

Abstract
Slow receptor dissociation kinetics has been implicated in the long clinical duration of action of the muscarinic receptor antagonist tiotropium. However, despite the potential benefits of new drugs with slow dissociation kinetics, the rate parameters of new compounds are seldom measured due to technical difficulties and financial implications associated with radiolabeling multiple ligands. Here we describe the development and optimisation of a medium throughput assay which is capable of measuring the kinetic parameters of novel, unlabelled compounds.
Radioligand binding studies were performed with membranes derived from CHO cells recombinantly expressing the human M3 muscarinic receptor.
Initial characterisation of the radioligand [3H]-NMS yielded on and off rates of 4.1±0.2 × 108 M−1 min−1 and 0.015±0.0005 min−1, respectively.
The specific binding of [3H]-NMS was measured over time in the presence and absence of several concentrations of unlabelled competitor compounds. These data were analysed using a competition kinetic model to provide on and off rates for the unlabelled competitor. Comparison of the kinetically derived Kd (koff/kon) with Ki values generated at equilibrium showed an excellent correlation (r2=0.99), providing good validation of the method.
The on and off rates were also used in theoretical computer simulations to successfully predict the effect of incubation time on apparent IC50 values.
This study demonstrates that a medium-throughput competition kinetic binding assay can be used to determine accurate on and off rates of unlabelled compounds, providing the opportunity to optimise for kinetic parameters early in the drug discovery process.
Keywords: Association rate (kon), competition binding, dissociation rate (koff), duration of drug action, equilibrium, kinetics, muscarinic acetylcholine receptor, tiotropium
Introduction
Long duration of action (preferably 24 h) is an important feature of drugs intended to treat chronic illness, enabling both prolonged efficacy (Tashkin, 2005) and a simple, once-daily dosage regime that gives high patient compliance (Smith et al., 1996). The duration of drug action depends on many pharmacokinetic factors, including absorption, distribution and clearance (Smith et al., 1996), and as a consequence, improving duration is frequently associated with optimisation of pharmacokinetic properties. Often regarded only as ‘pharmacodynamics', the direct kinetics of drug interaction with target protein is not conventionally considered, as commonly the dissociation rate of drugs from their target is too rapid to play a significant role in drug duration. Despite this, it has recently been demonstrated that the slow dissociation of drug from target protein is key to the long duration of the muscarinic antagonist, tiotropium bromide (Spiriva™) (Disse et al., 1999).
Structurally similar to ipratropium, the previous market-leader, tiotropium has become the anti-cholinergic drug of choice for the treatment of chronic obstructive pulmonary disease (Koumis & Samuel, 2005). Despite having similar pharmacokinetic profiles, tiotropium has a 24-h duration of action, compared to ⩽6 h for ipratropium (Maesen et al., 1995; Van Noord et al., 2000; Vincken et al., 2002). Studies using recombinant receptors have shown that [3H]-tiotropium has a much slower rate of dissociation from the M3 muscarinic receptor than ipratropium (34.7 and 0.26 h, respectively, Disse et al., 1993), and it has been concluded that this feature determines the difference in duration of action between the drugs (Disse et al., 1999). Data supporting this concept has also been obtained from in vitro studies in guinea-pig trachea, human bronchi (Takahashi et al., 1994; Barnes et al., 1995) and human lung (Haddad et al., 1994).
It is clear from this example that achieving slow dissociation kinetics for novel receptor antagonists represents an alternative approach for increasing duration drug of action. Despite this, the kinetics of drug-target interactions are seldom examined in the early stages of the drug discovery process and the kinetics of candidate drug compounds are almost never optimised. This is largely due to the technical difficulties associated with measuring rate parameters. The association and dissociation rates of compounds are traditionally assessed directly by monitoring the specific binding of a labelled form (often radiolabelled) of the ligand of interest. The process of labelling compounds is, however, both time-consuming and expensive, prohibiting the routine labelling of novel ligands during the lead optimisation phase of a drug discovery project where several hundred compounds may be synthesised. As a consequence, structure-activity relationships are often based only upon the equilibrium binding constant (Ki or KB), which does not provide quantitative information on the individual association and dissociation rates that compose it.
Determining the kinetic parameters of unlabelled ligands early in the drug discovery process may therefore be beneficial. Several different approaches have been previously described that potentially address this issue. An early study utilised tissue responses to estimate the kinetic parameters of acetylcholine antagonists by modelling their competition with agonist for a single site (Rang, 1966). The analytical solution for the rate of equilibration of two ligands competing for the same receptors was originally described by Colquhoun in 1968. More recently, Motulsky & Mahan (1984) described a theoretical analysis which used identical equations to derive rate constants for unlabelled ligands competing with radiolabelled antagonist. In this method, a kinetically characterised radioligand is added simultaneously with an unlabelled ligand to the receptor preparation of interest. As the pattern of radioligand binding over time is dependent on both the concentration and kinetic parameters of the competitor, the experimentally derived rate of specific radioligand binding can be fitted to provide the association and dissociation rates of the unlabelled compound.
A similar methodology has been employed previously to determine the association and dissociation rates of unlabelled muscarinic receptor ligands (Schreiber et al., 1985a, 1985b; 1987). The authors, however, made an important modification to the model of Motulsky & Mahan (1984), with the inclusion of an ‘antagonist-isomerisation' step to account for the two-phase association kinetics observed with the radiolabelled antagonists.
