

Abstract
Farnesoid X receptor (FXR), a bile-acid-activated transcriptional factor and a member of the hormone nuclear receptor superfamily, is essential in regulating bile-acid, cholesterol, and triglyceride homeostasis. Disruption of the FXR gene in mice results in a proatherosclerotic lipid profile with increased serum cholesterols and triglycerides. However, the role of FXR in foam-cell formation and atherosclerosis development remains unclear. Our current study showed that the peritoneal macrophages isolated from FXR-null mice took up less oxidized LDL-cholesterol (oxLDL-C), which was accompanied by a marked reduction of CD36 expression in these cells. This result appears to be FXR-independent, as FXR was not detected in the peritoneal macrophages. To assess to what extent FXR modulates atherosclerosis development, FXR/ApoE double-null mice were generated. Female mice were used for atherosclerosis analysis. Compared to ApoE-null mice, the FXR/ApoE double-null mice were found to have less atherosclerotic lesion area in the aorta, despite a further increase in the serum cholesterols and triglycerides. Our results indicate that deletion of the FXR gene could attenuate the atherosclerosis development, most likely resulting from reduced oxLDL-C uptake by macrophages. Our study cautions the use of serum lipid levels as a surrogate marker to determine the efficiency of FXR modulators in treating hyperlipidemia.
Keywords: FXR, nuclear receptor, atherosclerosis, cholesterol, cytokines
The abbreviations used are: ACAT: acyl coenzyme, A: acylcholesterol transferase, FXR: farnesoid X receptor, FAS: fatty-acid synthase, LOX-1: oxidized LDL receptor-1, LPL: lipoprotein lipase, LPS: lipopolysaccharide, LXR: liver X receptor, oxLDL-C: oxidized LDL cholesterol, PLTP: phospholipid transfer protein, PPARγ: peroxisome proliferator-activated receptor γ, SR: scavenger receptors, SREBP1c: sterol-response-element-binding protein 1c, VCAM-1: vascular cell adhesion molecule-1
INTRODUCTION
Some individuals are less prone to atherosclerosis despite high cholesterol intake or hypercholesterolemia with elevated LDL-cholesterol (LDL-C), suggesting that factors in addition to hypercholesterolemia contribute to the development of atherosclerosis. The current consensus is that atherosclerosis is an inflammatory disease. Although promoted by hypercholesterolemia, the development of atherosclerosis is initiated by foam-cell formation, (1, 2).
The sequence of events that lead to the generation of the earliest visible lesion of atherosclerosis, the fatty-streak lesion, starts from the formation of lipid-laden macrophages, the foam cells. Foam cells were formed from accumulation of oxidized LDL-C (oxLDL-C) that serves as an inflammatory stimulus and induces the expression of vascular cell adhesion molecule-1 (VCAM-1) on the endothelial cells in the main arteries (3). Expression of VCAM-1 assists in recruiting inflammatory cells from the circulation, and facilitates their subsequent transendothelial migration. The inflammatory cells are composed mainly of monocytes and T cells (4, 5). Once the monocytes penetrate and reside in the subendothelial space, they become activated and differentiate into macrophages. Macrophages are crucial for atherosclerosis because they express cell surface scavenger receptors (SR), such as CD36 that recognizes the altered molecular pattern on oxLDL-C. Binding of oxLDL-C to CD36 leads to the subsequent internalization of oxLDL-C, which provides a prerequisite for lipid accumulation in macrophages.
Macrophages use two mechanisms to reduce high cholesterol levels that are toxic to cells. First, excess free cholesterol undergoes re-esterification by acyl coenzyme A: acylcholesterol transferase (ACAT) to form cholesterol esters. Second, cholesterol efflux pathways are activated to transport free cholesterol out of the cell. Two efflux pathways exist, one is an active transport pathway that is mediated by the ABCA1 transporter and facilitated by ApoA-I, and the other one is a passive transport pathway facilitated by ApoE that transfer cholesterols to HDL, which then transports cholesterol back into the liver, a process termed reverse cholesterol transport. These two pathways collaborate to reduce the content of intracellular cholesterol (6).
Several lines of evidence show that the processes of uptake and/or efflux of cholesterol from macrophages are regulated by nuclear receptors, a superfamily of ligand-activated transcription factors that are involved in various developmental, physiological, and toxicological processes. The expression of CD36 is directly activated by peroxisome proliferator-activated receptor γ (PPARγ) (7). The expression of ABCA1 and ApoE is directly activated by liver X receptor (LXR), and indirectly via a PPARγ-LXR-ABCA1/ApoE cascade (8, 9). FXR is another member of the nuclear receptor supefamily that regulates cholesterol and bile acid metabolism and transport (10). Bile acids were identified as endogenous ligands for FXR and indeed FXR was found to be essential in regulating bile-acid homeostasis (11–14). Apart from playing a critical role in regulating bile-acid biosynthesis, metabolism, and transport, FXR is also important in regulating the homeostasis of cholesterol and triglycerides. There are two mechanisms by which FXR regulates cholesterol metabolism. One mechanism is to regulate the genes involved in bile-acid metabolism that catalyzes the elimination of cholesterol, since cholesterol is the precursor for bile-acid synthesis. The other mechanism is to directly regulate the genes involved in transport of cholesterol and bile acids. The emerging evidence demonstrates that FXR regulates several key components involved in maintaining cholesterol homeostasis, particularly those of HDL. FXR induces the expression of the phospholipid transfer protein (PLTP) (15), which encodes an enzyme involved in the transfer of phospholipids from very-low-density lipoproteins (VLDL) and LDL to HDL, indicating that activation of FXR induces PLTP levels to promote HDL formation. FXR also induces hepatic scavenger receptor B1 (SR-B1), which is the HDL-uptake receptor in liver and other peripheral tissues to remove cholesterol from the circulation; thus serves as an important component in reverse cholesterol transport (16). However, activation of FXR might result in reduction of HDL by suppressing ApoA-I, the major component of HDL, via binding as a monomer to a negative response element in the ApoA-I promoter region (17).
