

Abstract
Cyclodextrins (CDs) are nanostructures widely applied in biotechnology and chemistry. Owing to partially hydrophobic character, CDs interact with biological membranes. While the mechanisms of CDs interactions with lipids were widely studied, their effects on proteins are less understood. In the present study we investigated the effects of beta cyclodextrin (βCD) on GABAA receptor (GABAAR) gating.
To reliably resolve the kinetics of conformational transitions, currents were elicited by ultrafast gamma-aminobutyric acid (GABA) applications to outside-out patches from rat cultured hippocampal neurons. βCD increased the amplitude of responses to saturating GABA concentration ([GABA]) in a dose-dependent manner and this effect was accompanied by profound alterations in the current kinetics.
Current deactivation was slowed down by βCD but this effect was biphasic with a maximum at around 0.5 mM βCD. While the fast deactivation time constant was monotonically slowed down within considered βCD concentration range, the slow component first increased and then, at millimolar βCD concentration, decreased.
The rate and extent of desensitization was decreased by βCD in a dose-dependent manner.
The analysis of current responses to nonsaturating [GABA] indicated that βCD affected the GABAAR agonist binding site by slowing down the unbinding rate.
Modulation of GABAAR desensitization and binding showed different concentration-dependence suggesting different modualtory sites with higher affinity of the latter one.
All the βCD effects were fully reversible indicating that cholesterol uptake into βCD was not the primary mechanism.
We conclude that βCD is a strong modulator of GABAAR conformational transitions.
Keywords: GABAA receptor, β cyclodextrin, patch-clamp, ultrafast perfusion system, kinetics model
Introduction
Cyclodextrins (CDs) are nanostructures that attract increasing attention as a potent tool in for example, drug delivery, molecular recognition, modeling the catalytic enzymes and enhancing solubilization of lipophilic structures in aqueous media. CDs are cyclic components containing nanocavities designed as inclusion complexes for various low molecular weight compounds (Harada, 2001; Redenti et al., 2001; Douhal, 2004). Owing to hydrophilic exterior and hydrophobic nanocavities CDs may act as efficient ‘shuttles' for hydropohobic compounds (Harada, 2001; Redenti et al., 2001; Loftsson et al., 2004). However, it is likely that due to partially hydrophobic properties, CDs could interact with various components of cellular membranes and modulate their functions. Although most studies concentrated on CD interactions with lipids, it was found that a direct CD binding to proteins may also take place (e.g. Pajatsch et al., 1998; Kamionka & Dahl, 2001). CDs were shown to block connexins by direct interaction with the channel pore (Locke et al., 2004). However, in general, the nature and impact of direct CD–protein interactions are poorly understood. The best documented mechanism whereby CDs act as potent modulators of biological membranes is depletion of cholesterol (Kilsdonk et al., 1995; Yancey et al., 1996), a compound that is known as a key regulator of several membrane properties (Brown & London, 2000; Ottico et al., 2003; Fielding & Fielding, 2004). Cholesterol exerts its modulatory functions by controlling membrane rigidity and fluidity and by acting as a key constituent of so-called membrane lipid rafts (Brown & London, 2000). Alterations in cholesterol level in the membrane were found to profoundly affect functioning of membrane proteins including ionic channels (e.g. Bennett & Simmonds, 1996; Hajdu et al., 2003; Barbuti et al., 2004; Brady et al., 2004; Frank et al., 2004; Shu et al., 2004; Xia et al., 2004; Taverna et al., 2004), indicating that lipid microenvironment of membrane proteins plays a crucial regulatory role. For instance, properties of gramicidin channels are strongly sensitive to agents influencing membrane stiffness, including cholesterol (Chen & Gross, 1995; Lundbaek et al., 2004). The activation of nicotinic acetylcholine receptors requires the presence of cholesterol in the lipid environment of this channel (Fong & McNamee 1986, Addona et al., 1998). More recently, Lundbaek et al. (1996, 2004) have demonstrated that factors affecting lipid bilayer elasticity (e.g. micelle-forming drugs or cholesterol) may strongly affect the conformational transitions of sodium and calcium channels that are crucial in neuronal excitability. Ottico et al. (2003) have studied CD effect on cultured neurons and found that even a mild CD treatment (millimols of CD applied for tens of minutes) resulted in a substantial loss of main membrane lipid compounds (phosphatidylcholine, cholesterol, sphingolipids) giving rise to possible profound reorganization of membrane lipid domains. These findings altogether indicate that CDs may exert a variety of effects leading to a direct or indirect modulation of membrane proteins through several mechanisms. In particular, conformational transitions of proteins can be modulated by a number of factors that can be altered by CDs. In the present study, we pursued this issue and investigated the effect of CDs on the kinetics of neuronal GABAA receptor conformational transitions. GABAA receptors are ligand-activated channels that are responsible for neuronal inhibition in the adult brain and their gating is relatively well understood (e.g. Macdonald et al., 1989; Jones & Westbrook, 1995; McClellan & Twyman, 1999; Mozrzymas et al., 1999, 2003a, 2003b). The effect of CD on GABAARs was studied using electrophysiological tools by Shu et al. (2004), who found that CD does not affect GABA-evoked responses but modulated slow currents activated by a steroid. However, it is likely that due to relatively slow application system, CD effects on the receptor gating could have been difficult to detect. In the present study, to monitor the receptor gating at highest possible temporal resolution, current responses were elicited by ultrafast agonist applications (Jonas, 1995). We found that βCDs (cyclic heptamers of glucose) at relatively low concentrations (at which depletion of membrane cholesterol is expected to be minor) induced profound changes in GABAA receptor gating affecting mainly desensitization and binding kinetics.
Methods
Cell culture
Primary cell culture was prepared as already described (Andjus et al., 1997). Briefly, P2–P4 old Wistar rats were killed by decapitation, hippocampi were dissected, sliced, treated with trypsin, mechanically dissociated, and centrifuged twice at 40 × g, plated in the Petri dishes and cultured. Experiments were performed on cells between 10 and 15 days in culture.
