

Abstract
Newborn screening (NBS)—in which each newborn infant is screened for up to 50 specific metabolic disorders for early detection and intervention—is the first program of populationwide genetic testing. As a public health intervention, NBS has greatly improved the lives of thousands of affected children.
New technologies and new economic and social forces pose significant ethical and clinical challenges to NBS. Two primary challenges concern (1) accommodating clinical and ethical standards to rapid technological developments in NBS and (2) preparing public health systems to respond to the medical advances and social forces driving expansion of NBS programs.
We describe and analyze these challenges through consideration of 3 disorders: phenylketonuria, medium-chain acyl-CoA dehydrogenase deficiency, and cystic fibrosis.
NEWBORN SCREENING (NBS) is the first and largest example of systematic, populationwide genetic testing and has led to improved lives for thousands of affected children. Since the 1960s, a blood sample from virtually every infant born in the United States each year—roughly 4 million in 2004—is screened within the first weeks of life for specific metabolic, endocrine, and hematologic disorders. NBS programs are overseen by state public health departments and provide early diagnosis and referral for treatment of primarily presymptomatic, autosomal recessive diseases.1
These programs began with the detection of elevated phenylalanine levels in phenylketonuria (PKU) and now screen for up to 50 disorders.2 Together, including hearing screening, NBS may detect an inherited metabolic, endocrine, or hematologic disorder in as many as 1 in 500 to 1000 newborns in the United States (Therrell BL, PhD, National Newborn Screening and Genetics Resource Center, written communication, January 2004). More efficient technologies that permit simultaneous screening for multiple disorders, referred to as multiplex capabilities, as well as advances in the understanding of the genetic basis of disease, increasingly facilitate screening for many more disorders.3
Two questions have persisted throughout the history of NBS: (1) how do clinical and ethical standards accommodate the rapid developments made possible by new technologies? (2) How well are public health systems prepared to respond to both the medical advances and the political forces driving expansion of NBS programs?
Universal newborn genetic screening has been an enormous public health success. But, as with many successes, it also raises a number of significant ethical and clinical challenges, particularly with contemporary proliferation of screenable disorders through new technologies. The most pressing current challenges are illustrated by 3 disorders: phenylketonuria (PKU); medium-chain acyl-CoA dehydrogenase deficiency (MCAD); and cystic fibrosis (CF). Specifically, PKU screening resulted in questions regarding how policy is developed, the technical demands of screening, the extent of public health responsibility, and unanticipated consequences. The use of tandem mass spectrometry (MS/MS) for metabolic disorders such as MCAD has led to pressures to expand the list of screened conditions detectable by MS/MS and other technologies, despite considerable uncertainties about the clinical outcomes of these disorders. NBS for CF raises issues regarding fairness in DNA-based screening in diverse populations, non-medical benefits, and carrier identification.
THE 3 DISORDERS
Phenylketonuria
PKU is an autosomal recessive inborn error of metabolism with an incidence of approximately 1 in 15 000.4 Untreated PKU results in severe mental retardation, seizures, and other neurologic problems, usually requiring eventual institutionalization of the patient. Adherence to a special low-phenylalanine diet beginning in infancy prevents these devastating consequences. Robert Guthrie pioneered an early screening technique in the 1960s,4,5 assay of a dried blood spot from filter paper, which has led to population-based screening for affected newborns with consequent early diagnosis, referral, and initiation of dietary interventions. Currently, PKU screening in the United States detects several hundred new cases per year, cumulatively sparing thousands of affected children from severe mental retardation, as well as saving private and public resources by reducing the number of institutionalized PKU patients.4,5
Since the 1960s, the potential for disease prevention motivated professional and consumer organizations to apply legislative pressure to create NBS programs. By the 1970s, as a result of those early advocacy efforts, NBS programs for PKU screening were instituted throughout the United States within state public health departments.6 Although highly beneficial, NBS policy was not necessarily determined by the best scientific data available during that period. For example, public statements at times grossly overstated the impact of PKU disease prevention on the numbers of mentally retarded children and adults institutionalized at public expense.7
Today, in the absence of national standards for testing and follow-up that are based on rigorous scientific review, wide state-to-state variability still exists in the disorders screened and resources allocated to programs, as illustrated in Figure 1 ▶, and continues to be heavily influenced by local advocacy forces. For some disorders, commercial and parental pressures are forcing their rapid inclusion in NBS without sufficient data or data collection strategies to fully inform decisionmaking; this has just occurred in New York for Krabbe’s disease, a neurodegenerative disorder caused by deficiency of galactocerebroside, leading to the destruction of the nerve cells’ protective myelin covering.10 Optimal allocation of NBS resources also requires consideration of the risks and benefits for all, including children whose parents may be less accustomed to advocacy.