FXR appears to regulate serum and hepatic triglyceride levels. Activation of FXR induces the expression of ApoC-II and concurrently reduces expression of ApoC-III (18). ApoC-II is an activator, and ApoC-III is an inhibitor of lipoprotein lipase (LPL), which hydrolyzes triglycerides to free fatty acids to remove triglycerides from the blood. Disruption of the FXR gene in mice results in upregulation of the expression of two genes critical for hepatic lipogenesis, sterol-response-element-binding protein 1c (SREBP1c) and fatty-acid synthase (FAS) (19, 20), indicating that FXR inhibits the de novo lipogenesis in liver. Mice with a targeted disruption of the FXR gene clearly revealed a critical role for this receptor in maintaining the homeostasis of bile acids, cholesterol and triglycerides (13, 16).
Although disruption of the FXR gene in mice results in a proatherosclerotic lipid profile, the role of FXR in foam-cell formation is unclear. Elucidation of the role of FXR in the formation of foam cells as well as in the development of atherosclerotic plaques is critical in understanding the role of FXR in regulating lipid metabolism. This knowledge will also provide a scientific basis for future design of FXR modulators. Therefore, the current study was designed to investigate to what extent loss of FXR function in vivo affects the formation of foam cells, and furthermore, to elucidate the role of FXR in the development of atherosclerosis using the ApoE-deficient genetic model for accelerated atherosclerosis.
MATERIALS AND METHODS
Materials
Lipopolysaccharide (LPS) was purchased from Sigma (St. Louis, MD). Tissue culture reagents, unless otherwise indicated, were obtained from Invitrogen (Carlsbad, CA). Nutridoma HU was purchased from Roche (Indianapolis, IN). OxLDL was obtained from INTRACEL (Fredrick, MD), [3H]cholesterol was purchased from Perkin-Elmer Life and Analytical Sciences, Inc. (Boston, MA). ApoA-I and HDL3 were purchased from Calbiochem (San Diego, CA) and Sigma (St. Louis, MO), respectively. ELISA kits for cytokine measurement were obtained from Pierce (Rockford, IL).
Animals and Treatment
B6;129-Fxrtm1Gonz (FXR-null) mice were backcrossed to a C57BL/6 genetic background for 10 generations and genotyped by a PCR-based method as described previously (13). C57BL/6 mice were used as wild-type controls. The ApoE-null mice in a C57BL/6 genetic background were obtained from The Jackson Laboratory (Bar Harbor, ME). FXR/ApoE double-null mice were generated by cross-breeding between FXR-null and ApoE-null mice. All the animals were housed in a pathogen-free animal facility under a standard 12 hr light/12 hr dark cycle with access to chow and water ad libitum. All protocols and procedures were approved by the NCI Animal Care and Use Committee and are in accordance with the NIH and ALAC Guidelines. For the atherosclerosis study, female FXR/ApoE double-null and ApoE-null mice were randomly divided into two groups when weaned at three weeks of age (n=8 to 13/group). To develop spontaneous atherosclerosis, one group of mice from each strain was placed on a control diet and the tissues were collected at 20 weeks of age. To develop high-fat-induced atherosclerosis, the other group of mice from each strain was placed on an AIN-93G-modified 35% high fat (HF) diet (F4048, Bio-Serv, Frenchtown, NJ) for ten weeks and the tissues were collected at 13 weeks of age. Food intake and body weight were monitored every week. Overnight fasting blood was collected before the animals were killed and the tissues collected. Serum was separated by a Microtainer serum separating tube (BD Biosciences). Plasma levels of total cholesterol and total triglycerides were quantified by commercially available kits (Wako, VA).
Fast Protein Liquid Chromatography (FPLC)
FPLC separation of serum lipoproteins from pooled plasma samples (60 μl; n=8 to 14) was achieved by gel filtration using two Superose 6HR 10/30 columns (GE Bioscience) in series as previously described (13, 21).
Macrophage Isolation
For macrophage isolation, groups of 8- to 12-week-old female mice were used for all experiments. On day 0, mice were injected intraperitoneally with 2 ml of 3% thioglycolate (Becton Dickinson, Cockeysville, MD), and peritoneal macrophages were collected on day 4 as described (7). After centrifugation at 1000 × g for 5 min, the cells were resuspended into RPMI 1640 media containing 10% fetal bovine serum (FBS) and 100 U/ml of penicillin-streptomycin, 3 to 5 million cells were plated onto 100-mm Petri dishes. The non-adherent cells were removed and the culture media were changed fresh after 4 hrs incubation at 37 °C.
Cholesterol Loading and Efflux
Peritoneal macrophages were seeded at 50 to 70% confluence for 2 days (0.2 million/well in 12 well plate), then the cells were washed twice with 1X PBS, followed by incubation in RPMI 1640 supplemented with 1% Nutridoma HU, ox-LDL (50 μg/ml), and 2 μci/ml [3H] cholesterol. After a 48-hr incubation period, cells were washed twice with 1X PBS, and the medium was changed into RPMI 1640 supplemented with or without ApoA-I (50 μg/ml) or HDL3 (50 μg/ml) and incubated for another 24 hrs. The radioactivity of 3H was measured by liquid scintillation counting in centrifuged media as well as in cell samples lysed in 0.2 ml of 1M NaOH. The percentage of efflux is the ratio of counts in the supernatant to the total counts, and ApoA-I or HDL3–mediated cholesterol efflux were determined as the fraction of total radiolabeled cholesterol in the supernatant in the presence of ApoA-I or HDL3 after subtraction of values for ApoA-I- or HDL3-free media. Each experiment was carried out in triplicate and repeated four times.
Macrophage Lipid Analysis by Oil-Red O Staining
Peritoneal macrophages were collected (n=3) and seeded in 35 mm × 10 mm tissue culture dishes in RPMI 1640 with 1% Nutridoma HU at 1 million cell/dish density for 24 hrs before fresh media were changed with or without 50 μg/ml oxLDL. After another 24 hrs, the cells were washed twice with 1X PBS and fixed in 3.7% formaldehyde at room temperature for 1 hr. The cells were then washed three times with 1X PBS and stained with 2 ml of oil red solution for 1 hr at room temperature. For quantification of oil red O staining, 0.5 ml of isopropanol was added to each dish, and oil red was extracted into isopropanol. The degree of oil red staining was measured at 518 nm by a DU800 spectrophotometer (Beckman Coulter, Fullerton, CA).
