

Abstract
N-{2-[4-(4,9-diethoxy-1-oxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)phenyl]acetyl}benzene sulphonamide (GW627368X) is a novel, potent and selective competitive antagonist of prostanoid EP4 receptors with additional human TP receptor affinity.
At recombinant human prostanoid EP4 receptors expressed in HEK293 cells, GW627368X produced parallel rightward shifts of PGE2 concentration–effect (E/[A]) curves resulting in an affinity (pKb) estimate of 7.9±0.4 and a Schild slpoe not significantly different from unity. The affinity was independent of the agonist used.
In rings of phenylephrine precontracted piglet saphenous vein, GW627368X (30–300 nM) produced parallel rightward displacement of PGE2 E/[A] curves (pKb=9.2±0.2; slope=1).
GW627368X appears to bind to human prostanoid TP receptors but not the TP receptors of other species. In human washed platelets, GW627368X (10 μM) produced 100% inhibition of U-46619 (EC100)-induced aggregation (approximate pA2 ∼7.0). However, in rings of rabbit and piglet saphenous vein and of guinea-pig aorta GW627368X (10 μM) did not displace U-46619 E/[A] curves indicating an affinity of <5.0 for rabbit and guinea-pig prostanoid TP receptors.
In functional assays GW627368X is devoid of both agonism and antagonist affinity for prostanoid CRTH2, EP2, EP3, IP and FP receptors. At prostanoid EP1 receptors, GW627368X was an antagonist with a pA2 of 6.0, and at prostanoid IP receptors the compound increased the maximum effect of iloprost by 55%. At rabbit prostanoid EP2 receptors the pA2 of GW627368X was <5.0.
In competition radioligand bioassays, GW627368X had affinity for human prostanoid EP4 and TP receptors (pKi=7.0±0.2 (n=10) and 6.8 (n=2), respectively). Affinity for all other human prostanoid receptors was <5.3.
GW627368X will be a valuable tool to explore the role of the prostanoid EP4 receptor in many physiological and pathological settings.
Keywords: GW627368X, EP4, prostanoid, receptor, competitive, antagonist
Introduction
Prostanoids are a group of lipid hormone mediators that are derived from C-20 fatty acids by the action of cyclooxygenases 1, 2 and 3. They consist of the prostaglandins (PG) and the thromboxanes and they elicit a wide variety of biological responses through activation of G-protein-coupled receptors. The prostanoid receptor family consists of eight distinct rhodopsin-like receptor proteins each being the product of an individual gene (Coleman et al., 1994; Narumiya et al., 1999). These have been termed the DP, EP1, EP2, EP3, EP4, FP, IP and TP receptors. With the identification of PGD2 as a potent agonist at the chemoattractant receptor homologous molecule of TH2 cells (CRTH2) receptor (Hirai et al., 2001), the total number of prostanoid receptor subtypes is now nine.
In most cases, the myriad biological functions stimulated by PGs are transduced by activation of G-proteins. The prostanoid EP4 receptor falls into a group of receptors normally associated with elevation of intracellular cyclic adenosine monophosphate (cAMP) levels subsequent to Gs activation. However, in common with both IP and TP receptors (Hirata et al., 1994; Wise, 1999), we have generated unpublished data that suggests that EP4 receptors display promiscuous coupling towards both Gs and Gi G-proteins under certain conditions.
In pharmacological terms, EP4 receptors are most similar to EP2 prostanoid receptors. In many physiological settings, EP2 and EP4 receptors are colocated making identification of individual roles difficult. Recently, several agonists have been described which should help to discriminate between EP2 and EP4 receptors. These include selective EP2 agonists such as ONO AE1-259 (Yamamoto et al., 1999), ONO 8815 (Ogawa et al., 2000; Tani et al., 2000) and CP533,536 (Li et al., 2003), and EP4 agonists such as ONO-AE1-329 (Yamamoto et al., 1999), ONO AE1-437 (Sakata et al., 2000) and a series of agonists discovered by Merck (Billot et al., 2003).
Antagonists are key to definitive classification of receptors but identifying selective EP4 receptor antagonists has proved difficult. A commonly used EP4 antagonist, AH23848 (Brittain et al., 1985; Coleman et al., 1994), actually has highest affinity for TP receptors and does not discriminate between EP4 and other human and rat prostanoid receptors. This compound has been superceded by other more potent and selective prostanoid EP4 receptor antagonists such as EP4A (Machwate et al., 2001), ONO-AE2-227 (Mutoh et al., 2002), the diphenyloxazole ‘compound 8' and the Nδ-Z-ornithine ‘compound 11' described by Hattori et al. (2005), and the EP2 agonist/EP4 antagonist molecules described by Oxford et al. (2005).
N-{2-[4-(4,9-diethoxy-1-oxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)phenyl]acetyl}benzene sulphonamide (GW627368X; Figure 1), a potent EP4 receptor antagonist with additional affinity for human TP receptors, has previously been disclosed in poster form (Giblin et al., 2002; Wilson et al., 2003). Here we fully describe the binding and functional pharmacology of this molecule at recombinant human, and endogenously expressed animal, prostanoid receptors. This work represents the most complete characterisation of a potent and selective antagonist at human prostanoid EP4 receptors published to date. Indeed, to our knowledge GW627368X is the most selective EP4 receptor antagonist so far reported.
Figure 1.

The chemical structure of N-{2-[4-(4,9-diethoxy-1-oxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)phenyl]acetyl}benzene sulphonamide (GW627368X; Giblin et al., 2002).
Methods
Preparation of transiently expressing cell lines
Preparation of EP1, EP2, EP3I, EP4 Semliki Forest Virus (SFV) stocks
The coding regions of the human EP1 (GenBank L22647), EP2 (GenBank U19487), EP3I (GenBank X83857) and EP4 (GenBank L25124) receptors were inserted into pSFV using the method described by Marshall et al. (1997). pSFV/EP1–4 and the pSFV/helper constructs were linearised with SpeI, and cleaned using phenol/chloroform/isoamyl alcohol (25 : 24 : 1) followed by two volumes of chloroform/isoamyl alcohol (24 : 1) in phase lock tubes. The DNA was precipitated with 1/10 v v−1 3 M sodium acetate (pH 5.2) and two volumes of ethanol, then washed in acidified 70% ethanol before finally being resuspended in Tris/ethylenediaminetetra-acetic acid (EDTA) buffer (10 mM Tris HCl, 5 mM EDTA, pH 7.4) at 1 mg ml−1. Each DNA sample was checked to ensure that no circular DNA remained. The in vitro transcription reactions were performed with the SP6 Message Machine Kit (Ambion) according to the manufacturers instructions. A final GTP concentration of 4 mM was used to avoid [GTP] becoming rate limiting for these long transcripts. Final reaction volumes were all 200 μl. All incubations were for 2 h at 37°C. RNA was aliquoted and stored at −80°C. All reactions yielded sharp single bands when analysed by gel electrophoresis.
BHK cells were cultured in 175 or 500 cm2 flasks containing Dulbecco's-modified Eagle medium (DMEM)-HAM F12 +10% FBS +2 mM Glutamax to 70% onfluence and passaged with Trypsin 0.25% w v−1+0.6 mM EDTA. Cells were washed twice in PBS (calcium and magnesium free) and resuspended as a monosuspension in PBS at 1 × 107 cells ml−1. The cell suspension was then kept on ice. RNA stocks were defrosted on ice. Helper RNA and SFV-EPx RNA were mixed at different test ratios to optimise yields. Each electroporation was completed thus: 10 μl of RNA was added to 490 μl of cell suspension in a 0.2 cm3 electroporation cuvette on ice and subjected to two pulses of 25 μF (1.5 kV, time constants ∼0.75 ms). The resulting cell suspensions from three cuvettes were then transferred into a separate tube containing 50 ml of DMEM-HAM F12+10% FBS+2 mM Glutamax at 37°C and the resulting suspension cultured for 20 h at 27°C in a 175 cm2 tissue culture flask. The viral stocks were harvested and while still warm, α-chymotrypsin was added to give a final concentration of 500 μg ml−1. After 15 min incubation at 37°C, aprotinin was added to give a final concentration of 500 μg ml−1. The activated SFV stocks were then aliquoted and stored at −80°C.
Infection of CHOK1 cells
Cells were cultured at 37°C to 70% confluence in 1800 cm2 unbreakable roller bottles (0.25 r.p.m.; Cellon) containing DMEM-HAM F12+10% FBS+2 mM Glutamax. Aliquots of activated viral stocks were thawed and diluted 1/4 (v v−1) in medium at 37°C. Spent culture medium was removed from each bottle and 50 ml of virus+medium added before being incubated for 2 h (37°C, 0.5 r.p.m.). Each roller bottle was then supplemented with a further 350 ml of prewarmed medium containing 10−6 M indomethacin and incubated for 20 h (33°C, 0.25 r.p.m.). Each roller was then supplemented with a further 200 ml medium+10−6 M indomethacin and incubated for a further 20 h. Cells were harvested as follows: spent cell culture medium was discarded and cells washed with 400 ml PBS before treatment with Hanks'-buffered saline solution (HBSS) +0.6 mM EDTA (40 ml per roller bottle, 10–20 min, 2 r.p.m.). The resulting cell suspensions from several bottles were pooled together and centrifuged (300 × g, 10 min, 4°C), the supernatant discarded, and the cell pellet resuspended in 50 ml cold HBSS+0.6 mM EDTA for membrane preparation.