FIGURE 1—
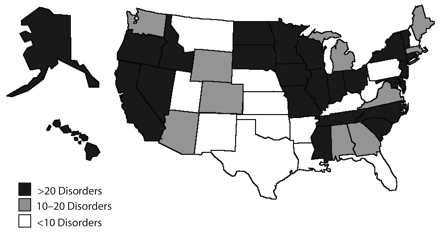
US newborn screening: mandated and implementeda American College of Medical Genetics–recommended core conditionsb by state (as of September 2005).
aOther disorders may be offered but are either nonmandatory or not implemented.
bCore conditions are 29 disorders recommended by the American College of Medical Genetics 2005 report Newborn Screening: Toward a Uniform Screening Panel and System.8
Source. National Newborn Screening and Genetics Resource Center.9
The federal government has only recently begun to provide the much-needed framework for the development of specific science-driven policies and standards for modern NBS, most notably through a 2005 report by the American College of Medical Genetics, Newborn Screening: Toward a Uniform Screening Panel and System, commissioned by the Department of Health and Human Services.8,11 This report is a significant step forward for NBS, despite concerns raised by some experts about its methodology, the change in ethical standards it proposes, and the membership of the task force.12,13 The report has been endorsed by the newly created Department of Health and Human Services secretary’s Advisory Committee on Heritable Disorders and Genetic Diseases in Newborns and Children (ACHDGDNC), which is charged with providing advice, recommendations, and “technical information to develop policies and priorities”8 on NBS, as authorized by Title XXVI of the Children’s Health Act of 2000, Screening for Heritable Disorders.8,14 The ACHDGDNC is focused on 3 critical areas for national and state application: laboratory standards and procedures; clinical follow-up; and education and training.
Technical dilemmas that arose during the early years of NBS still complicate screening for PKU and other disorders. For example, minimizing missed cases necessitates sufficient sensitivity of screening tests but results in high rates of false positives requiring significant resources for follow-up testing. An evaluation of the accuracy of the Guthrie test in systematic NBS was not published until 1974, some 10 years after state programs were initiated, and the results were unimpressive, with a sensitivity of 90% and only 5% specificity.7 NBS clearly provides substantial benefits for affected children and their families. However, evaluation of the efficacy of screening, including sorting out true positives from false positives, and appropriate clinical follow-up to prevent illness or complications, should be, but still is not, routinely performed.6,11 New technologies applied to NBS should facilitate more predictive screening approaches, for example, the addition of DNA-based mutation analysis for genotype–phenotype correlation.15
The extent and duration of public responsibility for children diagnosed by NBS remains controversial. A major problem is the long-term cost resulting from NBS beyond programs of sample collection, assay, and medical referral, for example, the cost of long-term treatment with expensive low-phenylalanine PKU formula. Despite existing laws defining therapeutic formula as a medical food, children and adults with PKU confront reimbursement barriers in both public and private systems that compromise adherence to therapy.16,17 Optimal outcomes require enduring support for the medical management of individuals identified by NBS; this support could be given through stable access to specialty medical facilities and public or private third-party payers.