Macrophage Cholesterol Analysis
Peritoneal macrophages from each mouse (n=6/group) were collected and seeded in 100 mm × 10 mm tissue culture dishes in RPMI 1640 with 1% Nutridoma HU at 1 million cell/dish density for 24 hrs before fresh media were changed with or without 50 μg/ml oxLDL. After another 24 hrs, the cells were washed twice with 1X PBS. The cells were lysed with 1 ml of 1 × PBS and 1% triton-X 100 and the protein concentrations were determined by BCA method. The cellular lipids were extracted according to the method published previously (13), and the total cholesterol levels were determined by a commercially available kit (Wako, VA).
Northern Blot Analysis of RNA
Peritoneal macrophages were collected and cultured as described in the previous section. The cells were washed once in 1X PBS, and total RNA were prepared using Trizol reagent (GIBCO–BRL, Grand Island, NY) and analyzed by electrophoresis of 5 μg total RNA in 0.22 M formaldehyde-containing 1% agarose gels. The cDNA probes and detailed northern blot analysis procedures were described (7). Probes were 32P-labeled by the random primer method using Ready-to-Go DNA labeling beads (Amersham-Pharmacia Biotech, Piscataway, NJ). The bands were quantified using a phosphoimager and the values expressed as fold of changes after corrected for β-actin RNA levels.
Flow Cytometry
The peritoneal macrophages were collected from wild-type and FXR-null female mice (n=3/genotype) as described in the previous section. 0.5 × 106 cells from each mouse were used and rat anti-mouse-FcγRIII/II-blocking reagent (BD Biosciences, San Jose, CA) was used to block non-specific staining. The cells were subsequently stained by anti-CD36-PE antibody (2 μg/half million of cells; Santa Cruz, Santa Cruz, CA) and analyzed by a flow cytometers (Becton Dickinson).
ELISA Assay for Cytokines
The peritoneal macrophages were collected and cultured as described in the previous section. The cell culture supernatants were collected after incubation with LPS (1 μg/ml) overnight (about 16 hrs), and kept frozen at − 80 °C until the cytokine levels (IFNγ, TNFα, IL-6, and IL-1β) were determined by ELISA assays according to the manufacturer’s instructions.
Aortic Lesion Analysis
The heart and the attached section of the ascending aorta were prepared and analyzed as described (22). Briefly, after blood was collected, the heart and aorta were carefully removed and transferred to 37 °C saline for 20 min. Afterwards, the samples were transferred to 4% PBS-buffered formaldehyde for 24 hrs before being permanently fixed in 10% PBS-buffered formaldehyde. Serial 10-μm-thick sections from the middle portion of the ventricle to the aortic arch were collected and mounted on poly-D-lysine-coated slides. In the region from the appearance to the disappearance of the aortic valves, every other section was collected. In all other regions, every fifth section was collected. Sections were stained with oil red and hematoxylin, counterstained with fast green, and lesion areas were examined by light microscopy followed by quantification and analysis with NIH image quantification software (http://rsb.info.nih.gov/nih-image).
Statistics
Unless otherwise stated, all values were expressed as the mean + SE. All data were analyzed by the unpaired Student’s t-test for statistical significance between WT or null mice. For the comparison among multiple groups, one-way ANOVA followed by Duncan’s test were performed.
RESULTS
OxLDL-C uptake in peritoneal macrophages
Deletion of the FXR gene in mice leads to disruption of the homeostasis of bile acids, cholesterols, and triglycerides. Macrophages are critical in cholesterol metabolism and alterations in the uptake, metabolism, and efflux of cholesterol in macrophages could affect foam-cell formation, a prerequisite for atherosclerosis development. To determine if deletion of the FXR gene affects foam-cell formation, we examined the accumulation of oxLDL-C in peritoneal macrophages from wild-type and FXR-null mice. OxLDL-C is a modified form of LDL which is considered the most relevant form of LDL to contribute to foam-cell formation. The uptake of oxLDL-C in peritoneal macrophages isolated from FXR-null mice was significantly reduced, as determined by oil-red staining (Fig 1A and B) and total cholesterol measurement (Fig 1C).
Fig 1. ox-LDL Cholesterol loading in peritoneal macrophages from wild-type and FXR-null mice.
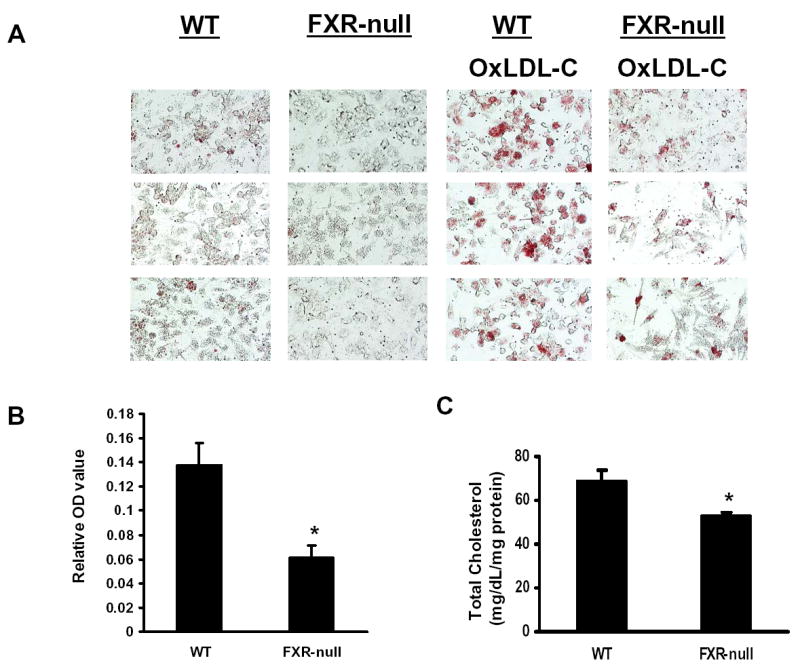
Peritoneal macrophages from wild-type or FXR-null mice were incubated with oxLDL, and the levels of cholesterol loading were determined by oil-red staining followed by spectrometric quantification as shown in Panel A and B. Panel A consists of the pictures taken randomly from three microscopic fields and panel B shows the quantification of oil-red staining following normalization by protein content. Panel C is the total cellular cholesterol levels after incubating with oxLDL. An asterisk indicates P<0.05.