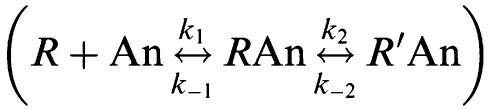 |
The aim of the present study was to test experimentally whether the simplest model of competition kinetics was sufficient to describe the interactions between a radioligand (A) and an unlabelled competitor (B) at the same receptor (R), according to the following mechanism:
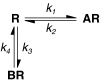
CHO-cells stably expressing recombinant M3 mACh receptor were used to ensure kinetic measurements were made at a single receptor subtype, and Ki values obtained from competition binding experiments at equilibrium were compared with the kinetically derived Kd (koff/kon) to validate the method.
Methods
Chemicals and reagents
l-[N-methyl-3H]Scopolamine methyl chloride ([3H]-NMS specific activity 80–90 Ci mmol−1) was obtained from Amersham Biosciences U.K. Ltd (GE Healthcare, Chalfont St Giles, U.K.). 96-deep well plates and 500 cm2 cell-culture plates were purchased from Fisher Scientific (Loughborough, U.K.) 96-well GF/B filter plates were purchased from Millipore (Watford, U.K.). MgCl2, atropine sulphate, clidinium bromide, ipratropium bromide, N-methyl scopolamine bromide, EDTA, NaCl and HEPES were obtained from Sigma Chemical Co. Ltd. (Poole, U.K.). All cell culture reagents were purchased from GIBCO (Invitrogen, Paisley, U.K.).
Cell culture techniques
Chinese hamster ovary (CHO) cells transfected with the cDNA encoding the human M3 (CHO-M3) muscarinic acetylcholine receptors were a kind gift from Professor S.R. Nahorski (Department of Cell Physiology and Pharmacology, University of Leicester, U.K.). CHO cells were grown in minimum essential medium (αMEM) supplemented with 10% new-born calf serum. Cells were maintained at 37°C in 5% CO2/humidified air. Cells were routinely split 1 : 10, using trypsin-EDTA to lift cells, and were not used in assays beyond passage 40.
Cell membrane preparation
CHO-cells expressing the M3 mACh receptor were grown to 80–90% confluency in 500 cm2 cell-culture plates. The cell-culture media was removed and the monolayer briefly washed with ice-cold HBS-EDTA (2 × 10 ml; 10 mM HEPES, 0.9% w v−1 NaCl, 0.2% w v−1 EDTA pH 7.4). All subsequent steps were conducted at 4°C to avoid receptor degradation. HBS-EDTA (10 ml) was added and left for approximately 20 min for the cells to detach from the culture substrate. The cells were then removed from the plates into a 30 ml centrifuge tube and subsequently centrifuged at 250 × g for 5 min to allow a pellet to form. The supernatant fraction was aspirated and 20 ml per 500 cm2 tray of wash buffer 1 (10 mM HEPES, 10 mM EDTA, pH 7.4) was added to the pellet. This was homogenised using a Polytron (20,000 r.p.m., 5 × 10 s bursts) and subsequently centrifuged at 50,000 × g at 4°C (Sovrall RC5). The supernatant fraction was discarded and the pellet rehomogenised and centrifuged as described above in wash buffer 2 (10 mM HEPES, 0.1 mM EDTA, pH 7.4). The final pellet was suspended in wash buffer 2 at a concentration of 3–5 mg ml−1. Protein concentration was determined by the method of Bradford (1976), using BSA as a standard and aliquots maintained at −80°C until required.
Common procedures applicable to all radioligand binding experiments
All radioligand experiments were conducted in 96-deep well plates, in assay binding buffer (10 mM HEPES, 1 mM MgCl2, pH 7.4). In all cases nonspecific binding (NSB) was determined in the presence of 1 μM atropine. After the indicated incubation period, bound and free [3H]-NMS were separated by rapid vacuum filtration using a FilterMate™ Cell Harvester (Perkin Elmer, Beaconsfield, U.K.) onto 96-well GF/B filter plates and rapidly washed three times with ice cold assay binding buffer. After drying (⩾4 h), 40 μl of Microscint™ 20 (Perkin Elmer, Beaconsfield, U.K.) was added to each well and radioactivity quantified using single photon counting on a TopCount™ microplate scintillation counter (Perkin Elmer, Beaconsfield, U.K.). Aliquots of [3H]-NMS were also quantified accurately to determine how much radioactivity was added to each well using liquid scintillation spectrometry on a LS 6500 scintillation counter (Beckman Coulter, High Wycombe, U.K.). In all experiments, total binding never exceeded more than 10% of that added, limiting complications associated with depletion of the free radioligand concentration (Hulme & Birdsall, 1992).
[3H]-NMS saturation binding
Binding was performed with a range of concentrations of [3H]-NMS (∼8–0.008 nM) to construct saturation binding curves, as described by Lambert et al. (1989). CHO-M3 cell-membranes (10 μg well−1) were incubated in 96-deep well plates at room temperature in assay binding buffer with (gentle agitation) for 18–20 h, to ensure equilibrium was reached. Owing to the low concentrations of [3H]-NMS employed, the assay volume was increased to 1.5 ml to avoid significant ligand depletion.
Determination of antagonist Ki
To obtain affinity estimates of unlabelled antagonists, [3H]-NMS competition experiments were performed at equilibrium. [3H]-NMS was used at a concentration of approximately 350 pM (∼40,000 c.p.m. in a volume of 1.5 ml) such that the calculated total binding never exceeded more than 10% of that added. [3H]-NMS was incubated in the presence of the indicated concentration of unlabelled antagonist and CHO-cell membranes (10 μg well−1) at room temperature with gentle agitation for 18–20 h.