Electrophysiological recordings
Currents were recorded in the outside-out mode of the patch-clamp technique using the EPC-7 amplifier (List Medical, Darmstadt, Germany) at a holding potential of −70 mV. The intrapipette solution contained (in mM) CsCl 137, CaCl2 1, MgCl2 2, 1,2-bis(2-aminophenoxy)ethane-N,N,N′-tetraacetic acid (BAPTA) 11, ATP 2, HEPES 10 (pH 7.2 with CsOH). The composition of the standard external solution was (in mM) NaCl 137, KCl 5, CaCl2 2, MgCl2 1, glucose 20, and HEPES 10 (pH 7.2 with NaOH). Two types of cyclodextrins were used (β-cyclodextrin and (2-hydroxypropyl)-β-cyclodextrin, Sigma, Poznan, Poland) but their effects on GABAARs were indistinguishable. CDs (at concentration up to 1.5 mM) were added in powder to the external solution and within this concentration range neither osmolarity or pH was affected at a detectable level.
To reduce the data scatter due to cell-to-cell variability the description of CD effect was based on comparison of kinetic parameters (e.g. amplitudes, 10–90% rise time, time constants of desensitization and deactivation) for currents recorded from the same patch. Stable recordings (<10% of rundown) were available for approximately 10–20 min. Since current responses were recorded every 1–2 min, the impact of rundown was minimal.
All experiments were performed at room temperature 22–24°C.
The current signals were low-pass filtered at 10 kHz with a Butterworth filter and sampled at 50–100 kHz using the analog-to-digital converter CED micro1401 (Cambridge, U.K.) and stored on the computer hard disk. The acquisition and analysis software were kindly given by Dr J. Dempster (Strathclyde University, Glasgow, U.K.).
GABA was applied to excised patches using the ultrafast perfusion system based on a piezoelectric-driven theta-glass application pipette (Jonas, 1995). The piezoelectric translator was from Physik Instrumente (preloaded HVPZT translator 80 μm, Waldbronn, Germany) and theta-glass tubing from Hilgenberg (Malsfeld, Germany). The open tip recordings of the liquid junction potentials revealed that 10–90% exchange of solution occurred within 40–80 μs. A minimum duration of drug application was 1 ms (when applying shorter pulses, often oscillations appeared). In experiments, in which, the effect of cyclodextrin was tested, this substance was present at the same concentrations in solutions supplied by both channels (wash and GABA-containing solution) of the theta-glass capillary. Before applying the agonist (in the presence or absence of βCD) the patch was exposed to the flux of washing solution for at least 2 min.
Analysis
The decay of the currents was fitted with a function in the form:
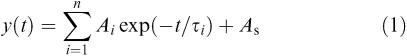 |
where, Ai are the fractions of respective components, As is the steady-state current and τi are the time constants. For normalized currents, ∑Ai+As=1. Deactivation time course was well fitted with a sum of two exponentials (n=2) and As=0. The weighted time constant of deactivation was calculated using the following formula: τmean=∑Aiτi. For the protocols aiming at description of desensitization onset (long applications of saturating GABA concentration) decaying phases of the currents were fitted with either one or two exponentials and As>0.
The recovery process in the double pulse protocol was estimated using the parameter defined as follows:
where R is the percentage recovery, I1 the first peak amplitude, Iend, the current value immediately before the application of the second pulse, I2, the second peak amplitude. The kinetic modelling was performed with the ChannelLab 2.0 software (developed by S. Traynelis for Synaptosoft, Decatour, GA, U.S.A.). This software converted the kinetic model (Figure 7) into a set of differential equations and solved them numerically assuming, as the initial condition, that at t=0, no bound or open receptors were present. The solution of such equations yielded the time courses of occupancies of all the states included in the model. The current time course was modelled as the time evolution of the sum of open state occupancies.
Figure 7.

Model simulations of βCD effect on the current responses to rapid applications of GABA. (a) Jones and Westbrook's model of GABAA receptor gating (Jones et al. 1998). Columns represent simulations of current responses evoked using various protocols in control conditions (left column), at 500 μM βCD (middle) and at 1500 μM βCD (right). For control conditions the following set of rate constants was used: kon=6 ms−1 mM−1, koff=0.8 ms−1, β2=3 ms−1, α2=0.5 ms−1, β1=0.0015 ms−1, α1=1.5 ms−1, d2=12 ms−1, r2=0.07 ms−1, d1=0.014 ms−1, r1=0.0015 ms−1, P=0.004 ms−1, q=0.0005 ms−1 mM−1. For 500 μM βCD: kon=6 ms−1 mM−1, koff=0.3 ms−1, β2=3 ms−1, α2=0.5 ms−1, β1=0.0015 ms−1, α1=1.5 ms−1, d2=8 ms−1, r2=0.12 ms−2, d1=0.014 ms−1, r1=0.0015 ms−1, P=0.001 ms−1, q=0.0005 ms−1 mM−1. For 1500 μM βCD: kon=6 ms−1mM−1, koff=0.3 ms−1, β2=3 ms−1, α2=0.5 ms−1, β1=0.0015 ms−1, α1=1.5 ms−1, d2=6.5 ms−1, r2=0.135 ms−1, d1=0.014 ms−1, r1=0.0015 ms−1, P=0.001 ms−1, q=0.0005 ms−1 mM−1. (b–d) Simulated current responses to brief applications of saturating [GABA] (10 mM) in control conditions (b) in the presence of 500 μM βCD (c) and in 1500 μM βCD (c). The increase in amplitude (compare to Figure 1) as well as slow down in the deactivation kinetics (compare to Figure 3) in the presence of βCD is clearly reproduced. (e–g) Simulated current responses to long applications of saturating [GABA] (10 mM) in control conditions (e) in 500 μM βCD (f) and in 1500 μM βCD (g). Decrease in the rate and extent of desensitization) in the presence of βCD is properly reproduced (compare to Figure 4). (h–j) Simulated current responses to paired pulses of brief and saturating [GABA] (10 mM) in control conditions (h) in 500 μM βCD (i) and in 1500 μM βCD (j). Acceleration of the recovery in the presence of βCD is reproduced (compare to Figure 5). (k–m) Simulated current responses to applications of nonsaturating [GABA] (10 μM) in control conditions (k) in 500 μM βCD (l) and in 1500 μM βCD (m). Note that, in agreement with experimental data, increase in amplitude in the presence of βCD is clearly larger than in the case of saturating [GABA] (compare Figures 1, 6 and Figure 7b–d and k–m). Moreover, in agreement with experimental evidence (Figure 6b) the increase in current amplitude at 500 μM βCD is predicted to be large while further increase in βCD concentration produces only a minor effect (compare l and m).