NBS for PKU has allowed a generation of children to lead markedly improved lives but has also created unforeseen consequences. Although affected children may be allowed to relax their dietary restrictions as adults, pregnancy is a critical time for affected women to adhere strictly to the low-phenylalanine diet. The developing fetus, even if genotypically unaffected, faces serious neurologic consequences if exposed to high maternal phenylalanine levels in utero.18 Several children severely affected by uncontrolled maternal PKU have been reported.18 More concerted efforts by obstetrical providers to expand referrals for maternal PKU to metabolic specialists are needed to ensure that the several thousand women with PKU who are now of childbearing age have access to dietary counseling and the resources necessary to protect the health of their children.17 Pregnancy or other adult conditions may alter the medical management of additional metabolic disorders originally detected by NBS, requiring renewed medical attention.
MCAD
MCAD is a disorder of fatty acid metabolism caused by the lack of an enzyme required to convert fat to energy.19 Symptoms usually manifest within the first 3 years of life and can include vomiting, lethargy, hypoglycemia, seizures, coma, apnea, and even sudden death. Long-term consequences can include developmental delay, failure to thrive, chronic muscle weakness, cerebral palsy, and behavioral disabilities. Symptoms of MCAD usually manifest when an infant undergoes metabolic stress such as fasting, fever, or infection. Treatment consists of steady food or glucose intake with a reduced-fat, high-carbohydrate diet that prevents metabolic crisis. Early identification and intervention significantly improve patient outcomes,20,21 a fundamental justification for including MCAD in NBS panels.
MCAD occurs with an approximate incidence of 1 in 20000.19 The highest frequency of MCAD mutations is among northern European Whites, in whom the most common mutation is the substitution mutation G985A. In this population, the carrier frequency of this mutation is 1 in 40 to 1 in 100 and the homozygote frequency is 1 in 6500 to 1 in 20000.19
MCAD is screened by detecting elevated blood levels of octanoylcarnitine by MS/MS. MS/MS generates a detailed metabolic profile and is currently used by about 70% of state NBS programs for screening for MCAD and a panel of up to 40 other metabolic disorders. Some of these other disorders, however, have no well-defined medical treatment.8,22
There is no clear genotype–phenotype correlation with the G985A mutation or other MCAD mutations, and significant clinical variability exists even within families.19 Some individuals who are homozygous for MCAD mutations do not manifest symptoms, indicating that the disease has incomplete penetrance. Only through a positive test result in a newborn do some families become aware of a diagnosis in their older children who had never had symptoms of the disease. Inadequate knowledge about the full clinical spectrum of MCAD and incomplete prognostic information require balancing the risk of emotional burden on the child and family and the cost of screening and treatment against the considerable benefits of potentially lifesaving interventions, along with what may be important genetic information for the family.19
Large-scale prospective studies are needed to ensure that early detection and treatment improve outcomes for children with MCAD and other rare disorders. Regional or nationwide systems for prospective diagnosis and treatment of affected children, modeled on the well-established system for children’s cancer, are 1 possibility. This strategy is especially necessary for other metabolic disorders about which much less is known. Many experts consider data accrual through NBS programs for the purpose of research or surveillance to be an appropriate public health activity. Although a traditional public health function, surveillance remains controversial because it may be beyond the explicit public mandate of NBS, namely to directly protect the health of an affected child. Additionally, the lack of written informed consent in most states’ NBS programs complicates the attainment of research goals, especially in the use of bloodspot samples for research.23 National policy, such as that determined by the ACHDGDNC, should clarify the scope of the public health mission of NBS in the context of expanding screening potential, enhancing genetic capabilities, and evolving public demand.