Cholesterol efflux rate in peritoneal macrophages
A decrease in cholesterol accumulation in macrophages could also be due to decrease in cholesterol uptake or increase in cholesterol efflux. Therefore, the rate of cholesterol efflux from peritoneal macrophages isolated from WT or FXR-null mice was determined. Cholesterol efflux mediated by ApoA-I mainly represents cholesterol excreted by the activate transporter, ABCA1, whereas cholesterol efflux mediated by HDL3 represents both the active transport as well as the passive diffusion that depends mainly on the levels of ApoE (23). We found no difference in the cholesterol-efflux rate between macrophages from WT or FXR-null mice (Fig 2). Therefore, the reduced accumulation of oxLDL-C in macrophages isolated from FXR-null mice was probably due to a decrease in cholesterol uptake.
Fig 2. Rate of Cholesterol efflux in peritoneal macrophages from wild-type and FXR-null mice.
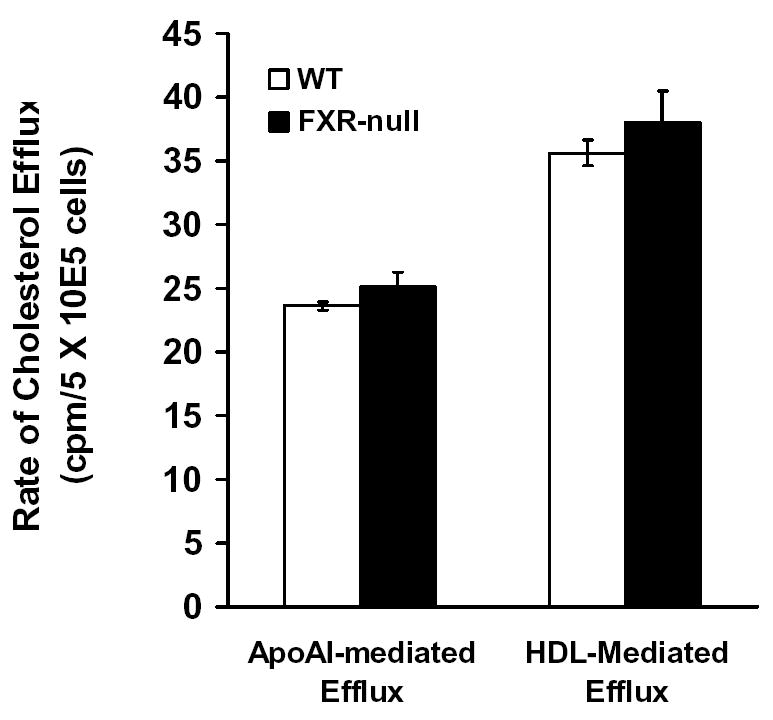
Peritoneal macrophages were isolated and incubated with [3H]cholesterol. The efflux of cholesterol was initiated by addition of ApoA-I or HDL3. The percentage of efflux is the ratio of counts in the supernatant to the total counts, and ApoA-I- or HDL3–mediated cholesterol efflux was determined as the fraction of total radiolabeled cholesterol in the supernatant in the presence of ApoA-I or HDL3 after subtraction of values for ApoA-I- or HDL3-free media.
Expression of genes involved in cholesterol homeostasis in peritoneal macrophages and CD36 expression
Uptake of oxLDL-C in macrophages was mainly carried out by CD36, a fatty acid transporter, although several other scavenger receptors (SR), such as SRA1, SRB1, and oxidized LDL receptor-1 (LOX-1), are also involved. The efflux of cholesterol to HDL in macrophages depends largely upon ABCA1 and ApoE. Thus, the mRNA levels of the aforementioned genes were determined by Northern blot analysis. Whereas SRA1, SRB1, LOX-1, and ABCA1 mRNA levels were similar (data not shown), those of CD36 and ApoE were markedly reduced in macrophages isolated from FXR-null mice compared to those from WT mice (Fig 3A). The protein levels of CD36 on the surface of the macrophages were determined by flow-cytometry analysis, which showed a significant reduction of macrophages expressing CD36 on their surface (Fig 3B). FXR was not detected in macrophages by RT-PCR analysis and by Northern blot analysis (data not shown) indicating that the reduction of CD36 and ApoE expression are due to an indirect mechanism following the deletion of the FXR gene. CD36 is a target gene of the nuclear receptor, PPARγ (7), and ApoE is a target gene of LXRα (24). Therefore, PPARγ and LXR mRNA levels were determined in the macrophages from FXR-null and wild-type mice, but no difference in the expression levels of these two nuclear receptors was found (Fig 3A).
Fig 3. The expression levels of genes involved in cholesterol metabolism in peritoneal macrophages.
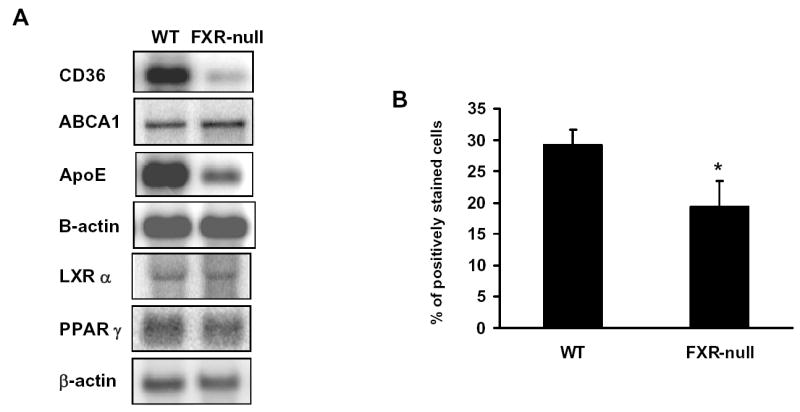
Peritoneal macrophages were isolated and pooled from a total of five wild-type and FXR-null mice. Total RNA were isolated and subjected to Northern blotting analysis (Fig A). The cell surface expression of CD36 was determined by flow cytometry (Fig B).
Levels of cytokines secretion from peritoneal macrophages
Besides the aforementioned nuclear receptors, the homeostasis of cholesterol in macrophage is also maintained by immunological factors, such as cytokines. For example, treatment of macrophages with IFNγ reduces the expression levels of CD36 and ApoE. Therefore, peritoneal macrophages isolated from WT or FXR-null mice were stimulated overnight with 1 μg/ml LPS, followed by the measurement of the secretion of cytokines from these cells by ELISA. The levels of secreted IL-1β tended to be lower in macrophages from FXR-null mice than those from WT mice, while levels of IL-6 secretion were similar between macrophages from the two mouse lines. However, the levels of TNFα and IFNγ secretion were markedly increased in FXR-null macrophages compared to WT macrophages (Fig 4).
