 0.75 to 1.0 × 106 per 20 ml in 35 × 10 mm culture dishes. The dishes were maintained in a CO2 incubator at 37°C and 5.0% CO2. Cells were allowed to attach for 2 h after which they were flooded with 2 ml of the previously described media. From day 2 onward, the inorganic phosphate (Pi) content of the "mineralizing" cultures was adjusted to 4 mM, while control, non-mineralizing cultures, were maintained with 1 mM Pi. Media was changed every other day.
The irreversible tripeptide apoptosis inhibitor, Z-VAD-fmk, was used at 25 and 50 μM concentrations based on concentrations reported in the literature to inhibit rat, bovine, and human chondrocyte apoptosis [Feng et al., 1999; Lee et al., 2000; Nuttall et al., 2000]. After pilot studies in which the concentration and day of addition (from day 5 to day 14) of the apoptosis stimulator staurosporine were investigated, a 40 nM concentration was used unless otherwise noted (see Fig. 1), with addition from day 7. This concentration was selected in part based on histochemistry and calcification assays as described previously [Kulyk, 1991; Borge et al., 1997]. The apoptosis inhibitor or the stimulator, unless otherwise noted, was added to the media before the start of calcification on day 7 and every other day with each change of media.
Fig. 1.
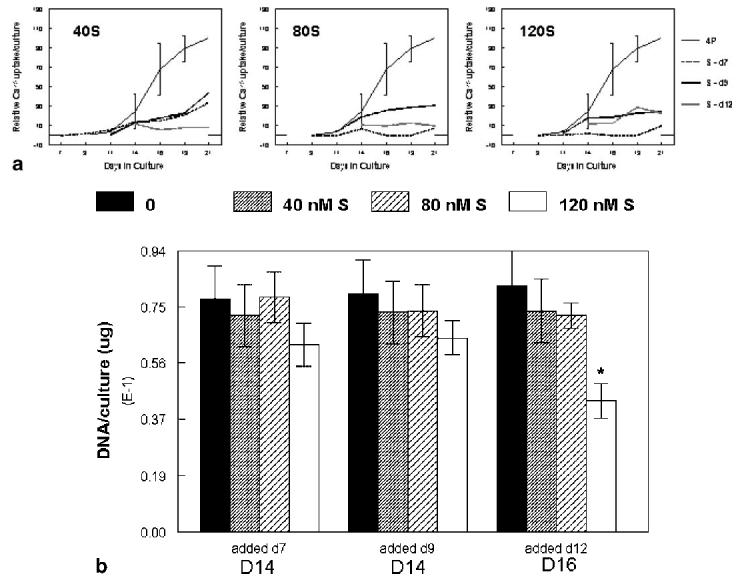
a: Pilot studies to determine effects of time of addition and concentration of staurosporine on 45Ca uptake. Mineral ion uptake was monitored in control mineralizing cultures (4P) and mineralizing cultures to which the apoptosis inducer was added at 40 (left), 80 (middle), or 120 (right) nM from day 7, 9, or 11. Error bars show mean ±SD for n = 3; duplicate experiments are provided for the other curves. All data was fitted by a non-linear sigmoid curve regression. b: DNA content (μg/culture) in the pilot study. Treatment groups are indicated on the x-axis; DNA measurements were performed at day 14 for cultures treated at day 7 or 9, and on day 16 for cultures treated on day 11. Error bars show mean ±SD for n = 3 determinations. *P < 0.05 relative to untreated control.
Calcification Assays
Calcium-45 uptake. Calcium-45 at a concentration of 0.5 mCi/ml was added to the culture dishes on day 5 and with every media change thereafter to monitor mineral accumulation. This uptake previously was correlated with total mineral accumulation [Boskey et al., 1992b]. At the indicated time points, cultures were washed in phosphate-buffered saline (PBS) and transferred to scintillation vials. Culture dishes were then flushed twice with 200 ml of 4N hydrochloric acid to dissolve any remaining mineral. The hydrochloric acid extract was then added to the scintillation vials. To solubilize the mineral in the cultures, vials were sealed and placed in a 60°C oven for 1 h and then cooled. Aquasol solution (5 ml) was then added to each vial and vortexed until the liquid was free of any cloudiness. Scintillation counting was performed on a Beckman scintillation counter. The amount of 45Ca uptake was corrected for uptake in similarly treated non-mineralizing controls [Boskey et al., 1992b] and then normalized to the uptake at day 21 in untreated mineralizing control (4P) cultures. All results were expressed as mean ±SD for a minimum of three experiments at each time point.
Fourier transform infrared (FT-IR) spectroscopy. After removal of the media and washing the cultures twice with PBS, the mineralized matrix within the culture dishes without 45Ca was air dried in the dish for at least 24 h. KBr (200 mg) was added to the dried cultures and KBr pellets prepared for spectroscopic analysis. Infrared spectra were collected using a Thermo-Nicolet Spectrometer 4700 under nitrogen purge. The background corrected spectra were base-lined (BioRad's Win-IR Pro 3.1), and areas under the phosphate n1,n3 peak (˜900-1,200/cm) and amide I peak (˜1,585-1,720/cm) calculated using the same software, and mineral/matrix ratio determined as the ratio of these two areas. Crystallinity was estimated from the relative intensities of bands at 1,030/cm (stoichiometric HA) and 1,020/cm (non-stoichiometric HA) [Boskey et al., 2003]. For illustrative purposes, spectra were normalized so that the area under the amide I peak was constant.
Measurement of Apoptosis
Flow cytometry. Flow cytometry was used to assess the percent apoptosis in treated and untreated mineralizing cultures from days 9-21. After media was removed from the unlabeled cultures, cells were released from the cultures via trypsinization as described above. The trypsin extract was then added, along with any residual culture material, to test tubes containing the previously removed culture media. The test tubes were centrifuged for 8 min at 4°C and 1,050 rpm to pellet the cells. Supernates were discarded and 2 ml of 1X Annexin-V Binding Buffer solution was added to each test tube. The pellet was then re-suspended by manual shaking. Aliquots of 100 ml of each suspension were mixed with 5 ml Annexin-V-Allophycocyanin and 5 ml propidium iodide (50 mg/ml), and incubated in the dark at room temperature for 20 min. Following incubation, another 400 ml of 1X Annexin-V Binding Buffer was added. Flow cytometry analysis was carried out on a FACS Calibur using Cell-QuestTM software. The numbers of apoptotic cells were calculated by counting the number of events showing high Annexin-V fluorescence. These events correlated with low amounts of forward scatter (FSC) and side scatter (SSC). The living cell population was defined as low to high SSC and moderate to high FSC. They were quantified based on low Annexin-V and propidium iodide fluorescence. Total apoptotic events divided by the total living plus apoptotic cell population were used to calculate percentage of apoptosis.