Determination of the observed association rate (kob) of [3H]-NMS
To determine the kon accurately, the kob was calculated at three different concentrations of [3H]-NMS (approximately 100, 300 and 1000 pM; exact concentration was calculated in each experiment using liquid scintillation counting). The experiment was initiated (t=0) by the addition of [3H]-NMS to CHO-M3 cell membranes (10 μg well−1) in assay binding buffer (final assay volume 500 μl) and incubated with gentle agitation. Free [3H]-NMS was separated at multiple time points to construct association kinetic curves. Care was taken to ensure that saturation for each concentration was reached before the experiment was terminated. After incubation, bound was separated from free by rapid filtration, plates were left to dry and radioactivity quantified (as previously described).
Determination of the dissociation rate (koff) of [3H]-NMS
The dissociation rate of [3H]-NMS was determined by allowing approximately 50 pM [3H]-NMS (exact concentration determined for each experiment using liquid scintillation counting) to reach equilibrium with CHO-M3 cell membranes (10 μg well−1) in a final volume of 500 μl. Equilibrium was verified by harvesting an identical plate before the start of the experiment and then comparing total binding on this plate to that on the proceeding (post t=0) plates. After equilibrium was reached (approximately 1 h), reassociation of [3H]-NMS was prevented by the addition of 1.5 ml atropine (1 μM final). The moment of atropine addition was considered as t=0 and bound [3H]NMS was measured at multiple time points post t=0. Care was taken to ensure that [3H]NMS was fully dissociated from the M3 receptor.
Competition kinetics between [3H]-NMS and unlabelled ligands
The kinetic parameters of unlabelled ligands were assessed using the equations described by Colquhoun (1968) and the theoretical method of Motulsky & Mahan (1984). Unlike methods in which one compound is pre-equilibrated with the receptor, this approach involves the simultaneous addition of both radioligand and competitor to receptor preparation, so that at t=0 all receptors are unoccupied.
Approximately 1 nM [3H]-NMS (a concentration which avoided ligand depletion in this assay volume) was added simultaneously with unlabelled compound (at t=0) to CHO-M3 membranes (10 μg well−1) in 500 μl assay buffer. The degree of [3H]-NMS bound to receptor was assessed at multiple time points by filtration harvesting and liquid scintillation counting, as described previously. Each time point was conducted on a separate 96-deep well plate incubated at room temperature with constant gentle agitation.
Three different concentrations of unlabelled competitor were tested to ensure the rate parameters calculated were independent of ligand concentration. All compounds were tested at 10-, 30- and 100-fold their respective Ki, except for tiotropium that was tested at 100-, 300- and 1000-fold Ki.
Data analysis
As the amount of radioactivity varied slightly for each experiment (<5%), data are shown graphically as the mean±s.d. for individual representative experiments, whereas all values reported in the text and tables are mean±s.e.m. for the indicated number of experiments. All experiments were analysed by either linear or non-regression analysis using Prism 4.0 (GraphPad Software, San Diego, U.S.A.). Competition displacement binding data were fitted to the equation described by Hill (1909) which gives the amount of radioligand bound b, with a Hill coefficient n, maximal specific binding bmax, at concentration z as:
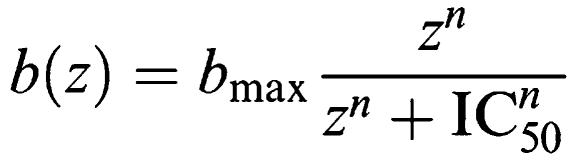 |
IC50 values obtained from the inhibition curves were converted to Ki values using the method of Cheng & Prusoff (1973). Dissociation data were fitted to a single phase exponential decay function and the t1/2 value obtained was transformed into a koff rate using Equation 1:
 |
[3H]-NMS association data were fitted to a single phase exponential association function to calculate an observed rate constant kob. The association rate constant, kon, was calculated using Equation 2, as described originally by Hill (1909):
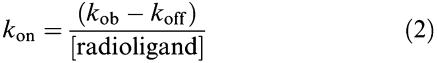 |
where the koff value used was predetermined from dissociation rate experiments.
Association and dissociation rates for unlabelled antagonists were calculated by fitting (least squares) the data from the competition kinetic experiments to a two-component exponential curve, with all parameters fixed apart from k3 and k4 (reproduced below).
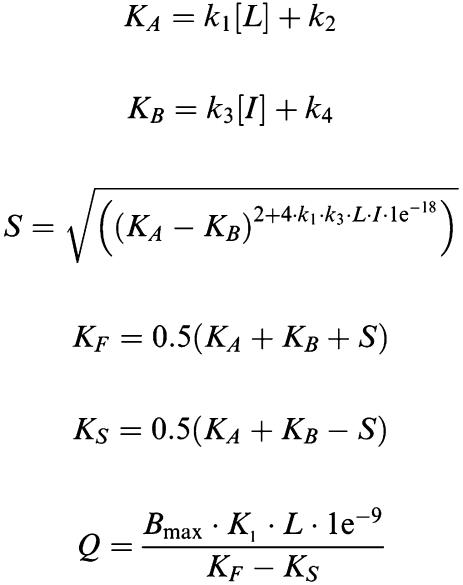 |
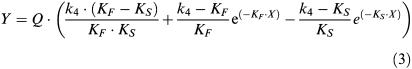 |
where X is the time (min), Y the specific binding (c.p.m.), K1 the kon [3H]-NMS, K2 the koff [3H]-NMS, L the concentration of [3H]-NMS used (nM), Bmax the total binding (c.p.m.), I the concentration unlabelled antagonist (nM).
Fixing the above parameters allowed the following to be calculated:
k3 is the association rate of unlabelled ligand (M−1 min−1) that is kon, k4 the dissociation rate of unlabelled ligand (min−1) that is koff.