Data are expressed as mean±s.e.m. and for comparison of data obtained from the same patch Student's paired t-test was used.
Results
In order to study the effect of βCD on conformational transitions of GABAA receptors, current responses to ultrafast applications of GABA were recorded. At sufficiently high (saturating) GABA concentrations, the occupancy of open receptors as well as activation rate reach their maximum values. When applying saturating agonist concentration, the receptors bind the agonist very quickly and the time course of current response is expected to be governed by transition rates between fully bound receptor conformations. Thus, the time course of current responses to saturating [GABA] have been found to be very sensitive to modulatory processes affecting these conformational transitions (e.g. Jones & Westbrook, 1997; Mozrzymas et al., 1999, 2003a, 2003b). Taking this into account, we measured the current responses to a saturating (10 mM) GABA concentration in control conditions and in the presence of βCD (Figure 1). As explained in Methods, in order to avoid excessive data scatter due to cell-to-cell variability, control currents and responses in the presence of βCD were recorded from the same patch. Surprisingly, current responses to 10 mM GABA in the presence of βCD had clearly larger amplitudes than respective controls and this effect was dose-dependent and highly significant (Figure 1a and b, P<0.05, paired t-test). In control conditions, the averaged amplitude of current responses to saturating [GABA] (at −70 mV) was 1133±163 pA, n=28. The effect of βCD on current amplitude was fully reversible within the considered βCD concentration range (in Figure 1a, an example is shown for 0.5 mM βCD). The minimum period of time between the test pulse (in the presence of βCD) and control one was at least 2 min (see Methods) to avoid accumulation of receptors in the desensitized state. After this time interval, current responses returned to the control level (Figure 1a), implying that 2 min were sufficient for reversal of βCD effects. The recovery from βCD-induced modulation is probably even faster but due to overlap with receptor desensitization, it is difficult to be precisely assessed. Successive GABA applications (separated by at least 2 min) in the continuous presence of βCD, elicited current responses with the same amplitude, indicating that βCD-induced modulation of GABAARs equilibrated within at most 2 min (data not shown). While the overall trend of βCD action was clear and robust, its effect both on amplitudes (Figure 1) and on current time course was characterized by a substantial cell-to-cell variability (e.g. increase in amplitude at 1.5 mM βCD ranged from 10 to 210% with a mean 168±11%, n=5, Figure 1b). βCD by itself did not elicit any detectable current.
Figure 1.

βCD enhances current responses to saturating [GABA]. (a) Typical consecutive current responses to 10 mM GABA in control conditions (left) in the presence of 0.5 mM βCD (middle) and in control (right). Note that control responses before and after recording in the presence of βCD are identical indicating that βCD effect is reversible. Insets above current traces indicate the concentration and time course of applied agonist. (b) Statistics showing dose-dependence of βCD effect on current amplitudes. Statistics was based on paired comparisons vs. control response for at least n=5 patches for each βCD concentration. Asterisks above bars indicate statistically significant effects.
In order to explore the mechanism underlying βCD effect on amplitudes of current responses to saturating [GABA] (Figure 1), the time course of currents elicited using various application protocols was analyzed. The onset rate of currents evoked by saturating [GABA] was slowed down by βCD but this effect was relatively weak reaching significance only at highest (1.5 mM) βCD concentration (in control conditions 10–90% rise time was 0.16± 0.01 ms, n=5, and in the presence of 1.5 mM βCD 0.23±0.01 ms, n=5, P<0.05, paired t-test, Figure 2). It is known, that the kinetics of the rising phase of currents (including those elicited by saturating agonist) can be controlled by several processes including opening/closing and desensitization rates (e.g. Mozrzymas et al., 2003a, 2003b). Thus, in order to further elucidate the mechanisms of βCD action on GABAA receptor gating, application of additional experimental protocols was required.
Figure 2.

βCD slows down the onset kinetics of current responses elicited by saturating [GABA]. (a) Typical normalized and superimposed current responses to 10 mM GABA in control conditions (thick line) and in the presence of 1.5 mM βCD (thin line). Inset above current traces indicate the concentration and time course of applied agonist. (b) Statistics of βCD effect on the 10–90% rise time. Asterisk above bar indicates a statistically significant effect.