Light and electron microscopy. Unlabeled mineralizing cultures were rinsed with PBS and fixed overnight with 2% paraformaldehyde and 0.5% glutaraldehyde in 0.05 M cacodylate buffer at pH 7.4. In some cases 0.7% ruthenium hexamine trichloride [Hunziker et al., 1983] was added to cultures to preserve proteoglycans. The majority of the microscopic analyses were performed at day 13 or day 14, the time point at which rapid proliferation is occurring and calcification is just commencing, although selective analyses were performed at days 11, 16, 19, and 21. Following fixation the cultures were removed from the dish. Several small pieces were removed from each culture for electron microscopy and the remaining sample was embedded in paraffin for light microscopy. Paraffin embedded samples were sectioned at 5-7 mm thickness, dried on Double Plus microscope slides, deparaffinized, and stained with Alcian Blue, pH 2.5 counterstained with Kernechtrot solution [Armed Forces Institute of Pathology, 1994]. Qualitative analysis was performed to determine cell size and chondrocyte nodule characteristics (amount of lacunae and presence of cells within lacunae).
For electron microscopy fixed samples were dehydrated in alcohols and embedded in Epon. Sections 0.5-1.0 mm thick were stained with toluidine blue and viewed under the light microscope. Thin sections (80-90 nm thick) were collected on copper grids, stained with lead citrate and uranyl acetate and viewed on a CM-12 Philips transmission microscope (FEI, Inc., Hillsboro, OR).
Terminal deoxynucleotidyl transferase (Tdt)-mediated dUTP nick-end labeling (TUNEL) assay. Deparaffinized sections prepared as above were used for TdT-mediated dUTP nick-end labeling staining to localize apoptotic cells. Briefly, in this method TdT binds to exposed ends of DNA fragments characteristic of apoptosis and catalyzes the template-dependent addition of biotin-labeled and unlabeled deoxynucleotides. Biotinylated nucleotides were detected using streptavidin-horseradish peroxidase (HRP) conjugate. Diaminobenzidine color reagent reacted with the labeled sample to generate an insoluble brown colored product at the site of DNA fragmentation.
Cell proliferation. Bromo-deoxyuridine uptake was used to compare proliferation in the presence and absence of staurosporine or ZVAD-fmk. Cultures were incubated 3 h at 37°C with 3 mM BrdU, rinsed in fresh media, fixed in 10% formalin for 24 h, washed, dehydrated, and paraffin-embedded. Sections were deparaffinized, hydrated, blocked with H2O2 for 30 min, treated with 2N HCl at 37°C for 30 min, treated with 0.1% pepsin for 45 min at 37°C, and blocked with serum-free protein. The presence of BrdU was detected by immunohistochemistry with a monoclonal antibody (G3G4) against BrdU. The primary antibody was used at 1:20 dilution in a humid chamber overnight. Streptavidin-avidin was the secondary antibody and diaminobenzidine was the color reagent. The percentage of BrdU-labeled cells was counted in five areas of 1 mm2 selected randomly for each condition using the Bioquant image analysis system (Nashville, TN).
DNA was measured in the cultures using the Hoechst dye fluorescence assay [Kim et al., 1988]. In brief, three micro-mass cultures per treatment were combined, washed with PBS, and digested in papain 18 h at 37°C. Aliquots of the papain digest (100 ml) were used for the DNA determination relative to a calf thymus DNA standard.
Statistical Analysis
Significant differences between treatment groups were determined using SigmaStat software by SPSS, Inc. One-way ANOVA with a post-hoc Dunnett's Test was used to determine differences relative to mineralizing control (untreated) cultures at each time point. Significance was accepted to be a P less than or equal to 0.05.
RESULTS
Pilot data used to select concentrations of staurosporine and time of staurosporine addition is presented in Figure 1a. While with 80 and 120 nM staurosporine added on day 7 there was no detectable 45Ca uptake, many of the cultures came off the dish because of the extensive cell death, making it impossible to do both temporal and replicate measurements. When added after day 9 or day 11/12 all of the cultures, independent of concentration, showed some 45Ca uptake. To determine whether the differences between staurosporine treated cultures and untreated mineralizing controls was a reflection of the difference in cell numbers, DNA was measured 2 days after staurosporine treatment, and compared to untreated mineralizing controls at the same time point. Because staurosporine addition was done after cell proliferation had plateaued, there were few significant differences in total DNA after induction of apoptosis (Fig. 1b). Thus with addition at day 7, there was less than a 5% change in DNA with 40 or 80 nM staurosporine, while 120 nM caused a non-significant 20% decrease at day 9. Addition at day 9 similarly caused a 6%, 13%, and 17% decrease in DNA content for treatment with 40, 80, and 120 nM, relative to controls at day 11. Addition at day 11 caused a 10% and 12% DNA decrease with 40 and 80 nM staurosporine, but a significant 47% decrease with 120 nM staurosporine, assessed 4 days after the treatment with the apoptosis inducer. Based on these cumulative results, it was decided to start the treatments before calcification commenced (day 7), and to use 40 nM staurosporine.
Radioactive calcium uptake for each individual time point did not differ significantly when mineralizing control (4P) and Z-VAD-fmk (4P+Z) treated cultures were compared from day 7 to day 21 (Fig. 2). Both culture groups showed maximal increases in radioactive calcium uptake between days 9 and 15 and gradually leveled off after that time. Although Z-VAD-fmk treated cultures had a numerically greater radioactive uptake at day 21 than untreated cultures, with the higher Z-VAD-fmk dose providing the highest yield, this was not statistically significant relative to the untreated mineralizing cultures. Rates of 45Ca uptake (initial slopes of the curves) were also not significantly different when mineralizing controls and the two concentrations of Z-VAD-fmk were compared. As shown in Figure 2, cells treated with 40 nM staurosporine (4P + S) from day 7 onward showed an initial significant increase in radioactive calcium uptake as compared to the other two groups. At both days 9 and 11, there was a significant increase in 45Ca uptake (P 1/4 0.01 and 0.02, respectively) relative to the untreated mineralizing controls at the same time point. While immediately after treatment the initial rate of calcium accretion was significantly faster than the untreated cultures, the cultures treated with 40 nM staurosporine had no change in radioactivity after day 13 showed decreases in lower 45Ca uptake, with significantly lower values compared to mineralizing control cultures. As noted in Figure 1, cultures treated with 80 and 120 nM staurosporine showed no additional uptake of 45Ca after the time of addition.