Modeling of [3H]-NMS competition binding
To simulate the change in IC50 values over time, Equation 3 was entered into a commercially available programme (Microsoft Excel™ XP, U.S.). The kinetic parameters (k1–4) were fixed to those determined in the results for [3H]-NMS and the appropriate unlabelled antagonist. The binding of [3H]-NMS was simulated in the presence of different concentrations of the unlabelled antagonist at the indicated time points. These families of IC50 curves were analysed in Prism, as described above, to provide simulated IC50 values.
Statistical analysis
Statistical analysis was commonly performed with a two-tailed unpaired Student's t-test. Analysis of multiple data sets was performed by one-way analysis of variance (ANOVA) followed by the appropiate post-test, as detailed in the text. Differences of P<0.05 were considered to be significant.
Results
Characterisation of M3 receptor-expressing CHO cell line
The expression level of the M3 CHO-cell line, assessed using [3H]-NMS saturation binding to CHO-cell membranes was 3.05±0.13 pmol mg−1 protein (n=21). From these studies the equilibrium dissociation constant (Kd) of [3H]-NMS was determined to be 38±1 pM (n=21) (data not shown).
[3H]-NMS competition binding
In the presence of a competing ligand, the time for a radioligand to come to equilibrium is always slower than in its absence (Rang, 1966; Hulme & Birdsall, 1992). Moreover, the time to reach equilibrium can be dramatically different depending on the kinetic parameters of the unlabelled compound (Motulsky & Mahan, 1984). Initial experiments were therefore conducted to examine the time taken for [3H]-NMS to reach equilibrium in the presence of a competing antagonist. It has been previously reported that [3H]-tiotropium dissociates comparatively slowly from M3 mACh receptors (Disse et al., 1993) and may therefore take a relatively long-time to approach equilibrium. To examine this possibility the competitive binding of tiotropium was compared to that of the non-selective muscarinic antagonist, atropine.
The competition curves for tiotropium shifted to the left as the incubation time was increased (Figure 1a). This is more clearly illustrated in Figures 1b and c, where the apparent IC50 values are plotted over time. For all time points shown except 18 h, there was a statistically significant difference between the respective IC50 when compared to that obtained for 20 h (P<0.05, ANOVA followed by Dunnett's post-test) indicating that equilibrium for tiotropium was reached at around 18 h. Unfortunately it was not possible carry out longer incubations as membrane degradation was observed at time points greater than 20 h, exemplified by a significant reduction in maximal binding.
Figure 1.

Example full IC50 curves for tiotropium over time (a). The change in pIC50 over time is shown over the full time course (b) and also at early time points (c). CHO-M3 cell membranes (10 μg well−1) were incubated with [3H]-NMS and a range of competitor concentrations at room temperature, NSB values were assessed in the presence of 1 μM atropine. Plates were then harvested at the indicated time points and IC50 values were obtained using a sigmoidal (variable-slope) function. Data are shown as means±s.e.m. for four experiments, each performed in triplicate. Log IC50 values which are statistically different from that obtained at 20 h are shown as *(P<0.05).
In contrast, IC50 values for atropine decreased at early time points (5–15 min), followed by a rapid increase in the IC50 before reaching a plateau (Figure 2a). Equilibrium (or at least the approach towards it) for atropine appeared to be achieved at between 1 and 2 h, as there was no significant difference between IC50 values measured after 1 h when compared to that obtained for 20 h. (P<0.05, ANOVA followed by Dunnett's post-test) (Figure 2b and c). Given these data, an assay incubation time of 18 h was used in all subsequent experiments as this provided a satisfactory compromise between attaining equilibrium while avoiding membrane degradation. The pKi values for the five antagonists tested under these conditions are summarised in Table 1.
Figure 2.

Example full IC50 curves for atropine over time (a). The change in pIC50 over time is shown over the full time course (b) and also at early time points (c). CHO-M3 cell membranes (10 μg well−1) were incubated with [3H]-NMS and a range of competitor concentrations at room temperature, NSB values were assessed in the presence of 1 μM atropine. Plates were then harvested at the indicated time points and IC50 values were obtained using a sigmoidal (variable-slope) function. Data are shown as means±s.e.m. for four experiments, each performed in triplicate. Log IC50 values which are statistically different from that obtained at 20 h are shown as *(P<0.05).
Table 1.
Binding affinity constants (pKi) and slope factors for antagonists at CHO-cells stably expressing the M3 mACh receptor
| Antagonist | pKi | Slope factor | n |
|---|---|---|---|
| Atropine |
9.71±0.03 |
1.05±0.001 |
18 |
| Clidinium |
9.60±0.02 |
0.94±0.03 |
10 |
| Ipratopium |
9.76±0.02 |
0.98±0.02 |
10 |
|
N-methyl scopolamine |
10.40±0.03 |
0.90±0.03 |
10 |
| Tiotropium | 11.10±0.02 | 1.2±0.01 | 22 |
Data are mean±s.e.m. for the indicated number of experiments.
Characterisation of [3H]-NMS kinetic parameters
The accuracy of rate constants calculated for unlabelled compounds is highly dependent upon the precision of the kinetic parameters defined for the radioligand. Initial experiments were therefore performed to fully characterise the rate parameters of [3H]-NMS. Several different concentrations of [3H]-NMS were used to experimentally determine its koff value. [3H]-NMS dissociation was monophasic and gave a half-time of dissociation (t1/2) of 47.2±2 min (n=8) (Figure 3a) which when applied to Equation 1 gave a koff of 0.015±0.0005 min−1 (n=8). This t1/2 value compares well to that reported by Trankle et al. (2001) (43.3±4.2 min). Preliminary experiments using an infinite dilution method have also validated this value (data not shown).