The synaptic agonist transient is believed to be very short lasting (hundreds of microseconds, Clements, 1996; Mozrzymas et al., 1999, 2003a; Mozrzymas, 2004) and therefore the decaying phase of synaptic currents is thought to reflect mainly the deactivation process (current time course following removal of free agonist). In our experiments, deactivation process was studied by analyzing the decaying phase of currents elicited by brief (1–3 ms) applications of saturating (10 mM) GABA. Variation in pulse duration within 1–3 ms did not affect the time course of deactivation process (data not shown). In control conditions, the current decay could be well described by two exponential components (τfast=2.8±0.14 ms, τslow=113.9±8.8 ms, Aslow=0.15±0.02, n=15) similarly to what observed in previous studies (e.g. Jones & Westbrook, 1995; Mozrzymas et al., 1999; 2003a, 2003b). In the presence of βCD, the deactivation kinetics was clearly altered (Figure 3). The mean decay time constant (τmean) strongly increased in the presence of βCD but this effect was not monotonic (Figure 3b). Indeed, as shown in Figure 3b, τmean showed a strong increase for βCD up to 500 μM but at higher concentration, the mean time constant clearly decreased. While the fast component of deactivation (τfast) showed a monotonic increase with βCD concentration (Figure 3c), the slow one (τslow) showed an increase for βCD up to 500 μM but above this concentration this trend was reversed (Figure 3d) and the percentage of this component showed a monotonic increase with βCD concentration (Figure 3e). A pronounced increase in the amplitude (Figure 1) combined with prolonged deactivation (Figure 3b) gave rise to a strong increase in the charge transfer but for high (1.5 mM) βCD concentration this trend was reversed (Figure 3f) similarly to what observed in the case of slow deactivation component (Figure 3d). The effects of βCD on the deactivation kinetics were fully reversible (data not shown).
Figure 3.

βCD strongly affects the deactivation kinetics of currents evoked by saturating [GABA]. (a) Typical normalized and superimposed current responses to 10 mM GABA in control conditions (thick line) and in the presence of 1.5 mM βCD (thin line). Inset above current traces indicate the concentration and time course of applied agonist. (b) Statistics of βCD effect on weighted average deactivation time constant (τmean). Note that the βCD effect on τmean is biphasic (increase in τmean is larger at 500 μM than at 1.5 mM βCD). In (c and d) Statistics of βCD effect on the fast (τfast) and slow (τslow) deactivation component are shown, respectively. Note that while βCD effect on τfast is monotonic, in the case of τslow it appears biphasic. (e) Dose-dependence of βCD effect on the percentage of the slow component (Aslow). (f) Statistics of βCD effect on charge transfer of current responses to 10 mM GABA. Asterisks above bars indicate statistically significant effects.
Deactivation kinetics was shown to strongly depend on desensitization (Jones & Westbrook, 1995) and it is thus interesting to check for the effect of βCD on this process. The kinetics of desensitization was studied by recording the current responses to prolonged (up to 100 ms) applications of saturating [GABA]. Long GABA applications resulted in appearance of slow desensitization components (e.g. at 300 ms application the current fading was clearly described by two components τfast=2.41±0.17 ms, τslow=126±7.5 ms, n=16, not shown). However, at sufficiently short GABA applications (20–50 ms) the desensitization onset could be fairly well described by one, fast exponential component. It is believed that such fast desensitization component is strongly involved in shaping the synaptic currents while the impact of slower ones on mIPSCs is negligible (Jones & Westbrook, 1995; Mozrzymas et al., 1999; 2003a, 2003b). Thus, in the present study, we restricted our analysis to the fast component of the desensitization process (in control conditions τDes=2.42±0.11 ms, ss/peak=0.128±0.005, n=8, fitting area set to 25 ms starting from the peak current). The time constant of desensitization onset was slowed down by βCD in a concentration dependent manner (Figure 4a and b). Moreover, the steady-state to peak ratio was increased with βCD concentration (Figure 4c). These data demonstrate that βCD decreases both the rate and extent of desensitization.
Figure 4.

βCD decreases the rate and extent of GABAAR desensitization. (a) Typical, normalized current responses to 10 mM GABA in control conditions (thick line) and in the presence of 1.5 mM βCD (thin line). Insets above current traces indicate the concentration and time course of applied agonist. (b and c) Show dose-dependence of βCD effect on the desensitization time constant (τDes) and on the steady-state to peak ratio (ss/peak), respectively. Asterisks above bars indicate statistically significant effects.
A characteristic property of GABAA receptors is that after a brief exposure to the agonist, the receptors tend to be trapped by the desensitized conformation not only during the exposure to the agonist but also after the neurotransmitter removal (Jones & Westbrook, 1995; Mozrzymas, 2004). Such effective receptor trapping in the desensitized conformation takes place due to combination of slow unbinding, fast desensitization and slow resensitization rates. In order to assess how quickly the receptors quit the desensitized state and become activable again, a standard paired-pulse protocol was applied. In the present study, we applied 2 ms pulses separated by variable interpulse gap. We found that the recovery process, observed in the paired pulse experiments, was clearly accelerated in the presence of βCD (Figure 5a and b).
Figure 5.

βCD accelerates the recovery of response to the second pulse in the paired pulse protocol. (a) Typical normalized and superimposed current responses to 10 mM GABA in control conditions (thick line) and in the presence of 1.5 mM βCD (thin line). Inset above current traces indicate the concentration and time course of applied agonist. (b) Statistics of the recovery parameter (see Methods) measured for various interpulse intervals.
The effect of βCD was tested also on current responses elicited by nonsaturating GABA concentration (10 μM, Figure 6). In control conditions, the averaged current amplitude elicited by 10 μM GABA (at −70 mV) was 465±66 pA, n=16. Surprisingly, at this GABA concentration, the enhancement of current at all βCD concentrations tested was larger than that observed at saturating [GABA] (Figure 1). Moreover, at variance to the effect on current amplitudes of responses to saturating [GABA], at 10 μM, the effect of βCD seems to be close to saturation at 500 μM (Figure 6, at 1.5 mM βCD no further significant increase is observed). The 10–90% rise time of currents elicited by 10 μM GABA showed a trend to slow down with increasing βCD concentration and this effect reached significance at 1.5 mM βCD (data not shown). A stronger increase in amplitude of currents elicited by 10 μM GABA in comparison to responses evoked by saturating [GABA] (at 500 μM βCD) indicates that this drug could enhance the agonist binding to GABAA receptor. However, it needs to be taken into consideration that any kinetic characteristics of current response represents a complex behavior that depends on transition rates describing all conformational changes available to the channel (Colquhoun, 1998; Mozrzymas et al., 2003b). Thus, in order to indicate more precisely which elements of GABAAR gating scheme are affected, tentative model simulations had to be performed.