Fig. 2.

Mineral ion accumulation in the presence and absence of modulators of apoptosis. The induction of apoptosis with 40 nM staurosporine (4P + S) from day 7 caused an immediate 45Ca uptake that declined relative to control mineralizing cultures (4P). Cultures in which apoptosis was inhibited with 50 mM Z-VAD-fmk had apparently higher but not significantly greater 45Ca accumulation per culture. Values were normalized to day 21 4P values and are mean ±SD for n = 5-7 independent experiments. *P < 0.05 relative to untreated mineralizing control cultures (4P).
Figure 3 presents the FTIR data describing mineral properties in the different treatment groups. Figure 3a shows typical infrared spectra of day 21 cultures. At day 15, only the staurosporine treated cultures contained hydroxyapatite mineral, and the crystals at this time point, based on detailed analyses of the spectra, were larger than those seen at later time points in any of the treatment groups and larger than those normally associated with chick cartilage calcification [Boskey et al., 1992a]. This is illustrated in the bar graphs of mineral/matrix ratio (Fig. 3b) and crystallinity (Fig. 3c). Mineral to matrix ratio increased with time in control mineralizing cultures, increased to a lesser extent in Z-VAD-fmk treated cultures, and decreased significantly in staurosporine treated cultures. In control and ZVAD-fmk treated cultures, crystallinity (crystal size and perfection) increased with time.
Fig. 3.
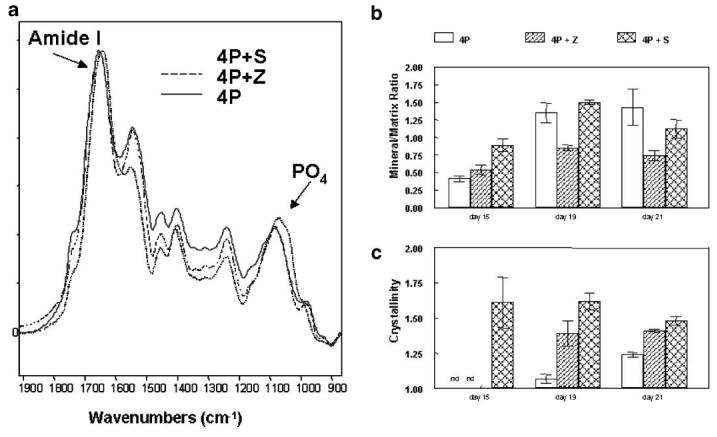
Infrared spectroscopic analysis of representative cultures: (a) Typical spectra from the three culture systems at day 21. All spectra were normalized so that the amide I bands were superimposed. The phosphate and amide I bands used for detailed analyses are indicated. b: Mineral:matrix ratios calculated from all spectra obtained as a function of time. Values are mean ±SD for n = 3-7 spectra from different cultures. c: Crystallinity, calculated from the peak intensity ratios of bands at 1,030 and 1,020/cm respectively. Values are mean ±SD for n = 3-7 spectra from different cultures.
Electron microscopy showed cells in the ZVAD-fmk treated cultures had greater amounts of normal appearing cells than those in the control cultures. At day 14, mineralizing control chondrocytes (Fig. 4a) showed large, uncondensed nuclei with abundant matrix production. The hypertrophic chondrocytes in these untreated mineralizing cultures (Fig. 4b) showed cellular swelling characteristic of this type of cell. EM analysis of staurosporine treated cells (Figs. 4c, d) showed a much more disorganized intracellular morphology. Apoptotic cells had characteristic condensed nuclei as well as decreased intracellular content (Fig. 4c). Necrotic cells (Fig. 4d) with numerous cytoplasmic inclusions were found in abundance in the staurosporine treated cultures consistent with the findings of flow cytometry (discussed below).
Fig. 4.
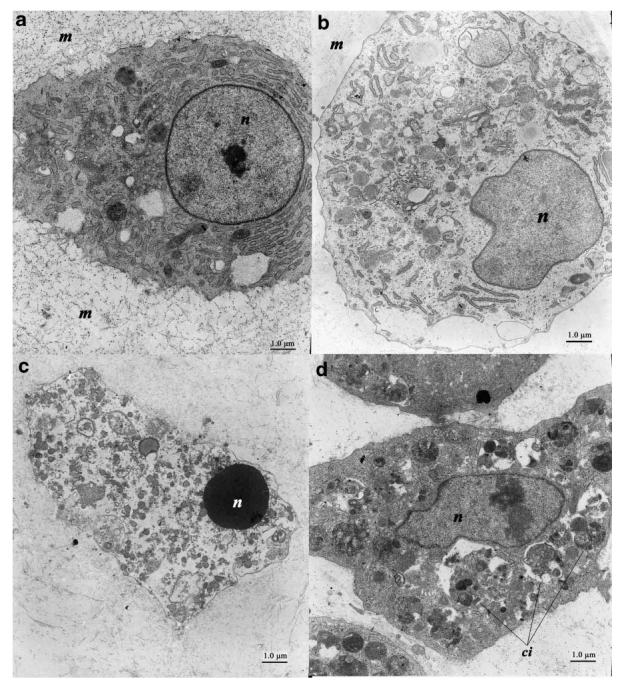
Electron microscopy of typical chondrocytes at day 13. a: A control chondrocyte showing a large, uncondensed nucleus (n) with abundant matrix (m) production. b: The control hypertrophic chondrocyte shows evidence of cellular swelling consistent with its physiologic role. These types of cells were also noted in Z-VAD-FMK treated cultures (not shown). c: Staurosporine treated sample shows a typical apoptotic cell with a condensed nucleus and relative lack of intracellular contents. d: Necrotic cells such as that shown here with numerous cytoplasmic inclusions (ci) were also seen in abundance in staurosporine treated cultures.