Figure 3.

Characterisation of the kinetic parameters of [3H]-NMS. To determine koff (a) CHO-M3 cell membranes (10 μg well−1) were preincubated with [3H]-NMS until equilibrium was obtained. Reassociation of [3H]-NMS was then prevented with the addition of 1 μM (final concentration) atropine at the indicated time points. Data was best fitted using a one-phase exponential decay function to produce a t1/2 estimate. This was converted into a koff value by using Equation 1, as detailed in the Methods. The kon (b) was determined by incubation of CHO-M3 cell membranes (10 μg well−1) with the indicated concentrations of [3H]-NMS for various time points. Data was fitted using a one phase exponential association function to yield an observed on-rate (kobs). The kon value was calculated using Equation 2, as detailed in the methods. As different concentrations of [3H]-NMS were used for each individual experiment both (a) and (b) are representative of ⩾4 separate experiments performed in quadruplicate. To verify these values, kob was plotted against the [3H]-NMS concentration employed (c). In this case data are shown the individual kob from three separate experiments performed in quadruplicate.
As the association rate of a ligand is, in part, dependent upon the concentration used, we constructed a family of association kinetic curves using a range of [3H]-NMS concentrations. Each association curve was monitored until equilibrium for that concentration (Ymax) was reached (Figure 3b). Using the value for koff described above, the mean kon value was calculated as 4.1±0.2 × 108 M−1 min−1 (this represents the mean from five separate experiments, each comprising ⩾3 different [3H]-NMS concentrations). If [3H]-NMS binding follows a simple law of mass action model, kob should increase in a linear manner with radioligand concentration (Hill, 1909). In this case, the slope of the line should equate to the association rate and extrapolation of the plot to the y intercept (at x=0) should equal the dissociation rate (Motulsky & Christopoulos, 2003). When the kob values were plotted against radioligand concentration (Figure 3c), the data were consistent with a straight line (r2=0.98), indicating that binding of [3H]-NMS to the M3 receptor was consistent with the law of mass action. Values obtained from this plot were 4.2 × 108 M−1 min−1 and 0.017 min−1 for kon and koff, respectively, which are in excellent agreement with those values given above. Furthermore the kinetically derived Kd (koff/kon) calculated from the mean values for individual experiments (37 pM) or from the linear plot (40 pM) are in good agreement which the value obtained from [3H]-NMS saturation experiments (38±1 pM).
Competition kinetic binding
This method models the binding between two ligands, one labelled and one unlabelled, competing for the same receptor site. With the association and dissociation constants of [3H]-NMS fully characterised, it was possible to calculate the kon (k3) and koff (k4) of the unlabelled compound using the equations detailed in the methods. The high affinity of [3H]-NMS (38±1 pM), the amount of protein added (10 μg well−1) and the assay volume (500 μl) restricted the lowest concentration of [3H]-NMS we were able to employ which was not subject to ligand depletion (approximately 1 nM).
Representative curves for the five ligands tested are shown in Figure 4a–e. To ensure that the each ligand displayed classical competitive and reversible binding, each antagonist was assayed at three different concentrations. On and off rates calculated at these three different concentrations were equivalent for each compound (i.e. no statistically significant difference; One way ANOVA with Bonferroni post-test), demonstrating that the model used was sufficient to describe the data. The reported values for each kon and koff (Table 2) were determined by first deriving the mean from the different concentrations taken from each individual experiment, and then calculating the mean±s.e.m. for ⩾4 separate such experiments. Data were obtained when using concentrations of 10, 30 and 100-fold Ki of antagonists that had faster or comparable dissociation rates to [3H]-NMS. The approach to equilibrium using similar concentrations of tiotropium was, however, so slow that membrane degradation became limiting. Higher concentrations of tiotropium were therefore employed to obtain suitable resolution of [3H]-NMS binding (i.e. 100, 300 and 1000-fold Ki).
Figure 4.

[3H]-NMS competition kinetics curves in the presence of atropine (a), clidinium (b), ipratropium (c), N-methyl scopolamine (d) and tiotropium (e). CHO-M3 cell membranes were incubated with ∼1 nM [3H]-NMS and either 0, 10-fold Ki, 30-fold Ki or 100-fold Ki. (In the case of tiotropium CHO-M3 membranes were incubated with 0, 100-fold Ki, 300-fold Ki or 1000-fold Ki compound). Plates were incubated at room temperature for the indicated time points and NSB levels determined in the presence of 1 μM atropine. Data were fitted to the equations to described in the methods to calculate kon and koff values for the unlabelled antagonists. (Data summarised in Table 2). As the total binding varied from experiment to experiment, data are presented as mean±s.d. from a representative of ⩾4 experiments performed in quadruplicate.
Table 2.