Preparation of HEK-DP, FP, IP and TP
The coding regions of the hDP gene (GenBank U31098 and U31332), the hFP gene (GenBank L24470), the hIP gene (GenBank L29016) and the hTP gene (GenBank D15056) were cloned into pcDNA3 (Invitrogen, San Diego, CA, U.S.A.) at the BamHI–NotI site. HEK293T cells (ECACC, Porton Down, U.K.) were grown to 80% confluence in minimum essential medium alpha+Glutamax supplemented with 10% heat-inactivated foetal bovine serum (HI-FBS) using 500 cm2 tissue culture-treated triple flasks (Nunc, Roskilde, Denmark). Cell culture was performed at 37°C under a 5% CO2/air atmosphere. The introduction of cloned pcDNA was achieved using a transfection mixture which was prepared as follows (quantities given are for a single triple flask): 0.8 ml of lipofectamine, and 45 μg of pcDNA were mixed and allowed to stand for 20 min at room temp. The DNA mixture was combined with 9 ml of Optimem and introduced to a culture flask from which medium had been aspirated. Flasks were returned to the incubator for 6 h at the end of which the transfection mixture was removed and 50 ml of normal culture medium reintroduced. Cells were allowed to grow for a further 48 h before being harvested.
Preparation of stable cell lines
CHO-NFAT-EP1 and FP
The coding regions of the hFP (GenBank L24470) and hEP1 (GenBank L22647) genes were cloned into pCIN3 (Invitrogen, San Diego, CA,. U.S.A.) at the EcoRI/NotI site and the BamHI fragment, respectively. The vectors were incorporated into CHO cells stably expressing the NFAT-luciferase gene using Lipofectamine®. Cells were grown for 24 h in DMEM-Ham F12 mix (DMEM-F12), supplemented with 10% FBS, 2 mM L-glutamine and 10 μg ml−1 puromycin. Subsequent culture was performed using fresh medium that additionally contained 1 mg ml−1 neomycin (G418 or geneticin). Individual clones were isolated by dilution cloning and tested for responses to PGE2 and PGF2α. A single clone of each receptor was selected for further study.
CHO-CRTH2, EP2, EP3I and IP
The coding regions of the hCRTH2 gene (GenBank AB008535), the hEP2 gene (GenBank X83868), the EP3I gene (GenBank X83857) and the hIP gene (GenBank L29016) were cloned into pcDNA3 (Invitrogen) and pCIN3 (hIP) vectors, at the BamHI–NotI site. Following the incorporation of plasmids into CHO cells using electroporation, cells were grown for 2 weeks under neomycin selection in Excel 301 medium containing 5% FBS and 400 μg ml−1 neomycin. CHO CRTH2 cells were additionally transfected with the chimeric Gα16z49 G-protein (Mody et al., 2000) and were cultured in the presence of 1 mg ml−1 neomycin and 400 μg ml−1 hygromycin B. Transfection of CHO cells with pCIN3-hIP was achieved using Lipofectamine® and were cultured in DMEM-F12, supplemented with 10% FBS, 2 mM L-glutamine and 400 μg ml−1 hygromycin for 24 h before being transferred to fresh medium additionally supplemented with neomycin (1 mg ml−1). Cells were separated using dilution cloning or flow cytometry in order to isolate individual clones in the wells of 96-well plates. Each clone was expanded and pharmacologically characterised. Single clones displaying the largest responses to PGD2, PGE2 or to iloprost were selected for further study.
HEK-EP4
HEK-293 cells expressing the recombinant human EP4 receptor were obtained from Receptor Biology Inc. (Beltsville, MD, U.S.A.).
Subsequent culture of all cells took place in DMEM-F12, containing 10% HI-FBS and 2 mM L-glutamine in 175 cm2 flasks. Cells were either passaged into fresh medium or used in an assay once 90% confluency (determined visually) had been achieved.
Membrane preparation
Membranes were prepared from cells grown in 1800 cm2 roller bottles or 175 cm2 tissue culture flasks as follows: the cells were harvested into HBSS containing 1 mM EDTA and centrifuged (250 × g, 5 min, 4°C). All subsequent steps were performed at 4°C. The cells were homogenised within a glass Waring blender for 2 × 15 s in 200 ml of 50 mM N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid] (HEPES) (pH 7.40)+10−4 M leupeptin+25 μg ml−1 bacitracin+1 mM EDTA+1 mM phenylmethylsulphonyl fluoride (PMSF)+2 μM Pepstatin A, (the latter 2 reagents added as freshly prepared 100 × and 500 × stocks, respectively, in ethanol). The blender was plunged into ice for 5 min after the first burst and for 30 min after the final burst to allow foam to settle. The material was spun at 500 × g for 20 min. The supernatant was removed and spun for 36 min at 48,000 × g. The resulting pellet was resuspended in the same buffer as above but not containing PMSF and Pepstatin A. The material was then forced through a 0.6 mm gauge needle, made up to the required volume, (usually 4 × the volume of the original cell pellet), and stored as frozen aliquots at −80°C. Protein concentration in membrane samples was determined using the Biorad Protein Assay kit and following the manufacturer's instructions (Bio-Rad Laboratories, Hercules, CA, U.S.A.).
Saturation analysis of radioligand binding
Assessment of binding signal
Incremental amounts of membrane were incubated with a fixed concentration of radioligand at or near its assumed Kd for each receptor type. V-bottom 96-well plates (Corning, Koolhovenlaan, The Netherlands) were prepared containing 25 μl [3H]-PGE2, 25 μl PGE2 (to determine non-specific binding (n.s.b.)), or 25 μl vehicle (to determine total binding). All reagents were diluted in assay buffer of the following composition: 50 mM HEPES, 10 mM MgCl2, adjusted to pH 7.4 with 1 M KOH(aq). The binding reaction was initiated by the addition of 50 μl of membrane suspension and proceeded for 120–180 min at room temperature. The reaction was terminated by rapid filtration through a pre-soaked 96-well GF/B glass fibre filtermat, which was subsequently dried and treated with Meltilex solid scintillant (Wallac, Turku, Finland). Results were obtained by scintillation counting (1450 Microbeta Trilux liquid scintillation counter, Wallac) using a suitable 1 min [3H] counting protocol in order to generate corrected counts per minute (c.c.p.m.). In each of two experiments, the mean of three data points were used to determine the concentration of membranes giving rise to a specific binding signal of 400–1000 c.c.p.m. on the linear part of the [membrane]/signal relationship.
Determination of radioligand Kd
Amounts of membrane giving rise to suitable specific binding signals (see above) were incubated with increasing concentrations of radioligand. The concentration range of each radioligand used varied according to the receptor being studied. A two-fold dilution series ranging from approximately 10-fold below to 10-fold above literature quoted Kd values was used. Binding reactions were conducted as described above in order to generate ccpm data representing total and n.s.b.. Experiments were performed in triplicate twice (except for EP2 where usable data were only generated in a single experiment).
Competition radioligand-binding studies
The ability of GW627368X to bind to prostanoid receptors was assessed by competitive radioligand displacement using membranes prepared as above and described in Table 2. A combination of filtration and scintillation proximity assay (SPA) formats were used, for which all membranes, beads, compounds and ligands were diluted/suspended in assay buffer of the following composition: 50 mM HEPES, 10 mM MgCl2, adjusted to pH 7.4 with 1 M KOH(aq). Reagents and concentrations suitable for these assays are described in Table 2.
Table 2.
The binding properties of GW627368X at human prostanoid receptors
| |
Reference compound |
GW627368X |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| pKi | nH | Max displacement (%) | n | pKi | nH | Max displacement (%) | n | ||
| DP |
PGD2 |
7.8±0.3 |
0.8±0.1 |
100 |
19 |
<5.0 |
|
|
2 |
| EP1 |
PGE2 |
7.3±0.3 |
0.9±0.3 |
100 |
49 |
<5.1 |
|
|
2 |
| EP2 |
PGE2 |
7.7±0.3 |
1.0±0.4 |
100 |
58 |
<5.1 |
|
|
2 |
| EP3 |
PGE2 |
8.2±0.2 |
1.1±0.2 |
100 |
69 |
<5.1 |
|
|
2 |
| EP4 |
PGE2 |
8.1±0.3 |
1.0±0.4 |
100 |
168 |
7.0±0.3 |
0.9±0.2 |
∼100 |
10 |
| FP |
PGF2α |
7.9±0.3 |
1.0±0.2 |
100 |
18 |
<5.1 |
|
|
2 |
| IP |
Iloprost |
7.5±0.3 |
1.2±0.3 |
100 |
28 |
<5.3 |
|
|
2 |
| TP | SQ29,548 | 8.9 | 0.9 | 100 | 2 | 6.8 | 1.1 | ∼100 | 2 |
Scintillation proximity assay
96-well SPA plates (Wallac, Turku, Finland) were prepared containing 25 μl of GW627368X, PGE2, PGD2, PGF2α, iloprost or U-46619 diluted in 0.5 log unit increments, vehicle (to define total binding), and unlabelled displacing ligand for determination of n.s.b. in appropriate wells. The appropriate radioligand was added to all wells (25 μl). The binding reaction was initiated by the addition of 50 μl of a mixture of wheatgerm agglutinin SPA beads (15 mg ml−1) and membrane suspension and allowed to proceed for 120 min at room temperature. Results were obtained by scintillation counting (1450 Microbeta Trilux liquid scintillation counter, Wallac) using a suitable SPA 1 min [3H] counting protocol in order to generate c.c.p.m. Data were generated in three separate experiments.
Filtration-binding assay
The low level of EP1 receptor expression coupled with its relatively low affinity for PGE2 prevented the use of SPA for this receptor. V-bottom 96-well plates (Corning) were prepared containing GW627368X, PGE2, [3H]-PGE2 and vehicle as described above. The binding reaction was initiated by the addition of 50 μl of CHO-EP1 membranes and then proceeded for 180 min at room temperature. The reaction was terminated by rapid filtration through a 96-well GF/B glass fibre filtermat which was subsequently dried and treated with Meltilex solid scintillant (Wallac). Results were obtained in the manner previously described with the use of a suitable filtermat-counting protocol. Data were generated in three separate experiments.