Private screening companies are becoming increasingly involved in NBS in various capacities in several states23,24 and compete with state public health laboratory services by offering a pay-for-service NBS for MCAD and up to 50 additional metabolic disorders, most of which are detectable by MS/MS, to parents.23 Companies usually report positive tests to public health authorities and refer the child for specialty medical care. Such companies should continuously exchange relevant information with the state NBS programs.23,24 Privatization has reduced state jurisdiction over public health policy through competitive marketing. Voluntary parental use of private screening resources creates 2-tier, nonuniversal screening, determined by selective parental knowledge of these options and ability to pay. On the other hand, competition from privatization has, in some cases, sped up the expansion of otherwise stagnant state programs. Thus, the growing impetus toward privatization has had potent and overall mixed effects on US NBS policy, intensifying the need for specific federal NBS guidelines for priorities and services that are rational, high quality, and equitable.
Cystic Fibrosis
CF is a disorder in which abnormal cationic transport across cell membranes causes highly viscous secretions, affecting many organ systems but primarily the gastrointestinal tract and lungs.25 Medical treatment includes replacement of digestive enzymes and aggressive use of antibiotics and other medications for improving lung function.25
Ten states currently screen for CF as part of standard NBS programs or as optional pilot screening.9,26 In most of these states, elevated levels of immunoreactive trypsinogen trigger confirmatory screening with a DNA mutation panel of the CF transmembrane conductance regulator (CFTR) gene27,28 performed off the Guthrie card to detect clinically significant mutations. Infants with 1 or 2 detected mutations are referred for diagnostic sweat testing.
NBS for CF has introduced widespread DNA-based testing to NBS. More than 1000 known mutations affect the CFTR gene.25 Most commonly, specific mutations are detected with sequence-specific hybridization probes for specific mutations.25,28
Different populations have been shown to carry different CFTR mutations in differing frequencies. The most common CFTR mutation is a small deletion called ΔF508. This mutation accounts for 70% of the CF mutations in White patients of northern European descent, 30% of CF mutations in affected Ashkenazi Jewish patients, and differing percentages in other populations.29 Some states use ΔF508 exclusively in their DNA-based CF screening.26
Because of the different frequencies of ΔF508 and other mutations among different ethnic/racial groups, the detection rate of DNA mutation testing is highly dependent on racial and ethnic background. Notably, DNA-based screening can detect specific mutations but cannot rule out other possible mutations. Mutations in individuals or communities with uncommon CF mutations may be missed, and thus these groups may be less well served by the current system of NBS. Screening for many more of the known CF gene mutations could help ameliorate these inequities,15 a goal to be balanced with the added burden generated by more false positives and identification of carriers.
Consensus approval of NBS for CF has recently occurred,30 largely because of data from the Wisconsin CF Neonatal Screening Project, which showed primarily nutritional benefits from early diagnosis.31,32 Potential benefits from NBS for CF, or for other disorders, however, may be indirect or not purely medical.6,33 Other potential benefits of screening that deserve consideration include informing parental reproductive decisions, decreasing costs and anxiety by streamlining the diagnostic process for an ill infant,22 identifying potential subjects for clinical trials for new interventions, and offering new interventions to asymptomatic patients.
Screening by DNA mutation analysis for CF and other disorders also reveals unaffected genetic carriers who are at risk of having children with CF if they pair with another carrier. Although carrier reporting is appropriately less urgent, some state programs do not even report the identification of carriers detected through NBS. Programs must responsibly convey the genetic information derived from screening, including carrier identification,30,34,35 so that the implications of this information can be responsibly communicated by the medical providers to the parents of these children.