Affinity values and kinetically derived parameters for unlabelled ligands
| Ligand | kon M−1 min−1 (k3) | koff min−1 (k4) | Kd pM (pKd) | Ki pM (pKi) |
|---|---|---|---|---|
| Atropine |
1.5±0.21 × 109 |
0.27±0.039 |
180±4 (9.71) |
9.71±0.03 |
| Clidinium |
1.1±0.07 × 108 |
0.029±0.002 |
259±7 (9.60) |
9.60±0.02 |
| Ipratopium |
4.83±0.2 × 108 |
0.07±0.005 |
160±4 (9.8) |
9.76±0.02 |
|
N-methyl scopolamine |
4.48±0.3 × 108 |
0.017±0.001 |
38±1 (10.35) |
10.39±0.03 |
| Tiotropium | 1.58±0.2 × 108 | 0.0015±0.0002 | 8±0.4 (11.1) | 11.10±0.02 |
Validation of the competition kinetics assay
If the method described herein produced accurate on and off-rates, the kinetically derived Kd (koff/kon) should be equal to the affinity constant (Ki) obtained from equilibrium competition binding experiments. When these two parameters were compared (Figure 5), an excellent correlation was obtained (r2=0.99). While it is possible that this ratio could be still obtained if the rate constants were incorrect by approximately the same factor, it is more likely that the kinetic values derived from the competition method are accurate. There was also an excellent correlation between the association and dissociation rates for [3H]-NMS measured directly (kon: 4.1±0.2 × 108 M−1 min−1, koff: 0.015±0.0005 min−1), and unlabelled N-methyl scopolamine calculated from the competition kinetics assay (kon: 4.48±0.3 × 108 M−1 min−1 and koff: 0.017±0.001 min−1).
Figure 5.

Correlation between pKi and kinetically derived pKd for the five test antagonists. pKi values were taken from [3H]-NMS competition binding experiments at equilibrium. The values comprising the kinetically derived Kd (koff/kon) were taken from the experiments in Figure 5. Data shown are mean±s.e.m. (n⩾4).
In addition to this internal validation, the off rates from this study compare well with those quoted in the literature, derived directly from the radiolabelled form of the antagonists. For example, the off rate for [3H]-ipratropium from the human M3 receptor was 0.044±0.0013 min−1 (Disse et al., 1993), compared to 0.07±0.005 min−1 calculated using the competition kinetics assay. Similarly, the off rate for [3H]-tiotropium was calculated to be 0.0033±0.00018 min−1 (Disse et al., 1993), compared to 0.0015±0.0002 min−1 from the present study.
The kinetic parameters described above were used to model the interaction of two ligands binding at a single, shared site over time. [3H]-NMS binding was simulated in the presence of various concentrations of either atropine or tiotropium to obtained IC50 curves at different time points. The simulated IC50 values were then compared to those obtained experimentally for the unlabelled ligands as described in Figures 1 and 2. There was a good correlation between the simulated and experimental IC50 values for tiotropium, with equilibrium reached at approximately 18 h (Figure 6a, Table 3). At early time points (<60 min), however, the actual IC50 values were higher than those predicted by the simulated data (∼100 pM). Similarly, there was a good correlation between actual and predicted IC50 values for atropine (Figure 7a and Table 3) except at very early time points (<15 min).
Figure 6.

Correlation between the change in IC50 over time (a) for tiotropium between experimental and simulated data. Experimental data was taken from that described in Figure 1. Simulated data was generated by entering Equation 3 was entered into a commercially available statistics programme (as detailed in the methods). The kinetic parameters (k1–4) were fixed to those determined in the results for [3H]-NMS and the appropriate unlabelled antagonist. The binding of [3H]-NMS was simulated in the presence of different concentrations of the unlabelled antagonist at the indicated time points. These families of IC50 curves were analysed in Prism, to provide simulated competition binding curves from which IC50 values were taken. Initial time points are also shown on a separate graph (b) to illustrate differences in early time points.
Table 3.
Comparison between experimental and simulated IC50 values over-time
|
Time (min) |
Atropine pIC50 (Log M) |
Tiotropium pIC50 (Log M) |
||
|---|---|---|---|---|
| Experimental | Simulated | Experimental | Simulated | |
| 5 |
9.04±0.06 |
9.34±0.003 |
8.52±0.03 |
8.62±0.002 |
| 10 |
9.11±0.05 |
9.33±0.009 |
8.75±0.02 |
8.84±0.01 |
| 15 |
9.20±0.06 |
9.24±0.01 |
8.82±0.02 |
8.94±0.009 |
| 20 |
9.11±0.02 |
9.15±0.01 |
8.90±0.02 |
8.90±0.01 |
| 25 |
8.99±0.04 |
9.07±0.01 |
8.87±0.02 |
9.00±0.01 |
| 30 |
8.97±0.01 |
9.01±0.01 |
8.88±0.02 |
9.06±0.01 |
| 45 |
8.87±0.03 |
8.88±0.01 |
8.92±0.01 |
9.14±0.01 |
| 60 |
8.78±0.03 |
8.80±0.01 |
8.97±0.01 |
9.20±0.01 |
| 120 |
8.69±0.03 |
8.69±0.01 |
9.16±0.02 |
9.38±0.01 |
| 180 |
8.62±0.02 |
8.67±0.01 |
9.31±0.03 |
9.50±0.01 |
| 360 |
8.60±0.05 |
8.66±0.01 |
9.64±0.03 |
9.69±0.01 |
| 1080 |
8.63±0.02 |
8.66±0.01 |
9.97±0.01 |
9.93±0.02 |
| 1200 | 8.65±0.02 | 8.66±0.01 | 9.98±0.04 | 9.94±0.008 |
Figure 7.

Correlation between the change in IC50 over time (a) for atropine between experimental and simulated data. Experimental data was taken from that described in Figure 1. Simulated data was generated by entering Equation 3 was entered into a commercially available statistics programme (as detailed in the methods). The kinetic parameters (k1–4) were fixed to those determined in the results for [3H]-NMS and the appropriate unlabelled antagonist. The binding of [3H]-NMS was simulated in the presence of different concentrations of the unlabelled antagonist at the indicated time points. These families of IC50 curves were analysed in Prism, to provide simulated competition binding curves from which IC50 values were taken. Initial time points are also shown on a separate graph (b) to illustrate differences in early time points.