Cyclic AMP assays
Cells were harvested by treatment with Versene, resuspended in fresh culture medium and plated out to yield approximately 1 × 105 cells per well of a 96-well plate for overnight culture. For assay, the culture medium was replaced with assay medium (CHO-EP2: DMEM-F12 containing 1 mM L-ascorbate and 300 μM isobutyl-methylxanthine (IBMX); HEK-EP4: DMEM-F12 containing 300 μM IBMX and 3 μM indomethacin) and incubated for 30 min. Following this, cells were incubated with agonists (CHO-EP2: three-fold dilution series; HEK-EP4: four-fold) for 15 min. The reaction was stopped by the aspiration of the assay medium and the addition of ice-cold ethanol. All incubations were carried out at 37°C in a 5% CO2 atmosphere. Care was taken to ensure the constancy of IBMX, indomethacin and vehicle (dimethyl sulphoxide (DMSO)) concentrations throughout these experiments. The amount of cAMP in each well was then determined by [125-I] cAMP SPA using a proprietary kit (Amersham, Bucks, U.K.) and according to the manufacturer's instructions. Data were generated in duplicate from three separate experiments.
Calcium influx assays
For experiments, cells were removed from flasks with Versene and plated into clear-bottomed, black-walled 96-well plates at ∼30,000 cells per well in 200 μl of growth medium. Immediately prior to assay, medium was replaced with 100 μl of assay buffer (145 mM N-methyl-D-glucamine, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose, 10 mM HEPES, 250 μM sulphinpyrazone, 2 mg ml−1 BSA, pH 7.4) containing 2 μM Fluo-4 AM and antagonist. Following incubation at 37°C for 90 min plates were washed twice with assay buffer at room temperature, antagonists replaced and plates transferred to a Fluorescence Imaging Plate Reader (FLIPR; Molecular Devices, Sunnywale, CA, U.S.A.) to monitor changes in fluo-4 fluorescence after addition of agonists. Data were generated in duplicate from three separate experiments.
Piglet and rabbit saphenous veins, and guinea-pig aorta
All animals used in this study were handled in accordance with the requirements of the U.K. Home Office, Animals (Scientific Procedures) Act, 1986, and all subsequently issued guidelines.
Large White piglets of either sex (3–6 days old) were humanely killed by captive bolt followed by exsanguination. The saphenous vein was removed from each hind leg of the animal and dissected free of connective and other adhering tissue. Rings of tissue of 5 mm length were suspended between tungsten wire hooks in 5 ml side-arm bubbling tissue baths for isometric force recording containing Krebs solution at 37°C, aerated with 95% O2+5% CO2. Krebs solution was of the following composition: NaCl 118 mM, NaHCO3 25 mM, KCl 4.8 mM, KH2PO4 1.2 mM, MgSO4 1.2 mM, glucose 11.1 mM, CaCl2 1.25 mM and contained indomethacin 3 μM (to block endogenous PG synthesis) and GR32191B 1 μM (to block TP receptors). Tissue was initially collected into Krebs solution additionally supplemented with the monoamine oxidase inhibitor pargyline (500 μM) such that a 30 min exposure to this agent was achieved. Changes in force were detected using FT03C force displacement transducers and recorded digitally on a MacLab data acquisition system running Chart v3.4.2 software (sampling frequency 0.66 Hz; AD Instruments, Hastings, U.K.).
An initial force of 1 × g was applied to each tissue ring for a period of 10 min at the end of which the bathing solution in each bath was replaced. A force of 2 × g was then applied for a period of 30 min followed by exposure to 80 mM KCl to establish the maximum level of force generated by each ring. This concentration of KCl had previously been shown to be maximally effective in these tissues (data not shown). Washout of vasoactive agents was achieved by four exchanges of bathing medium after which basal tone was allowed to re-establish for 10 min prior to the addition of the EP4 receptor antagonist GW627368X (1 nM–0.3 μM) or vehicle. In order to study the functional effects of prostanoids at relaxant receptors in whole tissues, tone must first be elevated with a suitable spasmogen. This was achieved by the addition of 1 μM phenylephrine (PE) which has previously been shown to represent an EC80 concentration of this compound (data not shown). Responses to PE were allowed to stabilise such that an overall antagonist incubation time of 60 min elapsed before the construction of agonist E/[A] curves. In order to maximise the number of experiments performed in tissues from each animal, a single agonist concentration–effect (E/[A]) curve was produced in each ring of tissue by the cumulative addition of compound at 0.5 log10 intervals.
Studies using rabbit saphenous vein and guinea-pig aorta were performed using an analogous procedure. New Zealand White rabbits of either sex (3.5 kg approximately) and Dunkin–Hartley guinea-pigs received a lethal dose of pentobarbital sodium (Euthanal®) prior to tissue excision.
Platelet aggregation
Platelets were obtained from healthy human volunteers using a standard venepuncture technique, in which blood was drawn by syringe through a 19-gauge needle into acid citrate dextrose anticoagulant (8.6:1.4 vv−1). Blood samples were centrifuged at 230 × g for 15 min to obtain platelet-rich plasma (PRP). Washed platelets (WP) were prepared from PRP by centrifugation at 900 × g for 10 min followed by resuspension in HEPES-modified Tyrode's buffer (HMTB; 138 mM NaCl, 2.9 mM KCl, 12 mM NaHCO3, 10 mM HEPES, 10 mM glucose, pH 7.4) containing 0.05 U ml−1 Grade VII apyrase (Sigma, Poole, Dorset, U.K.), 10 U ml−1 hirudin (Refludan; Berlex), and 1% platelet-poor plasma. Platelet counts were standardised to 2.5 × 105 platelets μl−1 with HMTB. CaCl2 was added to yield a final concentration of 1.25 mM. Platelet aggregation was then performed in a Chrono-Log aggregometer (Chrono-Log Corp., Havertown, PA, U.S.A.) using 250 μl of WP at 37°C with stirring (1000 r.p.m.). GW627368X (0.1, 1 and 10 μM) or DMSO vehicle was incubated with the platelets for 2 min prior to the addition of the prostanoid TP receptor agonist U46619 (Cayman Chemical, Ann Arbor, Michigan, U.S.A.). In each experiment, the concentration of U46619 was titrated so as to be just equal to EC100.
Data analysis
Curve fitting
A four-parameter logistic equation of the form:
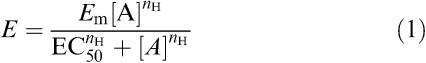 |
was fitted to data from tissue bath studies (grams tension normalised with respect to the PE response; %) and from cAMP assays (pmol cAMP per well determined from the mean of two replicate assay points). Thus, estimates of maximum effect (Em), curve mid-point (EC50), and Hill slope (nH) were obtained; other terms in the equation are effect (E) and concentration ([A]).
For data from radioligand competition-binding assays (c.c.p.m.) the following form of the equation was used:
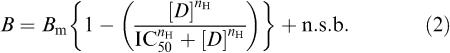 |
where B is the binding signal (in c.c.p.m.), Bm is the maximum signal before the addition of unlabelled displacing ligand, [D] is the concentration of the displacing ligand, IC50 is the concentration of displacing ligand required to produce a half-maximal reduction in binding signal, nH is the Hill coefficient, and n.s.b. is the non specific binding level (in c.c.p.m.).
Calculation of affinity estimates – saturation binding
The amount of specific radioligand binding to each receptor type was calculated as the difference between total and n.s.b. at each concentration. Three equations were fitted to data:
(1) A hyperbolic plus linear equation fitted to total binding data.
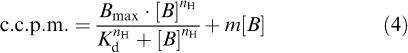 |
where c.c.p.m. are corrected counts per minute, Bmax is the maximum amount of radioligand binding under saturating conditions, [B] is the concentration of radioligand, Kd is the radioligand-binding dissociation constant, nH is the Hill slope, and m is the slope of the linear n.s.b. relationship.
(2) A linear equation fitted to n.s.b. data and using the value of m to constrain fitting to (4).
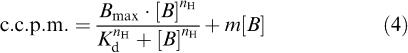 |
where n.s.b. is non-specific binding, m is the slope of the relationship, [B] is the concentration of radioligand and c is the intercept of the line on the c.c.p.m. axis which should equal background radiation.
(3) A hyperbolic equation fitted to specific binding data.
where terms are as previously defined.
Careful consideration of the values and their associated fitting errors obtained by each fit was made in order to arrive at robust affinity estimates.
Calculation of affinity estimates – competition binding
Where the Hill coefficient of a displacement-binding curve was not significantly different from unity, the Cheng & Prusoff (1973) correction was applied to IC50 values in order to estimate binding affinity values (pKi) according to the following equation:
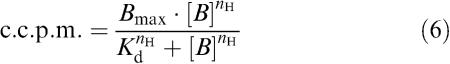 |
where KD is the dissociation-binding constant, and [L] is the concentration of the radioligand used.
Calculation of affinity estimates – antagonism
Constancy of agonist E/[A] curve shape i.p.o. increasing antagonist concentrations was assessed by computerised curve-fitting followed by Student's t-test on asymptotes and slopes. Computed EC50 values were fitted to a modification of the Schild equation suitable for nonlinear regression (Lew & Angus, 1985).
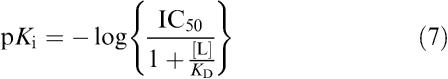 |
where the constant –log c is the difference between the agonist control curve EC50 and the antagonist pKb. Single agonist E/[A] curves were generated in each tissue therefore comparisons were made between data generated in different tissues from the same animal in order to obtain a single pKb estimate. For illustrative purposes, we have presented data graphically as Schild plots (Arunlakshana & Schild, 1959).
Individual estimates of curve parameters and affinity values were obtained from each experiment and then averaged to provide mean data. Quoted values are therefore the mean±standard deviation (s.d.) of n separate experiments, each derived from a separate animal, cAMP assay, or competition-binding experiment. As errors around slope estimates are log-normally distributed, slope data is expressed as the geometric mean with 95% confidence intervals.