Fig 4. The levels of cytokines secreted from peritoneal macrophages isolated from wild-type and FXR-null mice.
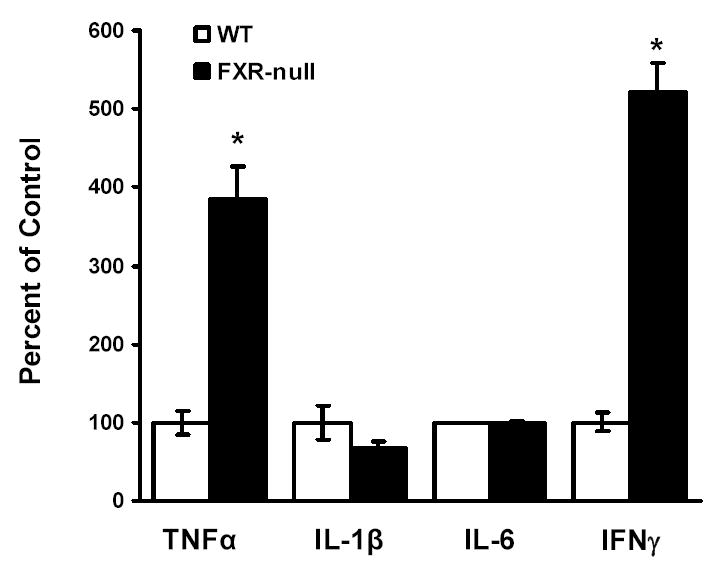
Peritoneal macrophages were isolated from wild-type and FXR-null mice. The cells were incubated with LPS (1 μg/ml) overnight. The supernatant from the cell culture was collected and the levels of cytokines, TNFα, IL-1β, IL-6 and IFNγ, were determined by ELISA analysis. An asterisk represents P<0.05.
Generation of FXR/ApoE double-null mice and characterization of their lipid profile
FXR is critical in regulating the homeostasis of cholesterol and triglycerides, and studies with the FXR-null mice clearly revealed that deletion of the FXR gene results in a proatherogenic lipid profile (13, 16). However, primary macrophages isolated from FXR-null mice showed a marked reduction of CD36 expression and oxLDL-C uptake, thus less form-cell formation, compared to wild-type mice. This indicates that deletion of the FXR gene may attenuate atherosclerosis development, as foam-cell formation is a prerequisite for atherosclerosis development. In order to determine the role of FXR in atherosclerosis, FXR/ApoE double-null mice (FA DN) were generated by cross-breeding FXR-null with ApoE-null mice, both on a C57BL/6 background. Following weaning, three-week-old male and female WT, FXR-null, ApoE-null and FA DN mice were randomly divided into two groups (n=8 to 10/group). One group of each strain was placed on a control chow diet until the mice were sacrificed at 20 weeks of age. The other group was fed a 35% high-fat diet for 10 weeks and the mice were sacrificed at 13 weeks of age. Fasting serum were collected right before the mice were sacrificed, and the serum lipid levels were determined. Fig 5 showed the serum total cholesterol and triglyceride levels in these mice, with panels A (control diet) and B (high-fat diet) for total cholesterols and C (control diet) and D (high-fat diet) for triglycerides. Regardless of the diet type and gender, FXR/ApoE double-null mice had the highest serum total cholesterols and triglycerides, followed by ApoE-null and FXR-null mice. The serum lipid contents and lipoprotein profiles were further analyzed by FPLC using pooled serum samples from females mice fed with the HF diet (Fig 6). For the result shown as FPLC analysis, the elution around 12–14 represents for VLDL, 18–22 for LDL, and 28–32 for HDL. Our result showed that deletion of the ApoE gene increased the fraction of VLDL in serum, which was elevated markedly with the deletion of the FXR gene. This was accompanied with a decrease in the fraction of LDL (around 20%) and a slight increase in HDL portion.
Fig 5. Analysis of serum total cholesterol and triglycerides in wild-type, FXR-null, ApoE-null and FXR/ApoE double-null mice.
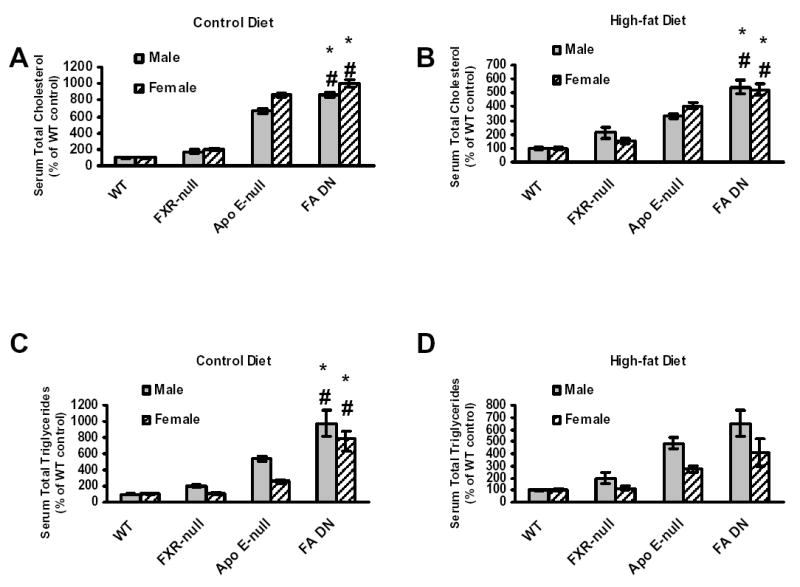
FXR/ApoE double-null mice were generated by cross-breeding of FXR-null and ApoE-null mice. Serum was collected from male and female animals fed with a control diet at 20 weeks of age, or a high-fat diet for 10 weeks at 13 weeks of age. The grey bar represents male mice and the striped bar for female mice. (A) is for serum total cholesterol on control diet, (B) is for serum total cholesterol on high-fat diet, (C) is for serum total triglyceride on control diet, and (D) is for serum total triglyceride on high-fat diet. The cholesterol concentrations, in mg/dL, for the control and high-fat fed male and female wild-type mice were: male wild-type = 52.9 +/− 3.4, female wild-type = 36.3 +/− 2.2 for the control diets, and male, wild-type = 92.5 +/− 9.3, female, wild-type = 64.1 +/− 5.0 for the high-fat diets. The triglyceride concentrations, in mg/dL, for the control and high-fat fed male and female wild-type mice were: male wild-type = 49.7 +/− 5.5, female wild-type = 37.1 +/− 3.2 for control diets, and male wild-type = 51.1 +/− 3.8, female wild-type = 36.8 +/− 4.6 for the high-fat diets. Data from each group are derived from 8 to 14 mice. An asterisk indicates P<0.05 between FXR-null and FXR/ApoE double-null mice. A # indicates P<0.05 between ApoE-null and FXR/ApoE double-null mice.