THE FUTURE
Newborn genetic screening has been a remarkable achievement for public health, providing populationwide detection of disorders that leads to improved clinical outcomes. Advances in medical genetics and testing technology permit the diagnosis of ever more diseases but also compel society to reconsider how NBS as a public health measure may best serve children, their families, and their communities. Powerful multiplex test technologies can identify children with anomalies that may—or may not—lead to disease. They can also find children for whom no treatment is yet available. Weighing the costs, risks, and benefits of screening in such cases is complex and will require consideration of the full range of costs and potential nonmedical benefits.33
New knowledge and NBS technologies raise additional challenges. For example, some experts advocate NBS for severe combined immunodeficiency, a uniformly fatal disorder if untreated and for which early diagnosis can lead to lifesaving bone marrow transplantation therapy.36 However, such treatments may not be within the moral compass of public health, as they are associated with risks of morbidity and mortality, expensive, and not universally available to all affected infants. Similar considerations arise with lysosomal storage disorders.37 Policy decisions will become increasingly complex and controversial regarding the use of NBS, understood as a public health measure to identify newborns with treatable diseases, when it is extended to include detecting genes with incomplete penetrance, genes for a given disease that only partially predict that disease, and disorders that cannot be successfully treated or for which treatment is available only to some newborns so identified.
In addition to the traditional single-gene disorders, conditions affected by multiple genes and gene–environment interactions raise additional possibilities for incorporating new disorders into NBS programs. Indeed, NBS for common complex diseases,38 such as asthma, is under consideration,39 with pilot screening for genetic susceptibility to diabetes40 already under way for early institution of prevention strategies for those at risk. These trends will likely push NBS programs and society to discuss the implications of revealing disease susceptibility rather than making a specific diagnosis in the first months of life.41 Some commentators have gone as far as to suggest a broad analysis of individual genomic variation from a NBS sample.42
NBS requires more resources to take advantage of current and future opportunities and challenges, including the creation of national consensus guidelines for screening and follow-up to establish universal minimal screening standards for the United States. Proposals to improve NBS, such as proposed test standards11 and key recommendations on NBS made by the American Academy of Pediatrics6 and the American College of Medical Genetics, 8 are already sparking broad debate within the world of public health. These national efforts require sufficient support from the federal health system for translation to state programs and should result in identifying and standardizing best screening policies and practices across the United States; providing adequate resources for programs to incorporate long-term specialty treatment, genetic counseling and referral, and educational outreach to consumers and health providers; and increasing research capacity for well-designed prospective studies on the predictive value and clinical effectiveness of expanded screening and treatment.
As a public health program, NBS has done enormous good. New technologies and new forces are pushing NBS into unfamiliar territory, to which NBS must actively react to set its course for a sound future.
Acknowledgments
Work on this article was supported by the National Human Genome Research Institute (award HG02579, Ethical Decision-Making for Newborn Genetic Screening).
We appreciate the generously shared expertise of Bradford Therrell, Kenneth Pass, Scott Grosse, and Mary Ann Baily, and technical assistance from Motoko Oinuma.
Peer Reviewed
Contributors N. S. Green led the conceptualization, writing, and editing of the article. S. M. Dolan assisted in the conceptualization and writing of the article. T.H. Murray greatly contributed to the conceptualization and editing of the article.
References
- 1.Newborn Screening: Characteristics of State Programs. Washington, DC: US General Accounting Office; 2003:1–47. Publication GAO-03–449.
- 2.Therrell BL. US newborn screening policy dilemmas for the twenty-first century. Mol Genet Metab. 2001;74: 64–72. [DOI] [PubMed] [Google Scholar]
- 3.Schulze A, Lindner M, Kohlmuller D, Olgemoller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111:1399–1406. [DOI] [PubMed] [Google Scholar]
- 4.National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16–18, 2000. Pediatrics. 2001;108:972–982. [DOI] [PubMed] [Google Scholar]
- 5.Healthy Children: Investing in the Future. Washington, DC: US Congress, Office of Technology Assessment; 1988. Publication OTA-H-345.
- 6.Serving the family from birth to the medical home. Newborn screening: a blueprint for the future—a call for a national agenda on state newborn screening programs. AAP Newborn Screening Task Force. Pediatrics. 2000; 106:383–422. [PubMed] [Google Scholar]
- 7.Paul DB. The history of newborn phenylketonuria screening in the US. In: Holzman NA, Watson MS, eds. Promoting Safe and Effective Genetic Testing in the United States: Final Report of the Task Force on Genetic Testing. Bethesda, Md: National Institutes of Health; 1997: 137–160.