Materials
Pargyline, indomethacin, PE, PGE2 (PGE2), PGD2, PGF2α, U-46619 (9,11-dideoxy-11α,9α-epoxy-methanoprostaglandin F2α), ethylinediaminetetra-acetic acid (EDTA), HEPES, PMSF, pepstatin A, leupeptin, bacitracin, Hanks'-buffered saline solution (HBSS), DMEM-Ham F12 mix (DMEM-F12), puromycin, Versene, Krebs' solution 10 × concentrate, and 3-isobutyl-1-methylxanthine (IBMX) were purchased from Sigma, Poole, Dorset, U.K. Potassium chloride (KCl), L-ascorbic acid, potassium hydroxide (KOH), magnesium chloride (MgCl2), ethanol and DMSO (all AnalaR grade) were obtained from BDH, Lutterworth, Leics, U.K. Lipofectamine®, FBS, HI-FBS, neomycin, hygromycin and 200 mM L-glutamine were purchased from Gibco-BRL Ltd, Paisley, U.K. Excel 301 medium was obtained from JRH Biosciences, Lenexa, KS, U.S.A. Radiolabelled PGs ([3H]-PGD2, [3H]-PGE2, [3H]-PGF2α and [3H]-iloprost trometamol salt), unlabelled iloprost and wheatgerm agglutinin – polyvinyl toluene SPA beads (WGA-PVT SPA beads) were purchased from Amersham, Bucks, U.K., while [3H]-[1S-[1α,2α(Z),3α,4α]]-7-[3-[[2-[(phenylamino)carbonyl]hydrazino]methyl]-7-oxabicyclo [2.2.1]hept-2-yl]-5-heptenoic acid ([3H]-SQ29,548) was purchased from NEN, Hounslow, U.K. GR32191B ([1R-[1α(Z),2β,3β,5α]]-(+)-7-[5-([1,1′-biphenyl]-4-ylmethoxy)-3-hydroxy-2-(1-piperidinyl) cyclopentyl]-4-heptenoic acid hydrochloride salt), and GW627368X were prepared in the Department of Medicinal Chemistry, Glaxo-Wellcome Research and Development, Stevenage, U.K.
Indomethacin, GW627368X and GR32191B were dissolved at 10 mM in DMSO. PGE2, PGD2 and PGF2α, were dissolved at 10 mM in 100% ethanol and stored at −20°C. Iloprost was supplied as a 1 mM solution in Tris buffer pH 8.0 and was stored in aliquots at −20°C. Radioligands were supplied as ethanolic solutions of differing concentrations which were all stored at −20°C. For tissue bath studies, dilutions of drugs and PE were made freshly on each day of study in Krebs solution containing indomethacin and GR32191B as described above. Diluted compounds were stored in the dark at 4°C for the duration of an experiment. Pargyline was dissolved at 0.5 M in dH2O and stored at −20°C. Potassium chloride was dissolved at 4 M in Krebs solution and stored at room temperature. For binding and cAMP assays, compounds and radioligands were freshly diluted into assay buffer as described above on each experimental occasion. IBMX was made up at 0.1 M in 0.1 M NaOH and added to DMEM-F12 for use. PMSF and Pepstatin A were used as × 100 and × 500 stocks, respectively, in ethanol.
Results
Characterisation of GW627368X
Competition radioligand binding at human prostanoid receptors
Membrane preparations containing a single recombinant human prostanoid receptor were characterised by non-linear curve fitting to saturation-binding data which revealed the presence of a single, saturable population of each receptor type (data for hEP4 and hTP are shown inset in Figure 2). Estimates of the radioligand dissociation-binding constants (Kd (nM)) and of the level of receptor expression are shown in Table 1. Competition-binding studies using a range of selective agonists and antagonists confirmed that each receptor possessed the expected pharmacology for that receptor type (data not shown).
Figure 2.

Mean radioligand displacement curves for GW627368X at human prostanoid EP4 (a) and TP (b) receptors and (inset) saturation curves for the radioligands [3H]-PGE2 and [3H]-SQ29548, respectively. pKi values quoted in Table 2 were generated by application of the Cheng–Prusofff correction to IC50 values as described in Methods.
Table 1.
Characteristics of membranes derived from cells transiently expressing prostanoid receptors and of the competition radioligand binding assays using them
| |
Membranes |
Assay |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Receptor | Vector | Bmax (pmol mg−1) | Kd (nM) | Protein (μg per well) | Assay type | Radioligand | Specific activity (TBq mmol−1) | [radioligand] (nM) | n.s.b. ligand | [n.s.b.] (μM) |
| DP |
pcDNA3 |
12.3 |
11.9 |
12.0 |
SPA |
[3H]-PGD2 |
5.96 |
1 |
PGD2 |
10 |
| EP1 |
SFV-1 |
3 |
12 |
11.3 |
Filtration |
[3H]-PGE2 |
6.07 |
10 |
PGE2 |
100 |
| EP2 |
SFV-1 |
4.3 |
19 |
7.8 |
SPA |
[3H]-PGE2 |
6.07 |
10 |
PGE2 |
100 |
| EP3I |
SFV-1 |
30 |
6 |
2.3 |
SPA |
[3H]-PGE2 |
6.07 |
3 |
PGE2 |
100 |
| EP4 |
SFV-1 |
1.1–8.8 |
3 |
0.7–2.4 |
SPA |
[3H]-PGE2 |
6.07 |
3 |
PGE2 |
100 |
| FP |
pcDNA3 |
12 |
8.2 |
18.0 |
SPA |
[3H]-PGF2α |
7.88 |
2.1 |
PGF2α |
10 |
| IP |
pcDNA3 |
6.5 |
13 |
15.0 |
SPA |
[3H]-iloprost |
0.63 |
15 |
iloprost |
1 |
| TP | pcDNA3 | 6.4 | 7 | 15.0 | SPA | [3H]-SQ29,548 | 1.80 | 6 | U-46619 | 1 |
Bmax: estimated receptor expression (pmol mg−1); Kd: the radioligand dissociation-binding constant (affinity) determined by saturation analysis (nM); protein: amount of protein per well for each assay (μg well−1); [radioligand]: concentration of radioligand (nM); n.s.b. ligand: the nonradiolabelled ligand used to displace radioligand from the receptors and therefore determine nonspecific binding; [n.s.b.]: concentration of n.s.b. ligand (μM).
GW627368X produced concentration-related displacement of radioligand from hEP4 and hTP receptors with equilibrium dissociation constants (pKi) values of 7.0±0.3 (n=10) and 6.8 (n=2), and slope (nH) values of 0.8 (0.7–1.0) and 1.1, respectively (Table 2 and Figure 2). The maximum level of radioligand displacement generated CCPM values indistinguishable from n.s.b. At all other prostanoid receptors, GW627368X, produced <50% displacement at 10 μM (n=4).
Effects on human recombinant EP4-mediated increases in cAMP
Basal cAMP production and PGE2 E/[A] curves at hEP4 receptors expressed in HEK293 cells were found to be altered in the presence of the nonselective COX1/2 inhibitor, indomethacin (3 μM; Figure 3a): curves were left-shifted and steepened (absence of indomethacin: pEC50=8.4±0.2, nH=0.6±0.3; presence of indomethacin (3 μM): pEC50=9.1±0.1, nH=1.0±0.3); basal cAMP production was reduced by 52%.
Figure 3.

Mean PGE2 (a) and GW627368X (b) concentration–effect (E/[A]) curves in HEK293 cells expressing recombinant human prostanoid EP4 receptors in the absence and presence of 3 μM indomethacin.
In this cell line, GW627368X was devoid of agonist activity and actually produced a significant and concentration-related reduction in basal cAMP levels (Figure 3; pIC50=6.3±0.1, nH=1.9±0.9). In the presence of indomethacin (3 μM) GW627368X reduced basal cAMP production to below detectable levels at the lowest concentration tested (0.1 nM). In untransfected HEK293 cells which were not obtained from the same source as the transfected cells, GW627368X (0.1 nM–100 μM) did not produce any alteration in basal cAMP turnover (data not shown). Subsequent experiments in this hEP4 receptor assay were conducted in the presence of 3 μM indomethacin.
In competition studies against PGE2 or 11-deoxy-PGE1 at recombinant human prostanoid EP4 receptors expressed in HEK293 cells, GW627368X (10 nM–1 μM) produced parallel rightward displacement of PGE2 E/[A] curves, with no significant change in Hill slope parameter or maximum effect (Figure 4). Using a modified method of Schild analysis (Lew & Angus, 1985) with PGE2 as agonist, a pKb estimate of 7.9±0.4 was generated with a regression slope of 1.2 (0.3–4.5; n=4). The requirements of simple competitive antagonism were not violated. In a separate study where the affinity of GW627368X was measured using both PGE2 and 11-deoxy-PGE1 as agonists, similar pKb estimates were obtained (data not shown).
Figure 4.

Antagonism of PGE2 concentration–effect (E/[A]) curves in HEK293 cells expressing recombinant human prostanoid EP4 receptors by GW627368X. (a) Mean PGE2 E/[A] curves in the presence of vehicle, and GW627368X 30, 100, 300 nM, 1, 3 and 10 μM n=3. (b) Schild plot using data displayed in (a). pKb and slope values quoted were generated by nonlinear regression as described in Methods.
Functional assays at other human recombinant prostanoid receptors
Functional assays in CHO cells were developed for hCRTH2, hEP1, hEP2, hEP3, hFP and hIP receptors. Mock-transfected CHO cells were devoid of responses to PGD2, PGE2 and PGF2α up to 10 μM agonist concentration, and to iloprost up to 1 μM agonist concentration. The natural agonists produced monophasic E/[A] curves at each receptor with Hill slopes in the range 0.7–1.6. Agonist potencies are shown in Table 3.
Table 3.