Fig. 6. Analysis of serum lipid profiles in high-fat-fed female wild-type, FXR-null, ApoE-null, and FXR/ApoE (FA) double-null mice.
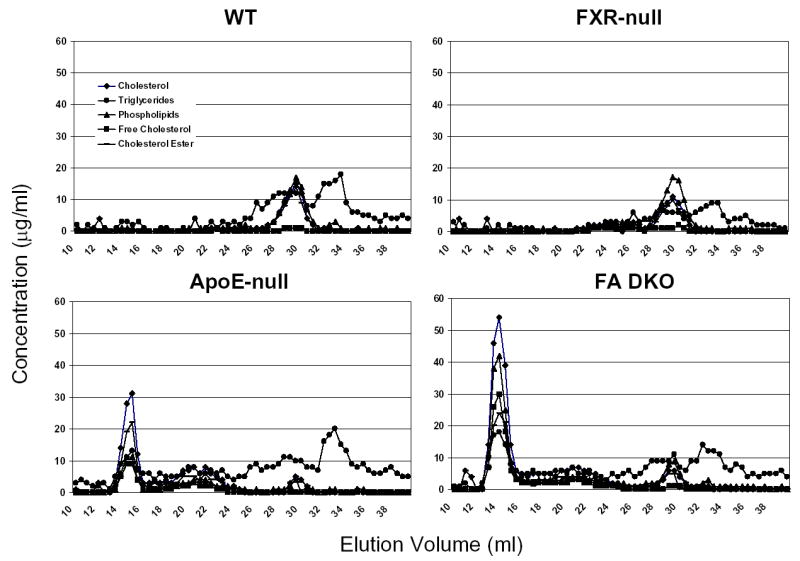
FPLC separation of serum lipoproteins from pooled plasma samples (60 μl, n=8 to 14) was achieved by gel filtration using two Superose 6HR 10/30 columns in series. The concentration of cholesterol, triglycerides, phospholipids, free cholesterol, and cholesterol ester was indicated in the y axes. FPLC analysis is shown for wild-type mice, FXR-null mice, ApoE-null mice, and FA DKO mice. Elution volume from 12–14 represents for VLDL, 18–22 for LDL, and 28–32 for HDL.
Analysis of aortic atherosclerosis development in FXR/ApoE double-null mice
To investigate the role of FXR in modulating atherosclerosis development, the aortas of female ApoE-null and FXR/ApoE double-null mice on the control diet (at 20 weeks of age) or on the high-fat diet (at 13 weeks of age) were analyzed and the degree of atherosclerotic lesion development was quantified by measuring the areas that were positive for staining of oil red (Fig. 7). Female mice were chosen to be the gender of focus in this experiment because a previous study indicated that female mice are more prone to atherosclerotic lesion development compared to male mice (25). On the control diet, a trend toward a decrease in the mean aortic lesion area in FXR/ApoE double-null mice compared to ApoE-null mice was noted (Fig 7A). When the animals were fed a high-fat diet, the atherosclerotic lesion areas in FXR/ApoE double-null mice were significantly reduced compared to that of ApoE-null mice (Fig 7B).
Fig 7. Analysis of aortic atherosclerotic lesion area for female ApoE-null and FXR/ApoE double-null mice (FA DN).
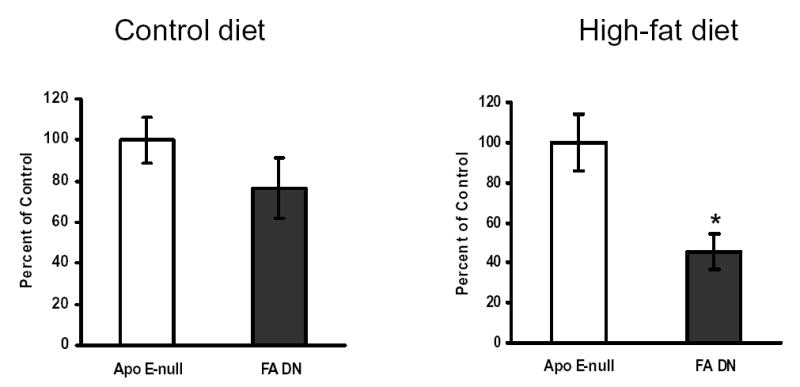
Aortas and hearts that were harvested from female ApoE-null and FXR/ApoE double-null mice at 20 weeks of age with a control diet (A) or at 13 weeks of age following a high-fat diet for 10 weeks (B). The numbers of mouse used per group are: 13 for ApoE-null mice on control diet, 9 for FA-DN mice on control diet, 8 for ApoE-null mice on high-fat diet, and 8 for FA-DN mice on high-fat diet. Five serial sections from each mouse were counted using oil-red O staining. Lesion areas were examined by light microscopy followed by quantification and analysis with NIH image quantification software. An asterisk indicates P<0.05 between ApoE-null and FXR/ApoE double-null mice.
DISCUSSION
In the current study, primary peritoneal macrophages isolated from the FXR-null mice were found to take up less oxLDL-C than those from the wild-type mice, which probably is due to the reduced expression of CD36 in macrophages isolated from FXR-null mice. CD36 is the main transporter in macrophages for the uptake of oxLDL-C. Although the FXR/ApoE double-null mice demonstrate a proatherosclerotic serum lipid profile, they had less atherosclerotic lesion area in the aorta. Our data clearly show that disruption of the FXR gene attenuates atherosclerosis development, confirming the concept that foam-cell formation leads to the initiation and progression of atherosclerosis.