Light microscopy (Fig. 5) revealed distinct differences among the treatment groups at day 14, confirming the presence of apoptotic cells in staurosporine treated cultures; with markedly fewer apoptotic cells in Z-VAD-fmk treated cultures as compared to controls. The nodules in the Z-VAD-fmk treated cultures also appeared slightly larger, however this was difficult to document in the whole-mount cultures. Alcian Blue staining (Fig. 5a-d) for mineralizing cultures (4P) and mineralizing cultures treated with 40 or 120 nM staurosporine (40S, 120S) or 25 nM Z-VAD-fmk showed abundant nodule formation within the cultures with strong staining for proteoglycan content. The staurosporine treated groups appeared to have fewer nodules and increased internodular space compared to mineralizing control cultures. The Z-VAD-fmk treatment also produced fewer nodules and more internodular space than control cultures however the nodules were always more intensely stained for proteoglycans and appeared larger. There was roughly the same proportion of hypertrophic chondrocytes per nodule in the untreated and Z-VAD-fmk treated cultures, and fewer such cells in the staurosporine treated cultures. Cell content was considerably greater within the internodular space and on the nodule surfaces of the Z-VAD-fmk treated groups compared to mineralizing controls and staurosporine treated cultures. H&E stained plastic sections (0.5 microns thick) showed considerable histologic variation among groups (Fig. 5e-h). The control mineralizing cultures had well defined nodules and considerable numbers of hypertrophic chondrocytes within each nodule. Following staurosporine treatment the nodules were less well defined and the numbers of hypertrophic cells decreased as the drug dosage increased (com-pare 40 nM (5 f) to 120 nM (5 g) staurosporine). The number of dense round cells increased with increasing dosage suggesting that many cells do not develop to the hypertrophic stage. The ZVAD-fmk treated group (5 h) was quite unique in that there also were fewer hypertrophic cells, but also few small dense cells within the nodules. This group is characterized by a significant number of cells lining the outer surface of the nodules as well as densely populating the internodular spaces. TUNEL staining (Fig. 5i-l) was used to estimate the number of apoptotic cells. The control culture contained very few such cells. Some apoptosis was seen in the zone of hypertrophic cells. All nuclei are counterstained in Figure 5i-l so they can be seen but should not be confused with TUNEL-positive staining. Only the large round dark nuclei are positive for TUNEL (see photos 5 j and k). Following the staurosporine treatment, the number of apoptotic bodies within each nodule increased with increasing dose. Since the number of hypertrophic cells decreased with increasing staurosporine dose, the apoptosis most likely occurred in non-hypertrophic cells. In contrast, with Z-VAD-fmk treatment even though the number of hypertrophic cells was decreased, there are few apoptotic bodies visible, even when compared to the mineralizing control culture.
Fig. 5.
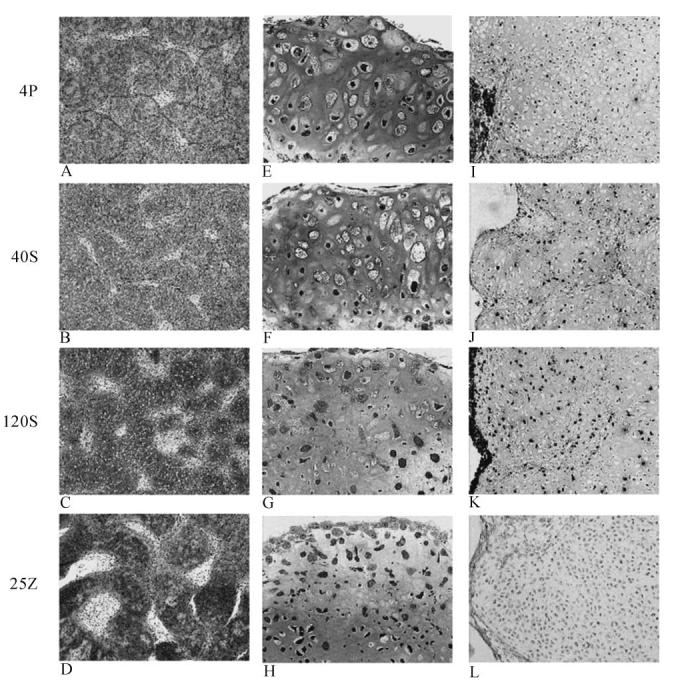
Light microscopy comparisons of 14-day chick cultures: Mineralizing controls (4P) (a, e, i), 40 nM Stauorsporine treated (b, f, j), 120 nM Staurosporine treated (c, g, k) and 25 nM Z-VAD-FMK treated (d, h, l). Alcian Blue stain (a-d; 4× original magnification): All cultures stain for proteoglycan content. The Z-VAD-fmk treated group showed strongest staining (5 d) and staurosporine treated cultures (5 b, c) showed less staining than control (5 a). Plastic epoxy sections (5 e-h; 20× magnification) stained with H&E showed distinctive differences within the hypertrophic cell population. Both treatment groups contained fewer hypertrophic cells compared to mineralizing control cultures. The Z-VAD-fmk treated group (5 h) was distinctive in that numerous cells lined the nodule surface and filled the internodular spaces. The TUNEL stain (5 i-l; 10× original magnification) showed that staurosporine contained more apoptotic cells than the mineralizing controls and higher doses of staurosporine resulted in greater apoptosis. The Z-VAD-fmk treated group (5 l) had fewer apoptotic cells than controls (5 i).
Bromo-deoxyuridine labeling (not shown) indicated that at 13 days, both within nodules as well as in the mesenchyme, there was extensive proliferation in mineralizing control cultures (average 54% uptake per field). In contrast, at the same time point in 40 nM staurosporine treated cultures, there was a marked decrease in BrdU uptake with only scant uptake in the mesenchyme and no evidence of uptake within the nodules (average 19% uptake per field).
Flow cytometry (Fig. 6) showed baseline apoptosis levels in control mineralizing cultures of 7-11% of cells throughout the time course of the experiment. Z-VAD-fmk treated cultures had 7-13% apoptotic cells, which was not significantly different from control samples. However, in cultures treated from day 7 with 40 nM staurosporine, apoptosis levels at day 9 were 35% and remained between 31% and 46% of total cells thereafter, significantly greater than untreated and Z-VAD-fmk treated cultures at all time points.
Fig. 6.

Flow cytometry using Annexin V to determine the percentage of apoptosis at various time points for mineralizing controls (4P), Z-VAD-fmk (25 nM) treated cultures, and 40 nM staurosporine treated cultures (4P + S) is shown. Cultures treated with the apoptosis inhibitor continuously from day 7 showed no significant difference compared to control. Cultures treated continuously with the apoptosis stimulator showed significant increase in apoptosis levels compared to control. (*P =0.05 relative to control mineralizing cultures).
DISCUSSION
The results of this study support our hypothesis that chondrocyte apoptosis is not an absolute requirement for initial calcification, at least in the avian micro-mass culture system studied here. This is corroborated by the observations that preventing apoptosis did not retard calcification, that giving sufficient apoptogen to kill the majority of the cells prevented calcification, and that cells with increased apoptosis while initially binding more calcium, did not form physiologic mineral and did not continue to accumulate calcium in their matrices.