The properties of GW627368X in functional assays at human recombinant prostanoid receptors (n=3–9)
|
Recombinant systems |
CRTH2 |
EP1 |
EP2 |
EP3I |
EP4 |
FP |
IP |
|---|---|---|---|---|---|---|---|
| GW627368X pA2/pIC501/pKb2 | <5.41 | 6.0 (n=2) | <5.0 | <5.01 | 7.9±0.22 | <4.5 | 5.6±0.8 |
| GW627368X max effect as % standard |
— |
0 |
43±9 |
— |
−5±1.8 |
0 |
5±2 |
| Standard agonist pEC50 ipo GW627368X |
— |
7.1±0.2 |
7.4±0.3 |
— |
7.3±0.3 |
7.1±0.3 |
8.9±0.7 |
| Standard agonist max effect i.p.o. GW627368X as % control (=100) |
— |
83±21 |
95±32 |
— |
103±25 |
46±9 |
155±53 |
| % Inhibition vs standard agonist EC80 |
12 |
— |
— |
37 |
— |
— |
— |
| | |||||||
| Cell type |
CHO |
CHO |
CHO |
CHO |
HEK |
CHO |
CHO |
| Readout |
Calcium flux (FLIPR) |
CRE-Luc reporter |
cAMP |
Calcium flux (FLIPR) |
cAMP |
Calcium flux (FLIPR) |
cAMP |
| Antagonist concentration (μM) |
10 |
10 |
10 |
10 |
10 |
30 |
10 |
| Standard agonist |
PGD2 |
PGE2 |
PGE2 |
PGE2 |
PGE2 |
PGF2α |
Iloprost |
| Standard agonist pEC50 | 7.6±0.1 | 8.7±0.2 | 7.5±0.3 | 8.4±0.2 | 10.3±0.1 | 7.2±0.3 | 9.8±0.2 |
ipo: in the presence of.
Data are mean±s.d.
GW627368X was devoid of agonist action at hEP1, hEP2, hFP & hIP receptors (Table 3). Furthermore, as an antagonist of the natural agonist at CRTH2, EP2 and EP3 receptors the compound produced no measurable effect at concentrations up to 10 μM. At prostanoid EP1 and IP receptors 10 μM compound produced rightward agonist E/[A] curve shifts resulting in pA2 estimates of 6.0 (n=2) and 5.6±0.8 (n=3), respectively. At prostanoid FP receptors, the compound produced no shift of PGF2α E/[A] curves but did depress the agonist curve asymptote by 54% at 30 μM.
Effects in piglet and rabbit saphenous veins, and in guinea-pig aorta
We have described the prostanoid receptor profile of piglet saphenous vein in a previous publication (Wilson & Giles, 2005). Briefly, relaxatory responses to PGE2 in this tissue are mediated predominantly by prostanoid EP4 receptors but, at high concentrations of PGE2, a significant population of prostanoid EP2 receptors can be detected. When precontracted with PE (1 μM) PGE2 produced well-sustained concentration-related relaxations which resulted in monophasic E/[A] curves (pEC50=8.7±0.5; nH=2.0 (0.9–4.2); α=104.1±2.7%; Figure 5).
Figure 5.

Antagonism of PGE2 concentration–effect (E/[A]) curves in piglet saphenous vein by GW627368X. (a) Mean PGE2 E/[A] curves in the presence of vehicle, and GW627368X 1, 3, 10, 30, 100, 300 nM, 1 and 3 μM (n=3). Tissues were incubated with antagonist for 60 min prior to exposure to PGE2 as described in Methods. (b) Schild plot using data displayed in (a). pKb and slope values quoted were generated by nonlinear regression as described in Methods. Data have been published previously in Wilson & Giles (2005).
In this EP4 receptor assay, GW627368X did not produce any change in basal or in PE-elevated tone (Table 4). However, occasional ‘spikes' of contraction were observed in 36% of antagonist-treated tissues (Wilson & Giles, 2005). In keeping with the requirements of simple competitive antagonism, parallel rightward displacement of PGE2 E/[A] curves was achieved with GW627368X (1–300 nM) yielding a pKb estimate of 9.2±0.2 with a slope of 0.8±0.3 (Figure 5, previously published in Wilson & Giles, 2005). No further rightward shift was obtained at concentrations of GW627368X above 300 nM, presumably due to activation of the EP2 receptor by PGE2.
Table 4.
The properties of GW627368X in functional isolated tissue bioassays at prostanoid receptors (n=3)
|
Receptor |
EP2 |
EP4 |
TP |
|---|---|---|---|
| Agonist | PGE2 | PGE2 | U46619 |
| Rabbit saphenous vein |
<5.0 |
— |
<5.0 |
| Piglet saphenous vein |
— |
9.2±0.1a |
<5.0 |
| Guinea-pig aorta |
— |
— |
<5.0 |
| Washed human platelets |
— |
— |
0% inhibition at 1 μM |
| 100% inhibition at 10 μM |
Activity at prostanoid EP4 receptors in piglet saphenous vein has been previously published (Wilson & Giles, 2005) but is included here for completeness. Values are pA2 excepta=pKb.
In the same tissue, the TP receptor agonist U-46619 produced concentration-related elevations of tissue tone resulting in monophasic E/[A] curves (pEC50=7.0±0.5; nH=3.6 (1.7–7.4), α=134±7). GW627368X (10 μM) did not alter U-46619 curve potency or maximum effect.
In rabbit saphenous vein the most potent response to PGE2 is via the EP2 receptor (Lydford et al., 1996). In PE (1 μM) precontracted rings of rabbit saphenous vein PGE2 produced well-sustained concentration-related relaxations which resulted in monophasic E/[A] curves (pEC50=8.99±0.2; nH=1.1 (0.8–1.4); α=102±3). GW627368X (10 μM) did not alter the PGE2 curve potency or maximum effect, nor did it produce any effect on PE induced tissue contraction. Similarly, the compound was devoid of activity vs U46619 induced contraction (TP receptor assay) in this tissue and also in guinea-pig aorta. Data are summarised in Table 4.
TP-mediated human platelet aggregation
GW627368X did produce antagonism of U-46619 induced washed human platelet aggregation (Table 4 and Figure 6). The antagonism was of an all-or-nothing nature, being 0% inhibition at 1 μM compound and 100% at 10 μM compound vs an EC100 of agonist (100–300 nM).
Figure 6.

(a) Representative platelet aggregometry tracings showing the effect of GW627368X (0.1–10 μM) on human platelet aggregation induced by the prostanoid TP receptor agonist U-46619 (EC100). Arrows indicate the time of U-46619 addition. Similar results were obtained with three donors. (b) Aggregometry data expressed as mean±s.e.m. and analysed by one-way ANOVA with subsequent post hoc comparison (Bonferroni); *** P<0.005.
Discussion
We have demonstrated that GW627368X is a potent and selective antagonist of human and porcine prostanoid EP4 receptors with additional affinity for human, but not porcine, prostanoid TP receptors. Furthermore, we have shown that it is possible to clone and express the nine established human prostanoid receptors in assay formats commensurate with the requirements of modern medium throughput, automated drug discovery. It is not our intention to provide a review of the binding and functional pharmacology of each receptor subtype we have used since this has been described extensively in the literature (for reviews see Coleman et al., 1994; Narumiya et al., 1999; Tsuboi et al., 2002). However, certain aspects of the characterisation of our assay systems do require discussion.
A wide body of literature has described the affinity of tritiated radioligands for prostanoid receptors and the Kd estimates we have generated by saturation binding agree well with those already published except for the prostanoid EP3 and DP receptor subtypes. At least eight human splice variants of prostanoid EP3 receptors exist (Kotani et al., 1995). We have cloned and expressed the EP3I (EP3A) variant to which [3H]-PGE2 bound with an affinity two- to 10-fold lower than values previously reported (6 nM, present study vs 0.6–2.6 nM (Yang et al., 1994; Abramovitz et al., 1995, respectively)). Pharmacological characterisation of our prostanoid EP3I clone produced an identical rank order of compound affinities to that previously reported by Adam et al. (1994) and Abramovitz et al. (2000) at the prostanoid EP3C receptor (data not shown). The reason for the difference in PGE2-binding affinity is therefore unclear. The most obvious difference between our methodology and that employed by other authors is our choice of the SFV-1/EP3I and CHO cell expression system for this receptor. SFV expression systems are known to generate large amounts of the target protein and to decrease synthesis of normal cellular constituents resulting in marked cytotoxicity (Lundstrom, 2002). Therefore, we may have unintentionally reduced synthesis of accessory proteins needed for high affinity binding of agonist radioligands to receptors. Alternatively, the lower PGE2 affinity we observed may have reflected an aspect of our assay methodology. For example, other authors have used alternative buffer systems, often at lower pH. The method we present here was optimised to generate the highest agonist affinity that we could achieve. In our hands there was little difference in PGE2-binding affinity when buffers of pH 7.4 and 6.0 were used and, arguably, data generated at pH 7.4 is more relevant to physiological situations.
Similarly, in contrast to the findings of Wright et al. (1998), [3H]-PGD2 bound to a single population of prostanoid DP receptors, with a Kd of 11.9 nM and a Bmax estimate of 12.3 pmol mg−1 protein. In the earlier study, biphasic radioligand binding resulted in the identification of high (Kd=0.5 nM, Bmax=0.3 pmol mg−1 protein) and low (Kd=5.9 nM, Bmax=13.4 pmol mg−1) affinity receptor states. Our data are most consistent with detection of the low affinity receptor state reported by Wright et al. but does not agree with the findings of Boie et al. (1995) who reported binding to a single receptor population with Kd of 1.5 nM and a Bmax of 1.2 pmol mg−1 protein. Again, this disagreement may relate to the amount of receptor protein expressed since no alteration in the sequence of our receptor clone could be detected.