CD36 expression increases as monocytes differentiate into macrophages (26). Surprisingly, Northern blot or reverse-PCR analysis did not detect FXR mRNA expression in mouse peritoneal macrophages (data not shown), and thus direct regulation of CD36 expression by FXR seems unlikely. However, expression of the CD36 gene in macrophage could be inhibited by increases in IFNγ, glucocorticoids, lipopolysaccharide (LPS), and transforming factor β (27–29). Furthermore, emerging evidence strongly suggests that expression of the CD36 gene in macrophages depends largely on levels of PPARγ, a ligand-activated transcription factor that belongs to the nuclear-receptor superfamily. Activation of PPARγ is involved in macrophage cholesterol metabolism, adipocyte differentiation, and suppression of inflammation (30, 31). Expression of CD36 in macrophage is minimal with deletion of the PPARγ gene and is strongly induced by PPARγ ligands (7, 32). Moreover, expression and activation of PPARγ is required to induce CD36 expression by several other factors, including phorbol esters, macrophage colony-stimulating factors, and IL-4 (33). The reduced PPARγ target gene expression could be due to a decrease in PPARγ activity or a reduction of endogenous PPARγ ligands. Our results showed that the expression levels of PPARγ were similar between wild-type and FXR-null macrophages; however, the activity of PPARγ could be modulated upon post-translational events, including interactions with co-activators and co-repressors, ligand availability, and phosphorylation status. For example, SHP, a target gene of FXR, augments PPARγ transcription activity in hepatocytes by inhibiting binding of co-repressors to PPARγ (34). Phosphorylation of PPARγ protein by mitogen-activated protein kinase decreases PPARγ transcription activity (35, 36). To what extent the activity of PPARγ is altered in macrophages of FXR-null mice remains unclear and elucidation of which will aid in understanding the mechanism by which CD36 is down-regulated in FXR-null macrophages.
The current study further confirms that the formation of foam cells precedes the initiation and development of the atherosclerotic lesion. Lack of FXR decreases the accumulation of cholesterol in macrophages, indicating that inhibition of FXR could reduce foam-cell formation, resulting in attenuation of the development of atherosclerotic lesions. However, the serum lipid profile of FXR-null mice indicates that loss of the FXR actually increases levels of cholesterol and thus results in a proatherosclerotic lipid profile (13). Although a balance between reduced foam-cell formation and increased serum cholesterol and triglycerides will ultimately determine the outcome of atherosclerosis development in FXR-null mice, current theory for the initiation of atherosclerosis development emphasizes the importance of foam-cell formation, rather than hypercholesterolemia. To understand to what extent FXR modulates atherosclerosis development is critical as there is a growing effort in designing and identifying FXR modulators to treat diseases that are caused by abnormal levels of bile acids and/or lipids, such as cholestasis and hyperlipidemia (37). The current study demonstrates that while FXR/ApoE double-null mice clearly have increased levels of serum cholesterol and triglycerides compared to either ApoE- or FXR-null mice, analysis of the aortic lesion area reveals that FXR/ApoE double-null mice have less atherosclerotic lesions compared to ApoE-null mice. Therefore our results indicate that activating FXR could facilitate rather than attenuate atherosclerosis development. On the contrary, inhibition of FXR perhaps through the use of an antagonist could reduce atherosclerosis development despite a proatherosclerotic serum lipid profile. In addition, the FXR/ApoE double-null mice showed a slight increase in HDL, which might also contribute to the reduction in atherosclerotic lesion formation.
Finally, human FXR has been detected in vascular smooth muscle cells and activation of FXR was shown to promote apoptosis in this cell type (38). The current study indicates that FXR could affect atherosclerosis development via multiple mechanisms including a role in modulating the immune response. A marked change in cytokine secretion from the peritoneal macrophages isolated from FXR-null mice was noted; both TNFα and IFNγ, two cytokines known to be critical in the participation of innate immune defenses mediated by macrophages, were increased compared to cells from wild-type mice. The role of FXR in the immune response is largely undefined, however, one study indicates that FXR is involved in regulating immune function (39).
After we submitted this manuscript, a paper from Peter Edwards’s group reported similar findings in FXR/LDLr double knockout mice with reduced atherosclerosis development and macrophage CD36 expression, in spite of a proatherosclerotic lipid profile (40). Token together, this emerging evidence suggests that FXR is involved in regulating foam-cell formation and arthrosclerosis development.
In summary, disruption of the FXR gene in mice reduces the uptake of oxLDL and foam-cells formation, which is likely due to a decreased CD36 expression. As a result, FXR/ApoE double-null mice exhibit less aortic atherosclerotic lesions, despite a proatherosclerotic serum lipid profile. Results from the current study further confirm the current concept that formation of foam cells is prerequisite in leading to atherosclerotic development. The information obtained from this study will be of great value in directing future efforts in the development of FXR modulators to treat hyperlipidemia and cholestasis. This study suggests that only selective but not pan-FXR modulators are beneficial in treating lipid disorders. In addition, this study suggests that using serum lipid levels alone as a biomarker to predict the efficiency of FXR modulators is not appropriate.
Acknowledgments
Supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health and the BIRCWH program of the University of Kansas Medical Center.