Based on both light and electron microscopic assessment, as well as TUNEL staining, cultures treated with Z-VAD-fmk had no detectable apoptosis throughout the culture period, while there were low levels in the untreated cultures. Since TUNEL staining tends to over-rather than under-estimate apoptosis [Gal et al., 2000], and as Z-VAD-fmk is known to inhibit chondrocyte apoptosis [Nuttall et al., 2000], it is suggested that Z-VAD-fmk treatment was effective. However, flow cytommetry based on annexin-V binding is known to demonstrate early apoptosis [van Engeland et al., 1998], and both untreated control and ZVAD-fmk treated cultures showed comparable, albeit low (< 10%) levels of apoptosis at all time points tested.
There are several reasons that we suspect the flow cytometry data may be misleading. The requirement that the cells be released from the matrices of the high density cultures before study introduces a potential for artifact. Additionally, it was difficult to achieve total lysis of all the cells, making it possible that a high proportion of already necrotic and apopotic cells were those studied. On average, each flow experiment included ˜ 1,000 cells, thus total numbers for apoptotic cells in the control and Z-VAD-fmk treated groups were < 100, pushing the sensitivity of the method. There were, in all experiments, many more dead cells than apoptotic cells. More importantly, while flow cytometry is routinely used to characterize apoptosis in articular chondrocytes, there is only one report of flow cytometry studying apotosis in growth plate chondrocytes [Mansfield et al., 2003] and this report only measured live and dead cells based on propidium iodide, and relied on TUNEL staining for measures of apoptosis.
The presence of phosphatidyl serine (PS) on the outer membrane of cells is the basis for the annexin-V labeling used to assess apoptosis [van Engeland et al., 1998]. Assessing apoptosis in hypertrophic growth plate chondrocytes based on annexin-V binding may be problematic, as hypertrophic chondrocytes have increased amounts of PS on their outer membranes [Wuthier, 1971; Boskey et al., 1980], increasing the binding of annexin-V to cells that are still viable, as hypertrophic chondrocytes are not generally apoptotic as shown by other methods [Kirsch et al., 2003]. The most suspect finding was that the Z-VAD-fmk treated cultures showed no microscopic evidence of apoptosis by day 21, while continuously demonstrated 10% apoptosis, similar to the untreated mineralizing controls.
The observation that both the Z-VAD-fmk treated cultures and the untreated cultures had the same proportion of apoptotic cells based on annexin-V labeling suggests that we may be observing the presence of equivalent fractions of PS-rich cells. The extracellular matrix vesicles derived from hypertrophic chondrocytes are richer in PS [Wuthier et al., 1985], but were excluded from the data based on their small size. Hence, despite the flow cytometry data, we suggest that Z-VAD-fmk treated cultures were not apoptotic, and note that these cultures mineralized. Furthermore, even if there was 10% apoptosis maintained from the start of the experiment until its conclusion, were apoptosis essential, one would expect the cultures with 50% (or almost 100% apoptosis (120 nM staurosporine)) to be mineralizing at rates similar, if not greater than, control samples. However, in cultures treated with high concentrations (120 nM) of staurosporine where induction of apoptosis was complete, there was no mineral accumulation throughout the culture period. The 45Ca uptake in cultures treated with 40 nM staurosporine occurred earlier than in the other culture groups, and the crystals formed were less physiologic than control and the Z-VAD-fmk treated cultures. It is likely that much of the 45Ca uptake in the staurosporine treated cultures arose from calcium binding to PS. Ca-PS salts, as distinct from complexes consisting of Ca, Pi, and PS, do not support the growth of hydroxyapatite mineral [Boskey and Dick, 1991]. It is also possible that with apoptotic cell death there is an efflux of both calcium and lysosomal enzymes from the cell, and these could contribute to pathologic rather than physiologic calcification. We have previously demonstrated this [Boskey et al., 1996] in this same culture system by freeze thawing cells.
The initial high mineral/matrix ratio noted in the infrared spectra is probably due to the absence of matrix as seen in the light and electron microscopic images, rather than a marked increase in mineral. Interestingly by day 21, the crystals in the 40 nM staurosporine treated cultures, though less abundant, were more physiologic in size based on IR analyses. An explanation for this is seen in the histology and TUNEL data, in which it is noted that not all the cells die, and it is likely that these deposit a physiologic matrix, which eventually has mineral deposited upon it. Thus, the average of the larger dystrophic crystals and any newly formed physiologic mineral gives the appearance of a more physiologic mineral. It is important to note, that we have previously shown that the mineral formed in the control mineralizing cultures resembles that in chick growth plates [Boskey et al., 1992]. The mineral to matrix values reflect only the relative amounts of mineral and matrix, and thus are not useful comparisons, but the crystallinity data indicates that the staurosporine treated cultures, especially at early time points, consists of very large crystals. With time, they average out to the size of the controls and the Z-VAD-fmk crystals, indicating there is still some physiologic mineralization occurring.
In the growth plate there are important spatial relationships among chondrocytes as they temporally progress from resting cells to proliferating and hypertrophic chondrocytes. We have demonstrated previously that the chick limb bud micro-mass culture system maintains these crucial relationships [Boskey et al., 1992b]. Terminally differentiated chondrocytes are generally believed to undergo apoptosis [Adams and Shapiro, 2002], although whether the calcified matrix contributes to the apoptotic process, or the apoptotic process induces the calcification has been in debate. The facts that in our system partial induction of apoptosis led to a less physiologic mineral deposition, that when the entire culture underwent complete apoptosis there was no mineral deposition, and that cultures in which apoptosis was inhibited tended to accumulate the greater amount of mineral, supports our hypothesis that apoptosis is not essential for calcification. It thus appears that apoptosis must follow initial cartilage calcification. Future studies will be needed to define what factors trigger these events.
Supporting the concept that apoptosis is not mandatory for calcification are two recent reports. In the first [Kirsch et al., 2003], histologic evidence of mineralization preceded the observation of TUNEL staining in the hypertrophic zone of the chick growth plate. The second described the phenotype in animals in which vascular endothelial growth factor A (VEGFA) was conditionally knocked out [Zelzer et al., 2004]. VEGFA is both an angiogenesis and a cell survival factor. In the growth plates of conditional VEGFA knockout mice there was extensive apoptosis especially in their extended hypertrophic zones. But the embryonic mice through E18.5 showed a failure in mineralization. Although the problem in these mice was a failure of vascular invasion, the absence of mineral deposition supports our underlying hypothesis.