It is interesting to note that the affinity of GW627368X at prostanoid hEP4 receptors measured in a radioligand-binding assay (7.0±0.3) is almost 10-fold lower than its antagonist affinity for the same receptor type determined in the functional cAMP assay (7.9). According to the two-state model of receptor behaviour (Leff, 1996), receptors are predicted to adopt one of two macroscopic conformations depending upon their activation state: activated or R* receptors (high agonist affinity) and inactivated or R receptors (low affinity for agonists). A dynamic equilibrium should exist between R and R* receptor states which gives rise to complex agonist-binding curves. We observed monophasic saturation binding of the radioligand to the human prostanoid EP4 receptors, and monophasic competition-binding curves with both unlabelled agonist (PGE2) and antagonist (GW627368X) in competition-binding experiments, in each case with slope values close to unity. Therefore, we assume that the experimental conditions we have employed have resulted in the labelling of receptors predominantly in a single affinity state. The low PGE2-binding affinity and lack of binding curve complexity we observed is suggestive of R state receptors: displacement by GW627368X should therefore have generated a pKi value in closer agreement with the functional pKb, also predicted to be the result of interaction with R state receptors. The explanation for the difference we observed is unclear, but we speculate that it may be due to the difference in the microenvironment of the receptor in our membrane-binding assays compared with a whole cell or tissue setting. In competition-binding studies, GW627368X does not discriminate between prostanoid EP4 and TP receptors having pKi values of 7.0 and 6.9, respectively. Radioligand displacement curves at these receptors had unit slopes and therefore conformed with the expectations of competition for a single binding site. Curve slopes such as these can also be generated by certain specialised forms of allosteric interaction (Birdsall et al., 1995; Lazareno & Birdsall, 1995). However, in this case, allosterism is unlikely to be occurring because functional data at prostanoid EP4 receptors (see below) is also consistent with simple competition. At other prostanoid receptors, GW627368X (10 μM) produced little or no radioligand displacement and is therefore about 75-fold selective over these receptors.
In functional assays, the potency of PGD2 at prostanoid hCRTH2 receptors, of PGE2 at hEP1, hEP2 hEP3 and hEP4 receptors, of PGF2α at FP receptors and of iloprost at IP receptors was in general agreement with published potency figures for these agonists. In most cases, the true level of receptor expression is unknown and so it is impossible to compare the ability of our functional cell lines to transduce agonist-binding signals into effect in a more meaningful manner. However, agonist rank orders of potency, and antagonist affinities, where available, were appropriate for each receptor subtype.
Competition analysis of GW627368X vs PGE2 in HEK cells expressing human prostanoid EP4 receptors and in rings of PSV in vitro have demonstrated that GW627368X at concentrations up to 300 nM is a competitive antagonist of prostanoid EP4 receptors. The affinity of GW627368X for human recombinant prostanoid EP4 receptors was at least 10-fold less than its affinity for the porcine prostanoid EP4 receptor. This is likely to be a reflection of interspecies differences in the molecular structure of the human and porcine receptors. However, we have been unable to find published sequence information for the porcine prostanoid EP4 receptor so we are unable to comment on specific amino-acid residue differences that may underlie the observed difference in affinity. Clearly, though, these data highlight specific areas of pharmacological behaviour that differ between human and porcine prostanoid EP4 receptors. This may be of particular importance in models relevant to the treatment of human heart conditions (for review see Hughes et al., 2003) where extensive use of porcine tissues is made. At concentrations of GW627368X of 1 mM and above, no further shift of PGE2 E/[A] curves was achieved resulting in dose ratios of zero. This complexity has been discussed by us in an earlier publication (Wilson & Giles, 2005) and is due to the presence of a prostanoid EP2 receptor population in this tissue, a finding confirmed by Jones & Chan (2005). Agonist independence of antagonist potency was demonstrated using 11-deoxy-PGE1 as agonist: in this study, identical pKb's were obtained irrespective of the agonist used, thus confirming the competitive nature of the interaction of GW627368X with prostanoid EP4 receptors.
The observed concentration-related decreases in basal cAMP generation produced in hEP4 HEK cells by GW627368X deserve some consideration. Responses such as these may have arisen through one (or more) of several mechanisms: (1) action at a cellular target unrelated to receptors but resulting in reduced cAMP production, for example inhibition of adenylate cyclase; (2) agonism at receptors coupled through Gαi to inhibition of adenylate cyclase (functional antagonism); (3) antagonism of endogenously synthesised PGE2 acting at prostanoid EP4 receptors or (4) inverse agonism at prostanoid EP4 receptors. Non receptor-mediated activity in the molecule would be expected to reveal itself as complexity in the analyses of competition. Since our Schild plots, generated in two very different assay systems, were linear and revealed no such complexity, this seems an unlikely explanation. Similarly, our characterisation data show that GW627368X is devoid of agonist activity at Gαi- and Gαq- coupled prostanoid receptors, and essentially devoid of antagonist activity at other Gαs coupled prostanoid receptors (the pA2 of 5.6 at prostanoid IP receptors we regard as an artefact of the increased iloprost maximum effect in the presence of GW627368X). While this does not rule out an effect at an endogenously expressed non prostanoid receptor, we can at least say that an action via an endogenous prostanoid receptor seems unlikely. Furthermore, the absence of responses on untransfected HEK cells suggests that the decreases in basal cAMP are linked to the expression of recombinant human prostanoid EP4 receptors. Interestingly, we observed that the COX1/2 inhibitor, indomethacin markedly altered the behaviour of both PGE2 and GW627368X in hEP4 HEK cells. The concentration of indomethacin we used (3 μM) was around 1000- and 500-fold greater than its IC50 at COX-1 and COX-2, respectively, measured in human cell-based functional assays (Palomer et al., 2002) and we would expect to achieve near total inhibition of COX1/2 enzyme activity under these conditions. Thus, we have shown that HEK cells synthesise significant amounts of PGE2 and it therefore follows that the reductions in basal cAMP seen with GW627368X treatment are largely due to inhibition of endogenously synthesised PGE2 activity. However, even in the presence of indomethacin, we still observed small decreases in basal cAMP generation in response to GW627368X suggesting that the antagonist is able to inhibit cAMP turnover by another mechanism. It is possible that more effective inhibition at all COX enzyme subtypes might totally abolish the effects of GW627368X on basal cAMP turnover but given the level of enzyme occupancy we achieved this seems unlikely. Basal effects of GW627368X were also observed in PSV treated with indomethacin. No obviously compound-related changes in basal tone were observed but spontaneous ‘spikes' of contractile activity were observed in 18/54 tissue rings treated with GW627368X possibly indicating the removal of prorelaxatory tone by the antagonist and suggestive of basal prostanoid EP4 receptor activation. Therefore, though our data does not prove that GW627368X is an inverse agonist at human prostanoid EP4 receptors, we cannot rule out this possibility.
The compound also produced considerable antagonism of human platelet aggregation induced with the TP receptor agonist, U-46619, with no direct stimulation of aggregation in its own right. U-46619 produces characteristically steep E/[A] curves in platelet aggregation studies, and thus the ‘on/off' nature of the antagonism observed is not unexpected given the antagonist concentrations used. The concentration of agonist selected, an EC100, would be expected to shift the antagonist pIC50 by c.0.95 log units relative to its true affinity. Assuming a unit agonist E/[A] curve slope and applying a modified Cheng–Prusoff correction to the binding pKi of 6.9, the approximate IC50 for GW627368X of between 6.0 and 5.0 is commensurate with an action at the prostanoid TP receptor. However, affinity for TP receptors appears to be limited only to human receptors since in functional assays using isolated animal tissues, GW627368X was devoid of activity at rabbit, piglet and guinea-pig TP receptors.
In functional assays we have shown that GW627368X is devoid of both agonism and antagonist affinity for prostanoid CRTH2, EP2, EP3, IP and FP receptors. At prostanoid EP1 receptors, GW627368X was an antagonist with a pA2 of 6.0, and at prostanoid IP receptors the compound increased the maximum effect of iloprost by 55%. The latter effect does not seem related to direct receptor activation since GW627368X had no effect on cAMP levels in the absence of iloprost. Similarly, an effect on adenylate cyclase or on phosphodiesterase enzymes seems unlikely since complexities of behaviour were not observed either by Schild analysis or in the prostanoid EP2 receptor assay, also performed in CHO cells. Furthermore, high selectivity over rabbit, guinea-pig and piglet prostanoid IP receptors has now been demonstrated elsewhere (Jones & Chan, 2005). These authors interpreted the increased maximum effect of another prostanoid IP receptor agonist, taprostene, in the presence of GW627368X as a ‘breakthrough effect' of the agonist (where the concentration of agonist is sufficient to overcome receptor blockade by GW627368X) at prostanoid EP4 receptors in mixed receptor population tissue strips. CHO cells have been shown to possess endogenously expressed prostanoid EP4 receptors which could also give rise to a breakthrough effect (Crider et al., 2000). However, E/[A] curves were monophasic in the presence and absence of GW627368X, and the effect observed was to increase the maximum response compared to that in the absence of antagonist where presumably any prostanoid EP4 receptor-mediated responses would still contribute. We also observed no effect of PGE2 on untransfected CHO cells (data not shown) so a breakthrough effect seems unlikely and we can offer no explanation for this phenomenon. At human prostanoid FP receptors, the compound produced no shift of PGF2α E/[A] curves but did depress the agonist curve asymptote by 54% at 30 μM. This effect was not concentration related and therefore is unlikely to be a receptor-mediated event. Overall, therefore, GW627368X is 100-fold selective for prostanoid EP4 receptors over other human prostanoid receptors in functional assays, except for prostanoid TP receptors.
We have demonstrated that GW627368X is a potent, competitive antagonist of prostanoid EP4 receptors with equal binding affinity, but differential functional affinity, for hEP4 and hTP receptors and, with the exception of human prostanoid TP receptors, is at least 100-fold selective over other human prostanoid receptors. The utility of GW627368X therefore lies in its application as a tool prostanoid EP4/TP receptor antagonist in a range of experimental settings, as exemplified in the work of Jones & Chan (2005). High affinity and selectivity for prostanoid EP4/TP receptors of this order is a significant advance over the classical prostanoid EP4 receptor antagonists AH23848 and AH22921 (Brittain et al., 1985; Coleman et al., 1994; 1995) and puts GW627368X into the class of recently discovered antagonists with high affinity and selectivity for prostanoid EP4 receptors (see Table 5 for brief comparison). Of these molecules, only EP4A can rightly claim to be antagonist at human prostanoid EP4 receptors, having a pKb of 8.4 determined in human middle cerebral artery. Furthermore, EP4A and GW627368X are the only molecules for which a competitive, or indeed, receptor-mediated, antagonism has been demonstrated. However, in addition to being a prostanoid EP4 receptor antagonist, EP4A possesses significant TP and EP3 receptor-binding affinity and as such appears to be less selective than our molecule. Indeed, as we have demonstrated in this paper, functional assays are often more revealing than binding assays, and so the full selectivity profile of EP4A has not yet been firmly established. Our molecule has a pKb of 7.9±0.2 (mean±s.d.) vs human prostanoid EP4 receptors and of 9.2±0.1 (mean±s.d.) vs porcine receptors, therefore GW627368X is one of the most potent and selective human prostanoid EP4 receptor antagonists on record. Finally, while considerable doubt remains over the inverse agonist properties of GW627368X, this aspect of the compound's behaviour may confer additional utility.