References
- 1.Curtiss LK, Kubo N, Schiller NK, Boisvert WA. Participation of innate and acquired immunity in atherosclerosis. Immunol Res. 2000;21:167–176. doi: 10.1385/IR:21:2-3:167. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8:1211–1217. doi: 10.1038/nm1102-1211. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb. 1993;13:197–204. doi: 10.1161/01.atv.13.2.197. [DOI] [PubMed] [Google Scholar]
- 4.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 5.Bourdillon MC, Poston RN, Covacho C, Chignier E, Bricca G, McGregor JL. ICAM-1 deficiency reduces atherosclerotic lesions in double-knockout mice (ApoE(−/−)/ICAM-1(−/−)) fed a fat or a chow diet. Arterioscler Thromb Vasc Biol. 2000;20:2630–2635. doi: 10.1161/01.atv.20.12.2630. [DOI] [PubMed] [Google Scholar]
- 6.Jessup W, Kritharides L. Metabolism of oxidized LDL by macrophages. Curr Opin Lipidol. 2000;11:473–481. doi: 10.1097/00041433-200010000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee YH, Ricote M, Glass CK, Brewer HB, Jr, Gonzalez FJ. Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol. 2002;22:2607–2619. doi: 10.1128/MCB.22.8.2607-2619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 9.Laffitte BA, Tontonoz P. Orphan nuclear receptors find a home in the arterial wall. Curr Atheroscler Rep. 2002;4:213–221. doi: 10.1007/s11883-002-0022-6. [DOI] [PubMed] [Google Scholar]
- 10.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 11.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 12.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 13.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 15.Urizar NL, Dowhan DH, Moore DD. The farnesoid X-activated receptor mediates bile acid activation of phospholipid transfer protein gene expression. J Biol Chem. 2000;275:39313–39317. doi: 10.1074/jbc.M007998200. [DOI] [PubMed] [Google Scholar]
- 16.Lambert G, Amar MJ, Guo G, Brewer HB, Jr, Gonzalez FJ, Sinal CJ. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J Biol Chem. 2003;278:2563–2570. doi: 10.1074/jbc.M209525200. [DOI] [PubMed] [Google Scholar]
- 17.Claudel T, Sturm E, Duez H, Torra IP, Sirvent A, Kosykh V, Fruchart JC, Dallongeville J, Hum DW, Kuipers F, Staels B. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J Clin Invest. 2002;109:961–971. doi: 10.1172/JCI14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Claudel T, Inoue Y, Barbier O, Duran-Sandoval D, Kosykh V, Fruchart J, Fruchart JC, Gonzalez FJ, Staels B. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 2003;125:544–555. doi: 10.1016/s0016-5085(03)00896-5. [DOI] [PubMed] [Google Scholar]
- 19.Moschetta A, Bookout AL, Mangelsdorf DJ. Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nat Med. 2004;10:1352–1358. doi: 10.1038/nm1138. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambert G, Amar MJ, Martin P, Fruchart-Najib J, Foger B, Shamburek RD, Brewer HB, Jr, Santamarina-Fojo S. Hepatic lipase deficiency decreases the selective uptake of HDL-cholesteryl esters in vivo. J Lipid Res. 2000;41:667–672. [PubMed] [Google Scholar]
- 22.Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 23.LI AGCK. The macrophage foam cell as a target for therapeutic intervention. Nature Medicine. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- 24.Laffitte BA, Repa JJ, Joseph SB, Wilpitz DC, Kast HR, Mangelsdorf DJ, Tontonoz P. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci U S A. 2001;98:507–512. doi: 10.1073/pnas.021488798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Navarro H, Nong Z, Amar MJ, Shamburek RD, Najib-Fruchart J, Paigen BJ, Brewer HB, Jr, Santamarina-Fojo S. The ligand-binding function of hepatic lipase modulates the development of atherosclerosis in transgenic mice. J Biol Chem. 2004;279:45312–45321. doi: 10.1074/jbc.M406495200. [DOI] [PubMed] [Google Scholar]
- 26.Huh HY, Pearce SF, Yesner LM, Schindler JL, Silverstein RL. Regulated expression of CD36 during monocyte-to-macrophage differentiation: potential role of CD36 in foam cell formation. Blood. 1996;87:2020–2028. [PubMed] [Google Scholar]
- 27.Han J, Hajjar DP, Tauras JM, Feng J, Gotto AM, Jr, Nicholson AC. Transforming growth factor-beta1 (TGF-beta1) and TGF-beta2 decrease expression of CD36, the type B scavenger receptor, through mitogen-activated protein kinase phosphorylation of peroxisome proliferator-activated receptor-gamma. J Biol Chem. 2000;275:1241–1246. doi: 10.1074/jbc.275.2.1241. [DOI] [PubMed] [Google Scholar]
- 28.Yesner LM, Huh HY, Pearce SF, Silverstein RL. Regulation of monocyte CD36 and thrombospondin-1 expression by soluble mediators. Arterioscler Thromb Vasc Biol. 1996;16:1019–1025. doi: 10.1161/01.atv.16.8.1019. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa T, Nozaki S, Nishida M, Yakub JM, Tomiyama Y, Nakata A, Matsumoto K, Funahashi T, Kameda-Takemura K, Kurata Y, Yamashita S, Matsuzawa Y. Oxidized LDL increases and interferon-gamma decreases expression of CD36 in human monocyte-derived macrophages. Arterioscler Thromb Vasc Biol. 1998;18:1350–1357. doi: 10.1161/01.atv.18.8.1350. [DOI] [PubMed] [Google Scholar]
- 30.Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144:2201–2207. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 31.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 32.Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D, Milstone DS, Mortensen RM, Spiegelman BM, Freeman MW. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat Med. 2001;7:41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- 33.Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 34.Nishizawa H, Yamagata K, Shimomura I, Takahashi M, Kuriyama H, Kishida K, Hotta K, Nagaretani H, Maeda N, Matsuda M, Kihara S, Nakamura T, Nishigori H, Tomura H, Moore DD, Takeda J, Funahashi T, Matsuzawa Y. Small heterodimer partner, an orphan nuclear receptor, augments peroxisome proliferator-activated receptor gamma transactivation. J Biol Chem. 2002;277:1586–1592. doi: 10.1074/jbc.M104301200. [DOI] [PubMed] [Google Scholar]
- 35.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem. 1997;272:5128–5132. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 36.Camp HS, Tafuri SR, Leff T. c-Jun N-terminal kinase phosphorylates peroxisome proliferator-activated receptor-gamma1 and negatively regulates its transcriptional activity. Endocrinology. 1999;140:392–397. doi: 10.1210/endo.140.1.6457. [DOI] [PubMed] [Google Scholar]
- 37.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, Moore LB, Wilson JG, Lewis MC, Jones SA, Willson TM. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem. 2000;43:2971–2974. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- 38.Bishop-Bailey D, Walsh DT, Warner TD. Expression and activation of the farnesoid X receptor in the vasculature. Proc Natl Acad Sci U S A. 2004;101:3668–3673. doi: 10.1073/pnas.0400046101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Pircher PC, Schulman IG, Westin SK. Regulation of complement C3 expression by the bile acid receptor FXR. J Biol Chem. 2005;280:7427–7434. doi: 10.1074/jbc.M411473200. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Wang X, Vales C, Ying Lee F, Lee H, Lusis AJ, Edwards PA. FXR deficiency causes reduced atheroscelross in LDLr −/− mice. Arterioscler Thromb Vasc Biol. 2006 doi: 10.1161/01.ATV.0000235697.35431.05. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]