Discussion
Since the theoretical description of the competitive kinetics binding assay there have been relatively few examples in the literature utilising and validating the technique. When determining the kinetics of ligand interaction with β-adrenergic receptor(s) endogenously expressed in L6 myoblasts, the simple model of Motulsky and Mahan was sufficient to describe the data (Affolter et al., 1985; Contreras et al., 1986). However, where it has been applied previously for muscarinic receptor antagonists, it was found that the model was not sufficient to describe the data, and that an additional receptor isomerisation step was required (Schreiber et al., 1985a, 1985b; 1987). In contrast, data from the present study fit well to the simple model of Motulsky and Mahan, and we have shown that by considered selection of both [3H]-NMS and competitor concentrations, the method is able to produce accurate kinetic values. One possible explanation for these discrepancies is that the studies of Schreiber and co-workers were conducted using a membrane preparation derived from rat brain cortex, which has subsequently been shown to expresses multiple muscarinic receptor subtypes (Vannucchi & Pepeu, 1995; Tice et al., 1996; Tayebati et al., 2001; Krejci & Tucek, 2002). As it is known that [3H]-NMS has distinct dissociation rates from different mACh subtypes (Trankle et al., 2001), it is possible that the two-phase kinetics observed was due to binding to multiple subtypes rather than the existence of an antagonist isomerisation step.
Figure 4a–e demonstrates two clear patterns of [3H]-NMS binding depending upon the dissociation rate of the competing ligand. The binding of [3H]-NMS essentially monitors the effect the competitor is having on the free receptor population (Hulme & Birdsall, 1992). When the radioligand dissociates more slowly than the competitor (k2<k4) the free concentration of receptor is effectively reduced and the time taken for [3H]-NMS to reach equilibrium is increased. The amount of receptors occupied by [3H]-NMS at equilibrium is reduced as the concentration of the competiting ligand is increased. When the radioligand dissociates more rapidly than the competitor (k2>k4), the immediate [3H]-NMS binding is unaffected by the presence of the competitor. Figure 4e shows that at the early time points [3H]-NMS binding overshoots its equilibrium. The timing of the subsequent decline to equilibrium is related to the dissociation rate of tiotropium.
An important variable to optimise in this assay was the concentration of competing ligand. We have found that the equations described are able to generate reproducible and accurate rate constants when the competition kinetics curves have been left to reach (or at least approach) equilibrium. If the concentration of competitor is too low, there will be little influence on radioligand association rate. This will lead to a high degree of error in rate estimates for the unlabelled ligand. Conversely, if the concentration of competing ligand greatly exceeds that of the radioligand, the amount of specific radioligand binding may be too small to allow meaningful data analysis.
We have also found that the time intervals at which the specific [3H]-NMS binding is assessed is crucial to produce accurate rate constants for the unlabelled compounds. Binding of the radioligand follows two distinct phases. The first phase measures binding of radioligand to unoccupied receptors, which in our system occurs rapidly (within the first few minutes). The second phase is dependent upon competition with unlabelled ligand and thus proceeds more slowly. The amount of radioligand bound at the point of transition from first to second phase (i.e. full receptor occupancy) is important for defining the observed on rate of unlabelled ligand. Failure to have sufficient measurements at these important early time points will lead to a low degree of confidence in subsequent rate parameter estimates.
In addition to generating kinetic parameters for unlabelled ligands, Motulsky & Mahan (1984) identified another practical application of the model: predicting the time at which equilibrium is reached between a radiolabel and competitor. When the dissociation rate of the competitor is faster than that of the radioligand (k4>k2) and the concentration of the radioligand is high, the time required for the IC50 curve to reach equilibrium is 1.75/k2. Using the off rate determined for [3H]-NMS, the time taken for the atropine IC50 curve to reach equilibrium would be predicted to be 1.9 h. This is in excellent agreement with the experimental data, which showed no significant change in the IC50 value after 1 h. The simulated data also predict the biphasic behaviour observed at the very early time points. As the initial phase largely measures the binding of radioligand to unoccupied receptors, the degree of competition is greatly reduced at these very early time points. This manifests as a higher initial IC50 that decreases with time as full competition occurs (approaching full occupancy). However, as atropine has a faster observed on rate than [3H]-NMS, it then overshoots its equilibrium occupancy, appearing to have a higher affinity than at equilibrium.
The simulated data, however, predict a much faster time to the lowest IC50 value; around 6 min as opposed to 15 min observed with the experimental data. This is likely due to diffusion rate artefacts. One of the assumptions of Motulsky & Mahan (1984) is that the ligands are ‘simultaneously exposed to the receptors'. Although the 96 well plates are shaken, this may not aid diffusion as readily as a stirred system, resulting in delayed exposure of ligand to receptor. The rate of diffusion is also likely to be different for each ligand.
When the dissociation of the competitor is slower than that of the radioligand (k4≪k2), initial radioligand binding overshoots its equilibrium occupancy. This results in a lower apparent affinity for the unlabelled ligand at early time points. With such ligands, an IC50 concentration of competitor is predicted to reach equilibrium with a high concentration of radioligand at 1.75/k4 (Motulsky & Mahan, 1984). Using these equations with the dissociation rate determined for tiotropium (0.0015 min−1), the time taken for an IC50 concentration to reach equilibrium is calculated to be 19.4 h. As can been seen in Figure 1, the experimentally derived IC50 plateaus at around 18–20 h, in good agreement with these predictions.