Table 5.
Key pharmacological features of recently discovered potent and selective prostanoid EP4 receptor antagonists
| |
Binding |
Functional |
|||||
|---|---|---|---|---|---|---|---|
| Human pKi | Rodent pKi | Human EP4 pKb | Human selectivity | Animal EP4 pKb | Animal EP4 pIC50 | Animal selectivity | |
| GW627368X |
EP4 7.0 |
— |
7.9 |
EP1 pA2 6.0 |
9.2 (porcine) |
— |
TP <5.0 rabbit and guinea-pig |
| |
TP 6.8 |
|
Competitive |
DP not tested |
⩾8.7 (rabbit)a |
|
|
| |
Other <5.3 |
|
Inverse agonist? |
TP pIC50 5.0–6.0 |
|
|
|
| |
|
|
|
Other <5.6 |
|
|
|
| EP4Ab |
EP4 7.6 |
EP4 7.5 (rat) |
8.4c |
— |
— |
7.0 (rat) |
— |
| |
EP3 5.7 |
|
Competitive |
|
|
|
|
| |
TP 6.2 |
|
|
|
|
|
|
| |
Other <5.3 |
|
|
|
|
|
|
| ONO-AE2-227d |
— |
EP4 8.5 (murine) |
— |
— |
— |
8.0 (murine) |
— |
| |
|
EP3 7.7 (murine) |
|
|
|
|
|
| |
|
Other <5.5 (murine) |
|
|
|
|
|
| Compound 8e |
EP4 9.5 |
EP4 9.1 (rat) |
— |
— |
— |
>8.0 (murine) |
— |
| |
Other <5.5 |
|
|
|
|
|
|
| Compound 11e |
EP4 9.0 |
EP4 8.3 (rat) |
— |
— |
— |
7.0–8.0 (murine) |
— |
| Other <5.0 | |||||||
Systematic compound names are given in Abbreviations.
Data taken from Jones & Chan (2005).
Data taken from Machwate et al. (2001).
Data taken from Davis et al. (2004).
Data taken from Mutoh et al. (2002).
Data taken from Hattori et al. (2005).
Abbreviations
- BHK
Syrian Hamster Kidney
- cAMP
cyclic adenosine monophosphate
- CHO
Chinese Hamster Ovary
- compound 8
3-{[(1S)-2-(4,5-diphenyl-1,3-oxazol-2-yl)-2-cyclohexen-1-yl]methyl}-N-[(phenylmethyl)sulfonyl]benzamide
- compound 11
6-[(N∼2∼-(1-benzofuran-2-ylcarbonyl)-N∼5∼{[(phenylmethyl)oxy]carbonyl}-L-ornithyl)amino]hexanoic acid
- DMEM-F12
Dulbecco's modified Eagle medium-Ham F12 mix
- DMSO
dimethyl sulphoxide
- E/[A]
concentration–effect curve
- EDTA
ethylenediaminetetra-acetic acid
- EP4A
(4′-[3-butyl-5-oxo-1-(2-trifluoromethyl-phenyl)-1,5-dihydro-[1,2,4]triazol-4-ylmethyl]-biphenyl-2-sulfonic acid (3-methyl-thiophene-2-carbonyl)-amide
- GW627368X
N-{2-[4-(4,9-diethoxy-1-oxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl) phenyl]acetyl}benzene sulphonamide
- HBSS
Hanks'-buffered saline solution
- HEK
human embryonic kidney 293 cells
- HEPES
N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid]
- HI-FBS
heat-inactivated foetal bovine serum
- HMTB
HEPES-modified Tyrode's buffer
- IBMX
isobutylmethylxanthine
- n.s.b.
non-specific binding
- ONO-AE2-227
2-[2-{2-(1-naphthyl)propanoylamino}phenyl]methylbenzoic acid
- PE
phenylephrine
- PG
prostaglandin
- PMSF
phenylmethylsulphonyl fluoride
- PRP
platelet-rich plasma
- SFV
Semliki Forest Virus
- SPA
scintillation proximity assay
- WP
washed platelets
References
- ABRAMOVITZ M., ADAM M., BOIE Y., CARRIERE D.D., GODBOUT C., LAMONTAGNE S., ROCHETTE C., SAWYER N., TREMBLAY N.M., BELLEY M., GALLANT M., DUFRESNE C., GAREAU Y., RUEL R., JUTEAU H., LABELLE M., OUIMET N., METTERS K. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim. Biophys. Acta. 2000;1483:285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- ABRAMOVITZ M., ADAM M., BOIE Y., GRYGORCZYK R., RUSHMORE T.H., NGUYEN T., FUNK C.D., BASTIEN L., SAWYER N., ROCHETTE C., SLIPETZ D.M., METTERS K.M. Human prostanoid receptors: cloning and characterization. Adv. Prost. Thromb. Leuk. Res. 1995;23:499–504. [PubMed] [Google Scholar]
- ADAM M., BOIE Y., RUSHMORE T.H., MULLER G., BASTIEN L., MCKEE K.T., METTERS K.M., ABRAMOVITZ M. Cloning and expression of three isoforms of the human EP3 prostanoid receptor. FEBS Letts. 1994;338:170–174. doi: 10.1016/0014-5793(94)80358-7. [DOI] [PubMed] [Google Scholar]
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BILLOT X., CHATEAUNEUF A., CHAURET N., DENIS D., GREIG G., MATHIEU M.C., METTERS K.M., SLIPETZ D.M., YOUNG R.N. Discovery of a potent and selective agonist of the prostaglandin EP4 receptor. Bioorg. Med. Chem. Letts. 2003;13:1129–1132. doi: 10.1016/s0960-894x(03)00042-8. [DOI] [PubMed] [Google Scholar]
- BIRDSALL N.J., COHEN F., LAZARENO S., MATSUI H. Allosteric regulation of G-protein-linked receptors. Biochem. Soc. Trans. 1995;23:108–111. doi: 10.1042/bst0230108. [DOI] [PubMed] [Google Scholar]
- BOIE Y., SAWYER N., SLIPETZ D.M., METTERS K.M., ABRAMOVITZ M. Molecular cloning and characterization of the human prostanoid DP receptor. J. Biol. Chem. 1995;270:18910–18916. doi: 10.1074/jbc.270.32.18910. [DOI] [PubMed] [Google Scholar]
- BRITTAIN R.T., BOUTAL L., CARTER M.C., COLEMAN R.A., COLLINGTON E.W., GEISOW H.P., HALLETT P., HORNBY E.J., HUMPHREY P.P., JACK D., KENNEDY I., LUMLEY P., MCCABE P.J., SKIDMORE I.F., THOMAS M., WALLIS C.J. AH23848: a thromboxane receptor-blocking drug that can clarify the pathophysiologic role of thromboxane A2. Circulation. 1985;72:1208–1218. doi: 10.1161/01.cir.72.6.1208. [DOI] [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- COLEMAN R.A., MALLETT A., SHELDRICK R.L.G.EP4-receptors and cyclic AMP in pig venous smooth muscle: evidence with agonists and the EP4 antagonist, AH 22921 Advances in Prostaglandin, Thromboxane and Leukotriene Research 1995New York: Raven; 241–246.eds. Samuelsson, B., Paoletti, R. & Ramwell, P. Vol. 23. pp [PubMed] [Google Scholar]
- COLEMAN R.A., SMITH W.L., NARUMIYA S. International Union of Pharmacology VIII. Classification of prostanoid receptors: properties, distribution and structure of the receptors and their subtypes. Pharmacol. Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- CRIDER J.Y., GRIFFIN B.W., SHARIF N.A. Endogenous EP4 prostaglandin receptors coupled positively to adenylyl cyclase in Chinese hamster ovary cells: pharmacological characterization. Prost. Leuk. Essent. Fatty Acids. 2000;62:21–26. doi: 10.1054/plef.1999.0120. [DOI] [PubMed] [Google Scholar]
- DAVIS R.J., MURDOCH C.E., ALI M., PURBRICK S., RAVID R., BAXTER G.S., TILFORD N., SHELDRICK R.L., CLARK K.L., COLEMAN R.A. EP4 prostanoid receptor-mediated vasodilatation of human middle cerebral arteries. Br J. Pharmacol. 2004;141:580–585. doi: 10.1038/sj.bjp.0705645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIBLIN G.M.P., WILSON R.J., FOORD S.M., SWARBRICK M., WALKER A., BAMFORD M., ROOMANS S., MASON A., MILLER N., JONES H., SHANAHAN S., RASMUSSEN S., SMITH L., SPALDING D., RANSHAW L., FENWICK R., ANCLIFF R., SAEZ V., FRYE S., STRATTON S., LEWELL X., CARTWRIGHT K.A., RHODES S., ROBERTS N., GREEN R.A Novel, selective, non-prostanoid EP4 receptor antagonist 2002. Poster at 224th ACS Natl Meet (August 18–22, 2002, Boston) Abst MEDI-306
- HATTORI K., TANAKA A., FUJI N., TAKASUGI H., TENDA Y., TOMITA M., NAKAZATO K., KATO Y., KONO Y., MURAI H., SAKANE K. Discovery of diphenyloxazole and Nδ-Z-ornithine derivatives as highly potent and selective human prostaglandin EP4 receptor antagonists. J. Med. Chem. 2005;48:3103–3106. doi: 10.1021/jm050085k. [DOI] [PubMed] [Google Scholar]
- HIRAI H., TANAKA K., YOSHIE O., OGAWA K., KENMOTSU K., TAKAMORI Y., ICHIMASA M., SUGAMURA K., NAKAMURA M., TAKANO S., NAGATA K. Prostaglandin D2 selectively induces chemotaxis in T-helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J. Exp. Med. 2001;193:225–261. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRATA T., USHIKUBI F., KAKIZUKA A., OKUMA M., NARUMIYA S. Two thromboxane A2 receptor isoforms in human platelets: opposite coupling to adenylate cyclase with different sensitivity to Arg60 to Leu mutation. J. Clin. Invest. 1994;97:949–956. doi: 10.1172/JCI118518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGHES G.C., POST M.J., SIMONS M., ANNEX B.H. Translational physiology: porcine models of human coronary artery disease: implications for preclinical trials of therapeutic angiogenesis. J. App. Physiol. 2003;94:1689–1701. doi: 10.1152/japplphysiol.00465.2002. [DOI] [PubMed] [Google Scholar]