Figure 6.

βCD strongly enhances the current responses to non saturating [GABA]. (a) Typical current responses to 10 μM GABA in control conditions (thick line) and in the presence of 1.5 mM of βCD. Currents were recorded from the same patch. (b) Statistics of the βCD effect on the amplitudes of current responses to 10 μM GABA. Asterisks above bars indicate statistically significant effects.
Model simulations
In order to further elucidate which conformational transitions were mostly affected by βCD, model simulations were performed using the Jones and Westbrook's model (Figure 7a, Jones et al., 1998). Although simplified, this model is known to properly reproduce the basic properties of GABAA receptor gating. The effects of βCD were clear and robust but they were characterized by a large cell-to-cell variability that is represented by relatively large error bars in the (Figures 1, 2, 3, 4, 5 and 6). Taking this into account, the task of the model simulations was to verify the qualitative trends of proposed mechanisms rather than a regular optimization of the model rate constants. Basing on our recent report (Mozrzymas et al., 2003a), we assumed that in control conditions the desensitization rate is the fastest transition in the GABAA receptor gating scheme (see figure legend of Figure 7). The observation that βCD strongly attenuates both the rate and extent of desensitization (Figure 4) suggests a decrease in the rate constant d2. A reduction of this rate constant would be expected to reproduce a slow down of the desensitization time constant, increase in the steady-state to peak ratio (Figure 4c) and an increase in amplitude of currents elicited by saturating [GABA] (Figure 1). The present finding that reduction in receptor desensitization rate is associated with a robust increase in the amplitude of currents elicited by saturating GABA further confirms that the desensitization rate is considerably faster than the opening rate. Thus, if d2>β2 then, after application of saturating [GABA], most of receptors would enter the desensitized state A2D. However, a reduction of d2 (assuming β2 constant) would favor entrance into the open state, giving rise to increased current amplitude (at saturating agonist concentration). However, a reduction in the desensitization rate would lead to an acceleration of the deactivation kinetics (Jones & Westbrook, 1995) contrary to our experimental observations (Figure 3). It needs to be pointed out, however, that at low [GABA] (Figure 6) βCD produced a larger enhancement of current than at saturating agonist concentration (Figure 1) indicating that βCD could affect binding/unbinding kinetics. Our experiments do not provide a direct means to precisely assess the βCD effect separately on binding and unbinding but a robust CD-induced prolongation of deactivation (while desensitization is reduced) would suggest a marked slow down of the unbinding rate. However, there is a tendency to saturate the βCD effect on binding/unbinding at concentrations close to 500 μM (Figure 6) while at higher βCD concentration, a further slow down of desensitization is observed (Figure 4). This could explain a biphasicity of βCD effect on the deactivation kinetics (Figure 3). We tested this hypothesis by simulating currents with minimum assumptions that at 500 μM βCD, both koff and d2 are decreased while an increase of βCD concentration to 1.5 mM further decreases desensitization rate d2 without affecting the binding/unbinding processes. Figure 7 shows that these minimum assumptions allow to qualitatively reproduce the observed trend with respect to current amplitudes (compare Figure 1 with Figure 7b–d), desensitization (compare Figure 4 with Figure 7e–g) and deactivation kinetics (compare Figure 3 and Figure 7b–d). A proper reproduction of βCD-induced acceleration of the recovery process in the paired pulse experiments (compare Figures 5 and 7h–j) was achieved by increasing the recovery rate r2 and by slowing down the unbinding rate from both fully bound open A2R* and desensitized A2D states and, as mentioned above, by slowing down of the desensitization rate d2. In addition, when using the rate constants for 0.5 and 1.5 mM βCD, it was possible to fairly reproduce a considerably larger βCD-induced enhancement of current amplitudes evoked by 10 μM GABA than in the case of responses elicited by saturating [GABA] (compare Figures 1, 6 to Figure 7a–c, k–m). However, a weak point of our simulations was the lack of reproduction of βCD-induced slow down of the current responses to 10 μM GABA. We may suspect that in the case of long GABA applications (1 s, Figure 6) such βCD effect could result from modulation of a slow desensitization component that was not included in the considered model.
Altogether these model simulations further indicate that the major effect of βCD on GABAAR gating is to decrease desensitization and enhance binding (mainly due to strong reduction of the unbinding rate).
Discussion
The major finding of the present report is that βCD, a compound commonly believed to act as an inert factor increasing the solubility of hydrophobic substances, is able to strongly affect the kinetics of conformational transitions of GABAA receptors. βCD-induced alterations of recorded current responses as well as model simulations indicate that the major βCD effect is to modulate desensitization and agonist binding/unbinding. As mentioned in Results, while the qualitative trends in βCD-induced modulation of GABA-evoked currents time course were clear, the extent of modulation showed a substantial cell-to-cell variability (see Results and large error bars in Figures 1, 2, 3, 4, 5 and 6). The reason for such large data scatter is not clear. It may be speculated that it reflects a heterogeneity of GABAA receptors in different neurons. Alternatively, since CD is likely to strongly interact with lipid phase of the membrane, it may be hypothesized that such diversity of CD action reflects differences in receptor microenvironment in different neurons. Elucidation of this problem will require determination of the site and molecular mechanism of CD action.