In the present study, we used different measures of apoptosis including: light and electron microscopy, TUNEL staining, and flow cytometry. These methods showed different absolute values for the amount of apoptosis but confirmed the relative effects of staurosporine and Z-VAD-fmk. Differences in the absolute values can be attributed to the ways in which cells are handled for each analysis. In processing cells for flow cytometry (in which control and Z-VAD-fmk treated cultures showed equivalent and measurable extents of apoptosis, some cells will invariably die as they are subjected to the 1 h processing protocol outside of the incubator before fixation. All measures were taken to ensure consistent conditions between flow cytometry experiments to minimize variability, but the increased proportion of dead and apoptotic cells in the control and Z-VAD-fmk treated cultures relative to that observed in other methods, suggests that there may be a processing artifact. Histologic analyses including TUNEL staining were consistent with each other showing Z-FAD-fmk treated cultures with the least apoptosis, and staurosporine treated cultures with the most. This could be attributed to the fact that processing outside of the incubator before fixation is minimal.
It is important to note that there may be species and cell dependent differences in the relationship between calcification and apoptosis. Parathyroid related protein (PTHrP) increases the expression of a protein (Bcl-2) that controls apoptosis in several cell types, including growth plate chondrocytes in vitro and in vivo, leading to delays in their maturation towards hypertrophy and apoptotic cell death. Overexpression of PTHrP under the control of the collagen II promoter in transgenic mice resulted in marked delays in skeletal development (and hence calcification) although a direct effect on calcification was not reported [Weir et al., 1996]. Deletion of the gene encoding Bcl-2 resulted in accelerated maturation of chondrocytes and concomitant shortening of long bones, though again without a reported direct effect on calcification [Amling et al., 1997]. Magne et al. [2003] reported that phosphate-induced calcification could be blocked by inhibiting apoptosis with 100 μM Z-VAD-fmk in a murine cell line that differentiates into chondrocytes. Whether the effects were due to necrosis caused by extremely high levels of phosphate, or by programmed cell death was not demonstrated. More recently, working with Li et al. [2005] we similarly found that induction of apoptosis in mouse osteoblast cultures caused the formation of physiologic mineral deposition. Whether the differences between the murine and the avian cultures are a species difference, or are associated with the inductive agent or cell-type remains to be determined. As noted above, mice conditionally null for VEGFA have increased apoptosis (and necrosis) and fail to mineralize [Zelzer et al., 2004], while mice in which insulin receptor substrate-1 was deleted showed increased apoptosis and increased cartilage calcification [Hoshi et al., 2004]. This suggests that the variations are system dependent, and reminds one that the deposition of physiologic mineral is a complex process that is likely to be under multiple controls. However, the data in this paper, as well as in other systems, indicates that chondrocyte apoptosis is not a mandatory precursor to calcification.
ACKNOWLEDGMENTS
This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06-RR12538-01 from the National Center for Research Resources, National Institutes of Health as well as the Musculoskeletal Integrity Core Center Grant Number AR46121, and Grant Number AR037661, National Institutes of Health. The authors would like to thank Ms. Orla O'Shea and Mr. Anthony LaBasieere for their assistance with the histology as well as Ms. Yukiji Fujimoto and Lyudmila Spevak for their assistance with infrared spectroscopy. Special thanks to Dr. Sergei Rudchenko for assistance with the flow cytometry.
Footnotes
Grant sponsor: NIH; Grant number: AR037661; Grant sponsor: National Center for Research Resources; Grant number: C06-RR12538-01; Grant sponsor: Musculoskeletal Integrity Core Center; Grant number: AR46121.
REFERENCES
- Adams CS, Shapiro IM. The fate of the terminally differentiated chondrocyte: Evidence for microenvironmental regulation of chondrocyte apoptosis. Crit Rev Oral Biol Med. 2002;13:465–473. doi: 10.1177/154411130201300604. [DOI] [PubMed] [Google Scholar]
- Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, Philbrick WM, Broadus AE, Baron R. Bcl-2 lies downstream of parathyroid hormone-related peptide in a signaling pathway that regulates chondrocyte maturation during skeletal development. J Cell Biol. 1997;136:205–213. doi: 10.1083/jcb.136.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armed Forces Institute of Pathology . In: Laboratory methods in histotechnology. Prophet EB, Mills B, Arrington JB, Sobin LH, editors. Washington DC: 1994. p. 19. [Google Scholar]
- Bab I, Sela J, Stein H. Transplantation of free perichondrial grafts into rabbit articular cartilage is associated with matrix vesicle calcification. Acta Anat (Basel) 1982;113:53–60. doi: 10.1159/000145537. [DOI] [PubMed] [Google Scholar]
- Borge L, Lemare F, Demignot S, Adolphe M. Restoration of the differentiated functions of serially passaged chondrocytes using staurosporine. In Vitro Cell Dev Biol Anim. 1997;33:703–709. doi: 10.1007/s11626-997-0128-9. [DOI] [PubMed] [Google Scholar]
- Boskey AL, Posner AS, Lane JM, Goldberg MR, Cordella DM. Distribution of lipids associated with mineralization in the bovine epiphysial growth plate. Arch Biochem Biophys. 1980;199:305–311. doi: 10.1016/0003-9861(80)90285-4. [DOI] [PubMed] [Google Scholar]
- Boskey AL, Camacho NP, Mendelsohn R, Doty SB, Binder-man I. FT-IR microscopic mappings of early mineralization in chick limb bud mesenchymal cell cultures. Calcif Tissue Int. 1992a;51:443–448. doi: 10.1007/BF00296678. [DOI] [PubMed] [Google Scholar]