- JONES R.L., CHAN K.M. Investigation of the agonist activity of prostacyclin analogues on prostanoid EP4 receptors using GW 627368 and taprostene: evidence for species differences. Prost. Leuk. Essent. Fatty Acids. 2005;72:289–299. doi: 10.1016/j.plefa.2004.12.002. [DOI] [PubMed] [Google Scholar]
- KOTANI M., TANAKA I., OGAWA Y., USUI T., MORI K., ICHIKAWA A., NARUMIYA S., YOSHIMI T., NAKAO K. Molecular cloning and expression of multiple isoforms of human prostaglandin E receptor EP3 Subtype generated by alternative messenger RNA splicing – multiple second messenger systems and tissue-specific distributions. Mol. Pharmacol. 1995;48:869–879. [PubMed] [Google Scholar]
- LAZARENO S., BIRDSALL N.J. Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol. Pharmacol. 1995;48:362–378. [PubMed] [Google Scholar]
- LEFF P. The two-state model of agonist action: challenges to pharmacological receptor theory. Proc. Western Pharmacol. Soc. 1996;39:67–68. [PubMed] [Google Scholar]
- LEW M.J., ANGUS J.A. Analysis of competitive agonist-antagonist interactions by nonlinear regression. Trends Pharmacol. Sci. 1985;16:328–337. doi: 10.1016/s0165-6147(00)89066-5. [DOI] [PubMed] [Google Scholar]
- LI M., KE H.Z., QI H., HEALY D.R., LI Y., CRAWFORD D.T., PARALKAR V.M., OWEN T.A., CAMERON K.O., LEFKER B.A., BROWN T.A., THOMPSON D.D. A novel, non-prostanoid EP2 receptor-selective prostaglandin E2 agonist stimulates local bone formation and enhances fracture healing. J. Bone Min. Res. 2003;18:2033–2042. doi: 10.1359/jbmr.2003.18.11.2033. [DOI] [PubMed] [Google Scholar]
- LUNDSTROM K. Semliki forest virus-based expression for versatile use in receptor research. J. Recept. Sig. Trans. 2002;22:229–240. doi: 10.1081/rrs-120014598. [DOI] [PubMed] [Google Scholar]
- LYDFORD S.J., MCKECHNIE K.C.W., LEFF P. Interaction of BW A868C, a prostanoid DP-receptor antagonist, with two receptor subtypes in the rabbit isolated saphenous vein. Prostaglandins. 1996;52:125–139. doi: 10.1016/0090-6980(96)00058-5. [DOI] [PubMed] [Google Scholar]
- MACHWATE M., HARADA S., LEU C.T., SEEDOR G., LABELLE M., GALLANT M., HUTCHINS S., LACHANCE N., SAWYER N., SLIPETZ D., METTERS K.M., RODAN S.B., YOUNG R., RODAN G.A. Prostaglandin receptor EP4 mediates the bone anabolic effects of PGE2. Mol. Pharmacol. 2001;60:36–41. doi: 10.1124/mol.60.1.36. [DOI] [PubMed] [Google Scholar]
- MARSHALL F.H., PATEL K., LUNDSTROM K., CAMACHO J., FOORD S.M., LEE M.G. Characterization of [3H]-prostaglandin E2 binding to prostaglandin EP4 receptors expressed with semliki forest virus. Br. J. Pharmacol. 1997;121:1673–1678. doi: 10.1038/sj.bjp.0701332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MODY S.M., HO M.K.C., JOSHI S.A., WONG Y.H. Incorporation of Gz-specific sequence at the carboxyl terminus increases the promiscuity of G16 toward Gi-coupled receptors. Mol. Pharmacol. 2000;57:13–23. [PubMed] [Google Scholar]
- MUTOH M., WATANABE K., KITAMURA T., SHOJI Y., TAKAHASHI M., KAWAMORI T., TANI K., KOBAYASHI M., MARUYAMA T., KOBAYASHI K., OHUCHIDA S., SUGIMOTO Y., NARUMIYA S., SUGIMURA T., WAKABAYASHI K. Involvement of prostaglandin E receptor subtype EP4 in colon carcinogenesis. Cancer Res. 2002;62:28–32. [PubMed] [Google Scholar]
- NARUMIYA S., SUGIMOTO Y., USHIKUBI F. Prostanoid receptors: structures, properties and functions. Phys. Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- OGAWA M., HARADA H., KADODE M., MATSUOKA H., MATSHUSHITA Y., SAGAWA K., YOSHIDA H., MARUYAMA T., TANI K., OHUCHIDA S., KONDO K.Development of ONO-8815, a potent and selective agonist of the prostaglandin E receptor EP2 subtype 2000. 11th International Conference on Advances in Prostaglandin and Leukotriene Research: Basic Science and New Clinical Applications. Florence, Italy, June 4th–8th, 2000
- OXFORD A.W., DAVIS R.J., COLEMAN R.A., CLARK K.L., CLARK D.E., HARRIS N.V., FENTON G., HYND G., STUTTLE K.A.J., SUTTON J.M., ASHTON M.R., BOYD E.A., BRUNTON S.A.EP2 receptor agonists 2005. WO 2005/080367 A1
- PALOMER A., CABRE F., PASCUAL J., CAMPOS J., TRUJILLO M.A., ENTRENA A., GALLO M.A., GARCIA L., MAULEON D., ESPINOSA A. Identification of novel cyclooxygenase-2 selective inhibitors using pharmacophore models. J. Med. Chem. 2002;45:1402–1411. doi: 10.1021/jm010458r. [DOI] [PubMed] [Google Scholar]
- SAKATA K., MARUYAMA T., SEKI A., YOSHIDA H., SHINAGAWA Y., KONDO K., OHUCHIDA S.The roles of PGE2 receptor EP4 subtype on cytokine regulation and its related diseases 2000. 11th International Conference on Advances in Prostaglandin and Leukotriene Research: Basic Science and New Clinical Applications. Florence, Italy, June 4th–8th, 2000
- TANI K., NAGANAWA A., ISHIDA A., KOBAYASHI K., SAGAWA K., OGAWA M., OHUCHIDA S.Synthesis and structure-activity relationships of potent and selective EP2 receptor agonists 2000. 11th International Conference on Advances in Prostaglandin and Leukotriene Research: Basic Science and New Clinical Applications. Florence, Italy, June 4th–8th, 2000
- TSUBOI K., SUGIMOTO Y., ICHIKAWA A. Prostanoid receptor subtypes. Prost. Other Lip. Med. 2002;68–69:535–556. doi: 10.1016/s0090-6980(02)00054-0. [DOI] [PubMed] [Google Scholar]
- WILSON R.J., GILES H. Piglet saphenous vein contains multiple relaxatory prostanoid receptors: evidence for EP4, EP2, DP & IP receptor subtypes. Br. J. Pharmacol. 2005;144:405–415. doi: 10.1038/sj.bjp.0706088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILSON R.J., GIBLIN G.M.P., FOORD S.M., SWARBRICK M., WALKER A., BAMFORD M., ROOMANS S., MASON A., MILLER N., JONES H., SHANAHAN S., RASMUSSEN S., SMITH L., SPALDING D., RANSHAW L., FENWICK R., ANCLIFF R., SAEZ V., FRYE S., STRATTON S., LEWELL X., CARTWRIGHT K.-A., K-A RHODES S., ROBERTS N., GREEN R. GW627368X: a Novel, potent and selective EP4 prostanoid receptor antagonist. Br. J. Pharmacol. 2003;138:84P. [Google Scholar]
- WISE H. Characterisation of chimeric prostacyclin/prostaglandin D2 receptors. Eur. J. Pharmacol. 1999;386:89–96. doi: 10.1016/s0014-2999(99)00725-6. [DOI] [PubMed] [Google Scholar]
- WRIGHT D.H., METTERS K.M., ABRAMOVITZ M., FORDHUTCHINSON A.W. Characterization of the recombinant human prostanoid DP receptor and identification of L-644,698, a novel selective DP agonist. Br. J. Pharmacol. 1998;123:1317–1324. doi: 10.1038/sj.bjp.0701708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO H., MARUYAMA T., SAKATA K., KOKETSU M., KOBAYASHI M., YOSHIDA H., SEKI A., TANI K., MARUYAMA T., KONDO K., OHUCHIDA S. Novel four selective agonists for prostaglandin E receptor subtypes. Prost. Lip. Med. 1999;59:150. [Google Scholar]
- YANG J.H., XIA M.H., GOETZL E.J., AN S.Z. Cloning and expression of the EP3-subtype of human receptors for prostaglandin E2. Biochem. Biophys. Res. Comms. 1994;198:999–1006. doi: 10.1006/bbrc.1994.1142. [DOI] [PubMed] [Google Scholar]