- 8.Watson M, Puryear M. Newborn Screening: Toward a Uniform Screening Panel and System [summary]. Pediatrics. In press.
- 9.National Newborn Screening and Genetics Resource Center. National Newborn Screening Status Report. Available at: http://genes-r-us.uthscsa.edu/nbsdisorders.pdf. Accessed September 15, 2005.
- 10.Governor Announces Historic Expansion of Newborn Screening [10/27/04]. Available at: http://www.huntershope.org/news/press_releases/albany_nbs_pr.asp. Accessed January 19, 2006.
- 11.Burke W, Atkins D, Gwinn M, et al. Genetic test evaluation: information needs of clinicians, policy makers, and the public. Am J Epidemiol. 2002;156: 311–318. [DOI] [PubMed] [Google Scholar]
- 12.Natowicz M. Newborn screening–setting evidence-based policy for protection. N Engl J Med. 2005;353:867–870. [DOI] [PubMed] [Google Scholar]
- 13.Botkin JR, Clayton EW, Fost NC, et al. Comment on a report of the American College of Medical Genetics report titled Newborn Screening: toward a uniform screening panel and system. Pediatrics. In press.
- 14.Title XXVI of the Children’s Health Act of 2000, Screening for Heritable Disorders, 42 USC §300b-10.
- 15.Green NS, Pass KA. Neonatal screening by DNA microarray: spots and chips. Nat Rev Genet. 2005;6: 147–151. [DOI] [PubMed] [Google Scholar]
- 16.Millner BN. Insurance coverage of special foods needed in the treatment of phenylketonuria. Public Health Rep. 1993;108:60–65. [PMC free article] [PubMed] [Google Scholar]
- 17.Brown AS, Fernhoff PM, Waisbren SE, et al. Barriers to successful dietary control among pregnant women with phenylketonuria. Genet Med. 2002;4: 84–89. [DOI] [PubMed] [Google Scholar]
- 18.Centers for Disease Control and Prevention. Barriers to dietary control among pregnant women with phenylketonuria–United States, 1998–2000. MMWR Morb Mortal Wkly Rep. 2002; 51:117–120. [PubMed] [Google Scholar]
- 19.Wang SS, Fernhoff PM, Hannon WH, Khoury MJ. Medium chain acyl-CoA dehydrogenase deficiency human genome epidemiology review. Genet Med. 1999;1:332–339. [DOI] [PubMed] [Google Scholar]
- 20.Pourfarzam M, Morris A, Appleton M, Craft A, Bartlett K. Neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency. Lancet. 2001; 358:1063–1064. [DOI] [PubMed] [Google Scholar]
- 21.Carpenter K, Wiley V, Sim KG, Heath D, Wilcken B. Evaluation of newborn screening for medium chain acyl-CoA dehydrogenase deficiency in 275 000 babies. Arch Dis Child Fetal Neonatal Ed. 2001;85:F105–F1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waisbren SE, Albers S, Amato S, et al. Effect of expanded newborn screening for biochemical genetic disorders on child outcomes and parental stress. JAMA. 2003;290:2564–2572. [DOI] [PubMed] [Google Scholar]
- 23.Atkinson K, Zuckerman B, Sharfstein JM, Levin D, Blatt RJ, Koh HK. A public health response to emerging technology: expansion of the Massachusetts newborn screening program. Public Health Rep. 2001;116:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holtzman NA. Expanding newborn screening: how good is the evidence? JAMA. 2003;290:2606–2608. [DOI] [PubMed] [Google Scholar]
- 25.Ratjen F, Doring G. Cystic fibrosis. Lancet. 2003;361:681–689. [DOI] [PubMed] [Google Scholar]
- 26.Green NS, Dolan SM, Oinuma M. Implementation of newborn screening for cystic fibrosis varies widely between states. Pediatrics. 2004;114:515–516. [DOI] [PubMed] [Google Scholar]
- 27.Gregg RG, Simantel A, Farrell PM, et al. Newborn screening for cystic fibrosis in Wisconsin: comparison of biochemical and molecular methods. Pediatrics. 1997;99:819–824. [DOI] [PubMed] [Google Scholar]
- 28.Comeau AM, Parad RB, Dorkin HL, et al. Population-based newborn screening for genetic disorders when multiple mutation DNA testing is incorporated: a cystic fibrosis newborn screening model demonstrating increased sensitivity but more carrier detections. Pediatrics. 2004;113: 1573–1581. [DOI] [PubMed] [Google Scholar]
- 29.The American College of Obstetricians and Gynecologists and American College of Medical Genetics. Preconception and Prenatal Carrier Screening for Cystic Fibrosis: Clinical and Laboratory Guidelines. Washington, DC: American College of Obstetricians and Gynecologists; 2001.