Although CDs are commonly used to deliver GABAAR-acting compounds (e.g. Wang et al., 1997), to our knowledge, the only study in which the βCD effect on GABAA receptors was addressed in electrophysiological experiments, was published by Shu et al. (2004). However, they found that βCD affected the steroid activated currents but responses elicited by GABA were not altered by this compound (Shu et al., 2004). The reason for this discrepancy is not clear. The currents recorded by Shu et al. (2004) were evoked by a relatively slow application system and it is possible that some aspects of βCD-induced modulation of receptor gating were beyond time resolution of their system. The fact that they observed a clear CD effect only on very slow currents activated by steroid could support this hypothesis. On the other hand, as pointed out by Shu et al. (2004), steroid-induced activation of GABAARs could result from a different molecular pathway that could show different sensitivity to CD.
The analysis of βCD effect on desensitization kinetics (Figure 4) and on amplitudes of current responses to nonsaturating [GABA] (Figure 6) suggests that desensitization and binding/unbinding are modulated by βCD with different potency. The molecular mechanism of such different βCD actions is not known. It may be speculated that modulation of these processes is mediated either by two different binding sites or that molecular determinants of binding and desensitization are differentially affected by βCD-induced alterations in the lipid environment. Perhaps the most obvious candidate to explain the described here modulation of GABAARs is βCD-mediated depletion of membrane cholesterol. At first glance, this possibility appears particularly plausible since, as already mentioned, membrane cholesterol was shown to strongly modulate several ionic channels in the plasma membrane (Chen & Gross, 1995; Bennett & Simmonds, 1996; Hajdu et al., 2003; Brady et al., 2004; Frank et al., 2004; Lundbaek et al., 2004; Shu et al., 2004; Taverna et al., 2004; Xia et al., 2004). In the case of acetylcholine receptor this effect is crucial as this channel cannot be activated in the absence of cholesterol (Fong & McNamee, 1986; Addona et al., 1998). Moreover, Bennett & Simmonds (1996) have shown that cholesterol may affect binding of different modulators to GABAARs. There are, however, several points arguing against any crucial role of cholesterol efflux in described here modulation of GABAARs gating by βCD. First of all, the βCD concentration range used in the present study (up to 1.5 mM) would be expected to induce a weak cholesterol efflux. Indeed, Kilsdonk et al. (1995) and Yancey et al. (1996) applied several millimols of CD for several minutes or even hours to observe measurable cholesterol uptake. It is noteworthy that in the present study, a detectable effect on GABAA receptor kinetics was observed at 150 μM of βCD, a concentration at which cholesterol efflux would be negligible. Moreover, in our experiments the membrane patches were exposed continuously to a rapid flux of solution containing βCD. In these conditions, we would expect an irreversible cholesterol depletion while the βCD effects on GABA-evoked currents were fully reversible. Yancey et al. (1996) have found that CD-induced cholesterol efflux was characterized by a biphasic time course with time constants of roughly a few tens of seconds (fast) and of several minutes (slow). If the observed changes in GABA-evoked current kinetics were due to the cholesterol depletion, one would expect that in consecutive recordings (separated by at least 2 min. interval), a modification in amplitude or current time course would show an evolution correlated with the kinetics of cholesterol efflux. However, consecutive current responses recorded in the presence of βCD were indistinguishable. Treatment of cellular membranes with CDs results in loss not only of cholesterol but also of sphingolipids and phospholipids (Kilsdonk et al., 1995; Ottico et al., 2003). However, since sphingolipids and especially phospholipids are taken up from membranes by CDs much less efficiently than cholesterol (Kilsdonk et al., 1995; Ottico et al., 2003) the efflux of these compounds seems unlikely to be responsible for the observed here βCD effects on GABAergic currents. Altogether, the above described arguments indicate that the efflux of cholesterol (or other compounds such as sphingolipids and phospholipids) is not the primary mechanism whereby βCD modulate the GABAA receptor gating. This, however, does not exclude any involvement of cholesterol in this process. It is well documented that cholesterol may exert different modulatory effects depending on its localization within the membrane (e.g. within specialized lipid microdomains or in close association with proteins). Thus it is conceivable that βCD might interact with cholesterol not necessarily by removing it from the membrane but for example, by affecting its localization. We may thus speculate that although βCD caused most likely only a weak cholesterol (or other lipids) efflux, it could effectively affect the lipid domains in the vicinity of the receptor by inducing local alterations in distribution of lipid compounds. In this context it is noteworthy that acetylcholine receptors (that fail to activate in the absence of cholesterol) require cholesterol presence close to lipid–protein interface (Addona et al., 1998). Thus the activity of this receptor is not that much sensitive to the average cholesterol content within the membrane but rather requires its presence in the specialized zones close to the receptor macromolecule. The hypothesis related to the influence of cholesterol on the channel microenvironment appears even more interesting in the light of studies of Lundbaek et al. (1996; 2004) who found that factors affecting membrane stiffness (including cholesterol) strongly modify the kinetics of gramicidine channel as well as voltage-gated sodium and potassium channels. Clearly, since conformational transitions of channels are associated with geometrical rearrangements of these macromolecules, it is expected that the rigidity of channel closest environment could affect its gating.
An alternative possibility could be that cholesterol binds to a protein, forming an easy removable pool of membrane bound cholesterol that, after βCD removal, can be recovered from the lipid environment. For instance, Ding et al. (1994) provided evidence for cholesterol binding sites on ATPases and Locke et al. (2004) have demonstrated that CD is able to block connexins by occlusion of channel pore.
It needs to be additionally emphasized that CDs may interact with applied modulators (e.g. steroids or cholesterol) via non-inclusion-mechanisms or form complexes (Loftsson et al., 2004). Creation by CDs a supramolecular structures seems compatible with a proposal of Shu et al. (2004) that CDs could act as molecular sponges for hydrophobic compounds supplied to the membranes.
Altogether, we conclude that CDs, at concentrations at which effective cholesterol uptake is unlikely, induce strong alterations in the kinetics of conformational transitions of GABAA receptors. Although the molecular mechanism of βCD effects is not clear, it seems particularly appealing to study in the future CD effects on the microenvironment of the protein hydrophobic coupling or a direct CD binding to the receptor macromolecule. Another important message coming from this study is that CDs cannot be regarded as inert solubilizers for hydrophobic compounds as they can interact with and modulate the membrane components (both lipids and proteins).