- Boskey AL, Stiner D, Doty SB, Binderman I, Leboy P. Studies of mineralization in tissue culture: Optimal conditions for cartilage calcification. Bone Miner. 1992b;16:11–36. doi: 10.1016/0169-6009(92)90819-y. [DOI] [PubMed] [Google Scholar]
- Boskey AL, Doty SB, Stiner D, Binderman I. Viable cells are a requirement for in vitro cartilage calcification. Calcif Tissue Int. 1996;58:177–185. doi: 10.1007/BF02526884. [DOI] [PubMed] [Google Scholar]
- Boskey AL, Stiner D, Binderman I, Doty SB. Type I collagen influences cartilage calcification: An immuno-blocking study in differentiating chick limb-bud mesenchymal cell cultures. J Cell Biochem. 2000;79:89–102. doi: 10.1002/1097-4644(2000)79:1<89::aid-jcb90>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Boskey AL, Paschalis EP, Binderman I, Doty SB. BMP-6 accelerates both chondrogenesis and mineral maturation in differentiating chick limb-bud mesenchymal cell cultures. J Cell Biochem. 2002;84:509–519. [PubMed] [Google Scholar]
- Boskey AL, Moore DJ, Amling M, Canalis E, Delany AM. Infrared analysis of the mineral and matrix in bones of osteonectin-null mice and their wildtype controls. J Bone Miner Res. 2003;18:1005–1011. doi: 10.1359/jbmr.2003.18.6.1005. [DOI] [PubMed] [Google Scholar]
- Chiang H, Kuo TF, Tsai CC, Lin MC, She BR, Huang YY, Lee HS, Shieh CS, Chen MH, Ramshaw JA, Werkmeister JA, Tuan RS, Jiang CC. Repair of porcine articular cartilage defect with autologous chondrocyte transplantation. J Orthop Res. 2005;23:584–593. doi: 10.1016/j.orthres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Daumer KM, Tufan AC, Tuan RS. Long-term in vitro analysis of limb cartilage development: Involvement of Wnt signaling. J Cell Biochem. 2004;93:526–541. doi: 10.1002/jcb.20190. [DOI] [PubMed] [Google Scholar]
- Desai BJ, Gruber HE. Anti-apoptotic actions of cytokines in mammalian cells. Proc Soc Exp Biol Med. 1999;221:1–13. doi: 10.1046/j.1525-1373.1999.d01-49.x. [DOI] [PubMed] [Google Scholar]
- Felisbino SL, Carvalho HF. Growth cartilage calcification and formation of bone trabeculae are late and dissociated events in the endochondral ossification of Rana catesbeiana. Cell Tissue Res. 2001;6:319–323. doi: 10.1007/s004410100446. [DOI] [PubMed] [Google Scholar]
- Feng L, Balakir R, Precht P, Horton WE., Jr. Bcl-2 regulates chondrocyte morphology and aggrecan gene expression independent of caspase activation and full apoptosis. J Cell Biochem. 1999;74:576–586. [PubMed] [Google Scholar]
- Frost HM, Jee WS. Perspectives: A vital biomechanical model of the endochondral ossification mechanism. Anat Rec. 1994;240:435–446. doi: 10.1002/ar.1092400402. [DOI] [PubMed] [Google Scholar]
- Gal I, Varga T, Szilagyi I, Balazs M, Schlammadinger J, Szabo G., Jr. Protease-elicited TUNEL positivity of non-apoptotic fixed cells. J Histochem Cytochem. 2000;48:963–970. doi: 10.1177/002215540004800709. [DOI] [PubMed] [Google Scholar]
- Galotto M, Campanile G, Robino G, Cancedda FD, Bianco P, Cancedda R. Hypertrophic chondrocytes undergo further differentiation to osteoblast-like cells and participate in the initial bone formation in developing chick embryo. J Bone Miner Res. 1994;9:1239–1249. doi: 10.1002/jbmr.5650090814. [DOI] [PubMed] [Google Scholar]
- Garofalo S, Kliger-Spatz M, Cooke JL, Wolstin O, Lunstrum GP, Moshkovitz SM, Horton MA, Yayon A. Skeletal dysplasia and defective chondrocyte differentiation by targeted overexpression of fibroblast growth factor 9 in transgenic mice. J Bone Miner Res. 1999;14:1909–1915. doi: 10.1359/jbmr.1999.14.11.1909. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- Gibson G. Active role of chondrocyte apoptosis in endochondral ossification. Microsc Res Tech. 1998;43:191–204. doi: 10.1002/(SICI)1097-0029(19981015)43:2<191::AID-JEMT10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Gibson GJ, Kohler WJ, Schaffler MB. Chondrocyte apoptosis in endochondral ossification of chick sterna. Dev Dyn. 1995;203:486–476. doi: 10.1002/aja.1002030409. [DOI] [PubMed] [Google Scholar]
- Gordon GV, Villanueva T, Schumacher HR, Gohel V. Autopsy study correlating degree of osteoarthritis, synovitis and evidence of articular calcification. J Rheumatol. 1984;11:681–686. [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Cole GM. Caspase inhibition protects nerve terminals from in vitro degradation. Neurochem Res. 2002;27:465–472. doi: 10.1023/a:1019840417796. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Homminga GN, Bulstra SK, Kuijer R, van der Linden AJ. Repair of sheep articular cartilage defects with a rabbit costal perichondrial graft. Acta Orthop Scand. 1991;62:415–418. doi: 10.3109/17453679108996635. [DOI] [PubMed] [Google Scholar]
- Hoshi K, Ogata N, Shimoaka T, Terauchi Y, Kadowaki T, Kenmotsu S, Chung UI, Ozawa H, Nakamura K, Kawaguchi H. Deficiency of insulin receptor substrate-1 impairs skeletal growth through early closure of epiphyseal cartilage. J Bone Miner Res. 2004;19:214–223. doi: 10.1359/JBMR.0301221. [DOI] [PubMed] [Google Scholar]
- Hunziker EB. Articular cartilage repair: Are the intrinsic biological constraints undermining this process insuperable. Osteoarthritis Cartilage. 1999;7:15–28. doi: 10.1053/joca.1998.0159. [DOI] [PubMed] [Google Scholar]
- Hunziker EB, Herrman W, Schenk RK. Ruthenium hexamine trichloride (RHT)-mediated interaction between plasmalemmal components and pericellular matrix proteoglycans is responsible for the preservation of chondrocytic plasma membranes in situ during cartilage fixation. J Histochem Cytochem. 1983;31:717–727. doi: 10.1177/31.6.6341460. [DOI] [PubMed] [Google Scholar]
- Johnson K, Pritzker K, Goding J, Terkeltaub R. The nucleoside triphosphate pyrophosphohydrolase isozyme PC-1 directly promotes cartilage calcification through chondrocyte apoptosis and increased calcium precipitation by mineralizing vesicles. J Rheumatol. 2001;28:2681–2691. [PubMed] [Google Scholar]