- 30.Grosse SD, Boyle CA, Botkin JR, et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Morbid Mortal Wkly Rep. 2004;53(RR-13): 1–36. [PubMed] [Google Scholar]
- 31.Farrell PM, Kosorok MR, Rock MJ, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics. 2001;107:1–13. [DOI] [PubMed] [Google Scholar]
- 32.Farrell PM, Li Z, Kosorok MR, et al. Bronchopulmonary disease in children with cystic fibrosis after early or delayed diagnosis. Am J Respir Crit Care Med. 2003;168:1100–1108. [DOI] [PubMed] [Google Scholar]
- 33.Clayton EW. What should be the role of public health in newborn screening and prenatal diagnosis? Am J Prev Med. 1999;16:111–115. [DOI] [PubMed] [Google Scholar]
- 34.Oliver S, Dezateux C, Kavanagh J, Lempert T, Stewart R. Disclosing to parents newborn carrier status identified by routine blood spot screening. Cochrane Database Syst Rev. October 2004:CD003859. [DOI] [PMC free article] [PubMed]
- 35.Kladny B, Gettig EA, Krishnamurti L. Systematic follow-up and case management of the abnormal newborn screen can improve acceptance of genetic counseling for sickle cell or other hemoglobinopathy trait. Genet Med. 2005;7:139–142. [DOI] [PubMed] [Google Scholar]
- 36.Lindegren ML, Kobrinksi L, Rasmussen S, et al. Applying public health strategies to primary immunodeficiency diseases: a potential approach to genetic disorders. MMWR Morb Mortal Wkly Rep. 2004;53(RR-1):1–29. [PubMed] [Google Scholar]
- 37.Meikle PJ, Ranieri E, Simonsen H, et al. Newborn screening for lysosomal storage disorders: clinical evaluation of a two-tier strategy. Pediatrics. 2004;114: 909–916. [DOI] [PubMed] [Google Scholar]
- 38.Merikangas KR, Risch N. Genomic priorities and public health. Science. 2003;302:599–601. [DOI] [PubMed] [Google Scholar]
- 39.Levy HL. Lessons from the past—looking to the future. Newborn screening. Pediatr Ann. 2003;32:505–508. [DOI] [PubMed] [Google Scholar]
- 40.Wion E, Brantley M, Stevens J, et al. Population-wide infant screening for HLA-based type 1 diabetes risk via dried blood spots from the public health infrastructure. Ann N Y Acad Sci. 2003; 1005:400–403. [DOI] [PubMed] [Google Scholar]
- 41.Ross LF. Minimizing risks: the ethics of predictive diabetes mellitus screening research in newborns. Arch Pediatr Adolesc Med. 2003;157:89–95. [DOI] [PubMed] [Google Scholar]
- 42.Finley Austin MJ, Kreiner T. Integrating genomics technologies in health care: practice and policy challenges and opportunities. Physiol Genomics. 2002; 8:33–40. [DOI] [PubMed] [Google Scholar]