Acknowledgments
Supported by the Polish State Committee for Scientific Research (KBN) grant (No. PBZ-MIN-001/P05/28).
Abbreviations
- βCD
beta cyclodextrin
- GABA
gamma-aminobutyric acid
- [GABA]
GABA concentration
- GABAAR
GABAA receptor
References
- ADDONA G.H., SANDERMANN H., JR, KLOCZEWIAK M.A., HUSAIN S.S., MILLER K.W. Where does cholesterol act during activation of the nicotinic acetylcholine receptor. Biochim. Biophy.s. Acta. 1998;1370:299–309. doi: 10.1016/s0005-2736(97)00280-0. [DOI] [PubMed] [Google Scholar]
- ANDJUS P.R., STEVIC-MARINKOVIC Z., CHERUBINI E. Immunoglobulins from motoneurone disease patients enhance glutamate release from rat hippocampal neurones in culture. J. Physiol. 1997;504:103–112. doi: 10.1111/j.1469-7793.1997.103bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARBUTI A., GRAVANTE B., RIOLFO M., MILANESI R., TERRAGNI B., DIFRANCESCO D. Localization of pacemaker channels in lipid rafts regulates channel kinetics. Circ. Res. 2004;94:1325–1331. doi: 10.1161/01.RES.0000127621.54132.AE. [DOI] [PubMed] [Google Scholar]
- BENNETT P.J., SIMMONDS M.A. The influence of membrane cholesterol on the GABAA receptor. Br. J. Pharmacol. 1996;117:87–92. doi: 10.1111/j.1476-5381.1996.tb15158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRADY J.D., RICH T.C., LE X., STAFFORD K., FOWLER C.J., LYNCH L., KARPEN J.W., BROWN R.L., MARTENS J.R. Functional role of lipid raft microdomains in cyclic nucleotide-gated channel activation. Mol. Pharmacol. 2004;65:503–511. doi: 10.1124/mol.65.3.503. [DOI] [PubMed] [Google Scholar]
- BROWN D.A., LONDON E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000;275:17221–17224. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- CHEN X., GROSS R.W. Potassium flux through gramicidin ion channels is augmented in vesicles comprised of plasmenylcholine: correlations between gramicidin conformation and function in chemically distinct host bilayer matrices. Biochemistry. 1995;34:7356–7364. doi: 10.1021/bi00022a008. [DOI] [PubMed] [Google Scholar]
- CLEMENTS J.D. Transmitter time course in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 1996;19:163–170. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- COLQUHOUN D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DING J., STARLING A.P., EAST J.M., LEE A.G. Binding sites for cholesterol on Ca(2+)-ATPase studied by using a cholesterol-containing phospholipid. Biochemistry. 1994;33:4974–4979. doi: 10.1021/bi00182a028. [DOI] [PubMed] [Google Scholar]
- DOUHAL A. Ultrafast guest dynamics in cyclodextrin nanocavities. Chem. Rev. 2004;104:1955–1976. doi: 10.1021/cr020669j. [DOI] [PubMed] [Google Scholar]
- FIELDING C.J., FIELDING P.E. Membrane cholesterol and the regulation of signal transduction. Biochem. Soc. Trans. 2004;32:65–69. doi: 10.1042/bst0320065. [DOI] [PubMed] [Google Scholar]
- FONG T.M., MCNAMEE M.G. Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry. 1986;25:830–840. doi: 10.1021/bi00352a015. [DOI] [PubMed] [Google Scholar]
- FRANK C., GIAMMARIOLI A.M., PEPPONI R., FIORENTINI C., RUFINI S. Cholesterol perturbing agents inhibit NMDA-dependent calcium influx in rat hippocampal primary culture. FEBS. Lett. 2004;566:25–29. doi: 10.1016/j.febslet.2004.03.113. [DOI] [PubMed] [Google Scholar]
- HAJDU P., VARGA Z., PIERI C., PANYI G., GASPAR R., JR Cholesterol modifies the gating of Kv1.3 in human T lymphocytes. Pflugers. Arch. 2003;445:674–682. doi: 10.1007/s00424-002-0974-y. [DOI] [PubMed] [Google Scholar]
- HARADA A. Cyclodextrin-based molecular machines. Acc. Chem. Res. 2001;34:456–464. doi: 10.1021/ar000174l. [DOI] [PubMed] [Google Scholar]
- JONAS P.Fast application of agonists to isolated membrane patches Single-channel. recording. 1995New York and London: Plenum Press; 231–243.ed. Sakmann, B. & Neher, E. pp [Google Scholar]
- JONES M.V., WESTBROOK G.L. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- JONES M.V., WESTBROOK G.L. Shaping of IPSCs by endogenous calcineurin activity. J. Neurosci. 1997;17:7626–7633. doi: 10.1523/JNEUROSCI.17-20-07626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES M.V., SAHARA Y., DZUBAY J.A., WESTBROOK G.L. Defining affinity with the GABAA receptor. J. Neurosci. 1998;18:8590–8604. doi: 10.1523/JNEUROSCI.18-21-08590.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAMIONKA A., DAHL M.K. Bacillus subtilis contains a cyclodextrin-binding protein which is part of a putative ABC-transporter. FEMS. Microbiol. Lett. 2001;204:55–60. doi: 10.1111/j.1574-6968.2001.tb10862.x. [DOI] [PubMed] [Google Scholar]
- KILSDONK E.P., YANCEY P.G., STOUDT G.W., BANGERTER F.W., JOHNSON W.J., PHILLIPS M.C., ROTHBLAT G.H. Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem. 1995;270:17250–17256. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- LOCKE D., KOREEN I.V., LIU J.Y., HARRIS A.L. Reversible pore block of connexin channels by cyclodextrins. J. Biol. Chem. 2004;279:22883–22892. doi: 10.1074/jbc.M401980200. [DOI] [PubMed] [Google Scholar]