- Kajta M. Apoptosis in the central nervous system: Mechanisms and protective strategies. Pol J Pharmacol. 2004;56:689–700. [PubMed] [Google Scholar]
- Kim YJ, Sah RL, Doong JY, Grodzinsky AJ. Fluorometric assay of DNA in cartilage explants using Hoechst 33258. Anal Biochem. 1988;174:168–176. doi: 10.1016/0003-2697(88)90532-5. [DOI] [PubMed] [Google Scholar]
- Kim R, Emi M, Tanabe K. Caspase-dependent and independent cell death pathways after DNA damage. Oncol Rep. 2005;14:595–599. [PubMed] [Google Scholar]
- Kirsch T, Nah HD, Shapiro IM, Pacifici M. Regulated production of mineralization-competent matrix vesicles in hypertrophic chondrocytes. J Cell Biol. 1997;137:1149–1160. doi: 10.1083/jcb.137.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch T, Wang W, Pfander D. Functional differences between growth plate apoptotic bodies and matrix vesicles. J Bone Miner Res. 2003;18:1872–1881. doi: 10.1359/jbmr.2003.18.10.1872. [DOI] [PubMed] [Google Scholar]
- Knopov V, Leach RM, Barak-Shalom T, Hurwitz S, Pines M. Osteopontin gene expression and alkaline phosphatase activity in avian tibial dyschondroplasia. Bone. 1995;16:329S–334S. doi: 10.1016/8756-3282(94)00048-5. [DOI] [PubMed] [Google Scholar]
- Kulyk WM. Promotion of embryonic limb cartilage differentiation in vitro by staurosporine, a protein kinase C inhibitor. Dev Biol. 1991;146:38–48. doi: 10.1016/0012-1606(91)90444-8. [DOI] [PubMed] [Google Scholar]
- Lee D, Long SA, Adams JL, Chan G, Vaidya KS, Francis TA, Kikly K, Winkler JD, Sung CM, Debouck C, Richardson S, Levy MA, DeWolf WE, Jr., Keller PM, Tomaszek T, Head MS, Ryan MD, Haltiwanger RC, Liang PH, Janson CA, McDevitt PJ, Johanson K, Concha NO, Chan W, Abdel-Meguid SS, Badger AM, Lark MW, Nadeau DP, Suva LJ, Gowen M, Nuttall ME. Potent and selective nonpeptide inhibitors of caspases 3 and 7 inhibit apoptosis and maintain cell functionality. J Biol Chem. 2000;275:16007–16014. doi: 10.1074/jbc.275.21.16007. [DOI] [PubMed] [Google Scholar]
- Li X, Liu P, Liu W, Maye P, Zhang J, Zhang Y, Hurley M, Guo C, Boskey A, Sun L, Harris SE, Rowe DW, Ke HZ, Wu D. Dkk2 has a role in terminal osteoblast differentiation and mineralized matrix formation. Nat Genet. 2005;37:945–952. doi: 10.1038/ng1614. [DOI] [PubMed] [Google Scholar]
- Magne D, Bluteau G, Faucheux C, Palmer G, Vignes-Colombeix C, Pilet P, Rouillon T, Caverzasio J, Weiss P, Daculsi G, Guicheux J. Phosphate is a specific signal for ATDC5 chondrocyte maturation and apoptosis-associated mineralization: Possible implication of apoptosis in the regulation of endochondral ossification. J Bone Miner Res. 2003;8:1430–1442. doi: 10.1359/jbmr.2003.18.8.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield K, Pucci B, Adams CS, Shapiro IM. Induction of apoptosis in skeletal tissues: Phosphate-mediated chick chondrocyte apoptosis is calcium dependent. Calcif Tissue Int. 2003;73:161–172. doi: 10.1007/s00223-002-1056-z. [DOI] [PubMed] [Google Scholar]
- Marcelli M, Shao TC, Li X, Yin H, Marani M, Denner L, Teng B, Cunningham GR. Induction of apoptosis in BPH stromal cells by adenoviral-mediated overexpression of caspase-7. J Urol. 2000;164:518–525. [PubMed] [Google Scholar]
- Mello MA, Tuan RS. High density micromass cultures of embryonic limb bud mesenchymal cells: An in vitro model of endochondral skeletal development. In Vitro Cell Dev Biol Anim. 1999;35:262–269. doi: 10.1007/s11626-999-0070-0. [DOI] [PubMed] [Google Scholar]
- Mirkes PE. 2001 Warkany lecture: To die or not to die, the role of apoptosis in normal and abnormal mammalian development. Teratology. 2002;65:228–239. doi: 10.1002/tera.10049. [DOI] [PubMed] [Google Scholar]
- Nuttall ME, Nadeau DP, Fisher PW, Wang F, Keller PM, DeWolf WE, Jr., Goldring MB, Badger AM, Lee D, Levy MA, Gowen M, Lark MW. Inhibition of caspase-3-like activity prevents apoptosis while retaining functionality of human chondrocytes in vitro. J Orthop Res. 2000;3:356–363. doi: 10.1002/jor.1100180306. [DOI] [PubMed] [Google Scholar]
- Reed JC. Apoptosis mechanisms: Implication for cancer drug discovery. Oncology (Huntingt) 2004;18(13 Suppl 10):11–20. [PubMed] [Google Scholar]
- Roach HI. New aspects of endochondral ossification in the chick: Chondrocyte apoptosis, bone formation by former chondrocytes, and acid phosphatase activity in the endochondral bone matrix. J Bone Miner Res. 1997;12:795–805. doi: 10.1359/jbmr.1997.12.5.795. [DOI] [PubMed] [Google Scholar]
- Sanders EJ, Parker E. Ablation of axial structures activates apoptotic pathways in somite cells of the chick embryo. Anat Embryol (Berl) 2001;204:389–398. doi: 10.1007/s004290100208. [DOI] [PubMed] [Google Scholar]
- van Engeland M, Nieland LJ, Ramaekers FC, Schutte B, Reutelingsperger CP. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry. 1998;31:1–9. doi: 10.1002/(sici)1097-0320(19980101)31:1<1::aid-cyto1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, Mooyer P, Noort Gv G, Kelley RI, Wilcox WR, Wanders RJ, Hennekam RC, Oosterwijk JC. Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol delta 14-reductase deficiency due to mutations in the lamin B receptor gene. Am J Hum Genet. 2003;72:1013–1017. doi: 10.1086/373938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci USA. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuthier RE. Zonal analysis of phospholipids in the epiphyseal cartilage and bone of normal and rachitic chickens and pigs. Calcif Tissue Res. 1971;8:36–53. doi: 10.1007/BF02010121. [DOI] [PubMed] [Google Scholar]
- Wuthier RE, Chin JE, Hale JE, Register TC, Hale LV, Ishikawa Y. Isolation and characterization of calcium-accumulating matrix vesicles from chondrocytes of chicken epiphyseal growth plate cartilage in primary culture. J Biol Chem. 1985;260:15972–15979. [PubMed] [Google Scholar]
- Zelzer E, Mamluk R, Ferrara N, Johnson RS, Schipani E, Olsen BR. VEGFA is necessary for chondrocyte survival during bone development. Development. 2004;131:2161–2171. doi: 10.1242/dev.01053. [DOI] [PubMed] [Google Scholar]