- LOFTSSON T., MASSON M., BREWSTER M.E. Self-association of cyclodextrins and cyclodextrin complexes. J. Pharm. Sci. 2004;93:1091–1099. doi: 10.1002/jps.20047. [DOI] [PubMed] [Google Scholar]
- LUNDBAEK J.A., BIRN P., GIRSHMAN J., HANSEN A.J., ANDERSEN O.S. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- LUNDBAEK J.A., BIRN P., HANSEN A.J., SOGAARD R., NIELSEN C., GIRSHMAN J., BRUNO M.J., TAPE S.E., EGEBJERG J., GREATHOUSE D.V., MATTICE G.L., KOEPPE R.E., 2nd, ANDERSEN O.S. Regulation of sodium channel function by bilayer elasticity: the importance of hydrophobic coupling. Effects of Micelle-forming amphiphiles and cholesterol. J. Gen. Physiol. 2004;123:599–621. doi: 10.1085/jgp.200308996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACDONALD R.L., ROGERS C.J., TWYMAN R.E. Kinetic properties of the GABAA receptor main conductance state of mouse spinal cord neurones in culture. J. Physiol. 1989;410:479–499. doi: 10.1113/jphysiol.1989.sp017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCLELLAN A.M.L., TWYMAN R.E. Receptor system response kinetics reveal functional subtypes of native murine and recombinant human GABAA receptors. J. Physiol. 1999;515:711–727. doi: 10.1111/j.1469-7793.1999.711ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOZRZYMAS J.W. Dynamism of GABAA receptor activation shapes the ‘personality' of inhibitory synapses. Neuropharmacology. 2004;47:945–960. doi: 10.1016/j.neuropharm.2004.07.003. [DOI] [PubMed] [Google Scholar]
- MOZRZYMAS J.W., BARBERIS A., MERCIK K., ZARNOWSKA E. Binding sites, singly bound states and conformation coupling shape GABA-evoked currents. J. Neurophysiol. 2003b;89:871–884. doi: 10.1152/jn.00951.2002. [DOI] [PubMed] [Google Scholar]
- MOZRZYMAS J.W., BARBERIS A., MICHALAK K., CHERUBINI E. Chlorpromazine inhibits miniature GABAergic currents by reducing the binding and by increasing the unbinding rate of GABAA receptors. J. Neurosci. 1999;19:2474–2488. doi: 10.1523/JNEUROSCI.19-07-02474.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOZRZYMAS J.W., ZARNOWSKA E.D., PYTEL M., MERCIK K. Modulation of GABAA receptors by hydrogen ions reveals synaptic GABA transient and a crucial role of the desensitization process. J. Neurosci. 2003a;23:7981–7992. doi: 10.1523/JNEUROSCI.23-22-07981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OTTICO E., PRINETTI A., PRIONI S., GIANNOTTA C., BASSO L., CHIGORNO V., SONNINO S. Dynamics of membrane lipid domains in neuronal cells differentiated in culture. J. Lipid. Res. 2003;44:2142–2151. doi: 10.1194/jlr.M300247-JLR200. [DOI] [PubMed] [Google Scholar]
- PAJATSCH M., GERHART M., PEIST R., HORLACHER R., BOOS W., BOCK A. The periplasmic cyclodextrin binding protein CymE from Klebsiella oxytoca and its role in maltodextrin and cyclodextrin transport. J. Bacteriol. 1998;180:2630–2635. doi: 10.1128/jb.180.10.2630-2635.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REDENTI E., SZENTE L., SZEJTLI J. Cyclodextrin complexes of salts of acidic drugs. Thermodynamic properties, structural features, and pharmaceutical applications. J. Pharm. Sci. 2001;90:979–986. doi: 10.1002/jps.1050. [DOI] [PubMed] [Google Scholar]
- SHU H.J., EISENMAN L.N., JINADASA D., COVEY D.F., ZORUMSKI C.F., MENNERICK S. Slow actions of neuroactive steroids at GABAA receptors. J. Neurosci. 2004;24:6667–6675. doi: 10.1523/JNEUROSCI.1399-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAVERNA E., SABA E., ROWE J., FRANCOLINI M., CLEMENTI F., ROSA P. Role of lipid microdomains in P/Q-type calcium channel (Cav2.1) clustering and function in presynaptic membranes. J. Biol. Chem. 2004;279:5127–5134. doi: 10.1074/jbc.M308798200. [DOI] [PubMed] [Google Scholar]
- WANG M.D., WAHLSTROM G., BACKSTROM T. The regional brain distribution of the neurosteroids pregnenolone and pregnenolone sulfate following intravenous infusion. J. Steroid. Biochem. Mol. Biol. 1997;62:299–306. doi: 10.1016/s0960-0760(97)00041-1. [DOI] [PubMed] [Google Scholar]
- XIA F., GAO X., KWAN E., LAM P.P., CHAN L., SY K., SHEU L., WHEELER M.B., GAISANO H.Y.A., TSUSHIMA R.G. Disruption of pancreatic beta-cell lipid rafts modifies Kv2.1 channel gating and insulin exocytosis. J. Biol. Chem. 2004;279:24685–24691. doi: 10.1074/jbc.M314314200. [DOI] [PubMed] [Google Scholar]
- YANCEY P.G., RODRIGUEZA W.V., KILSDONK E.P., STOUDT G.W., JOHNSON W.J., PHILLIPS M.C., ROTHBLAT G.H. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration of kinetic pools and mechanism of efflux. J. Biol. Chem. 1996;271:16026–16034. doi: 10.1074/jbc.271.27.16026. [DOI] [PubMed] [Google Scholar]