This long equilibration time raises an important issue, as we observed a substantial reduction in total radioligand binding after 20 h, indicative of receptor degradation. According to the calculations above, compounds with only moderately slower off-rates than tiotropium would take far longer to attain equilibrium (e.g. a compound with an off rate of 0.001 min−1 would take 29.2 h to equilibrate at IC50 concentration), which is probably unachievable in most systems.
It is therefore likely that in situations where compounds are being optimised for slow dissociation rates, conventional equilibrium assays may not be appropriate for assessing affinity. Instead, a kinetic assay such as that described here may be the only appropriate method for accurately measuring the affinity of slowly dissociating compounds. Furthermore, once kon and koff for unlabelled competitors are known it is possible to determine the time required for a competition binding experiment to come to equilibrium.
We have demonstrated that with the correct technical application of the theoretical method of Motulsky & Mahan (1984) it is possible to obtain accurate kinetic parameters of unlabelled compounds. Importantly, this assay is capable of testing large numbers of compounds, making it applicable to the early assessment of compound rate parameters during the lead optimisation phase of drug discovery programmes. This will enable chemical optimisation of kinetic parameters, which may result in the discovery of new drugs that utilise slow receptor dissociation for achieving long duration of action.
Abbreviations
- CHO
Chinese hamster ovary
- COPD
chronic obstructive pulmonary disease
- EDTA
ethylenediaminetetra-acetic acid
- HBS
HEPES-buffered saline
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- MEM
minimal essential medium
- mACh
muscarinic acetylcholine
- NMS
l-[N-methyl-]Scopolamine methyl chloride
- NSB
non-specific binding
References
- AFFOLTER H., HERTEL C., JAEGGI K., PORTENIER M., STAEHELIN M. (−)S-[3H] CGP-12177 and its use to determine the rate constants of unlabeled beta-adrenergic antagonists. Proc. Natl. Acad. Sci. U.S.A. 1985;82:925–929. doi: 10.1073/pnas.82.3.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARNES P.J., BELVISI M.G., MAK J.C., HADDAD E.B., O'CONNOR B. Tiotropium bromide (Ba 679 BR), a novel long-acting muscarinic antagonist for the treatment of obstructive airways disease. Life Sci. 1995;56:853–859. doi: 10.1016/0024-3205(95)00020-7. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CHENG Y., PRUSOFF W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- COLQUHOUN D. The rate of equilibration in a competitive n drug system and the auto-inhibitory equations of enzyme kinetics: some properties of simple models for passive sensitization. Proc. R. Soc. Lond. B. Biol. Sci. 1968;170:135–154. doi: 10.1098/rspb.1968.0030. [DOI] [PubMed] [Google Scholar]
- CONTRERAS M.L., WOLFE B.B., MOLINOFF P.B. Kinetic analysis of the interactions of agonists and antagonists with beta adrenergic receptors. J. Pharmacol. Exp. Ther. 1986;239:136–143. [PubMed] [Google Scholar]
- DISSE B., REICHL R., SPECK G., TRAUNECKER W., LUDWIG ROMINGER K.L., HAMMER R. Ba 679 BR, a novel long-acting anticholinergic bronchodilator. Life Sci. 1993;52:537–544. doi: 10.1016/0024-3205(93)90312-q. [DOI] [PubMed] [Google Scholar]
- DISSE B., SPECK G.A., ROMINGER K.L., WITEK T.J., JR, HAMMER R. Tiotropium (Spiriva): mechanistical considerations and clinical profile in obstructive lung disease. Life Sci. 1999;64:457–464. doi: 10.1016/s0024-3205(98)00588-8. [DOI] [PubMed] [Google Scholar]
- HADDAD E.B., MAK J.C., BARNES P.J. Characterization of [3H] Ba 679 BR, a slowly dissociating muscarinic antagonist, in human lung: radioligand binding and autoradiographic mapping. Mol. Pharmacol. 1994;45:899–907. [PubMed] [Google Scholar]
- HILL A.V. The mode of action of nicotine and curare, determined by the form of the contraction curve and the method of temperature coefficients. J. Physiol. 1909;39:361–373. doi: 10.1113/jphysiol.1909.sp001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HULME E.C., BIRDSALL N.J.M.Strategy and tactics in receptor-binding studies Receptor-Ligand Interactions. A Practical Approach 1992New-York: Oxford University Press; 63–176.ed. Hulme, E.C., pp [Google Scholar]
- KOUMIS T., SAMUEL S. Tiotropium bromide: a new long-acting bronchodilator for the treatment of chronic obstructive pulmonary disease. Clin. Ther. 2005;27:377–392. doi: 10.1016/j.clinthera.2005.04.006. [DOI] [PubMed] [Google Scholar]
- KREJCI A., TUCEK S. Quantitation of mRNAs for M(1) to M(5) subtypes of muscarinic receptors in rat heart and brain cortex. Mol. Pharmacol. 2002;61:1267–1272. doi: 10.1124/mol.61.6.1267. [DOI] [PubMed] [Google Scholar]
- LAMBERT D.G., GHATAORRE A.S., NAHORSKI S.R. Muscarinic receptor binding characteristics of a human neuroblastoma SK-N-SH and its clones SH-SY5Y and SH-EP1. Eur. J. Pharmacol. 1989;165:71–77. doi: 10.1016/0014-2999(89)90771-1. [DOI] [PubMed] [Google Scholar]
- MAESEN F.P., SMEETS J.J., SLEDSENS T.J., WALD F.D., CORNELISSEN P.J. Tiotropium bromide, a new long-acting antimuscarinic bronchodilator: a pharmacodynamic study in patients with chronic obstructive pulmonary disease (COPD). Dutch Study Group. Eur. Respir. J. 1995;8:1506–1513. [PubMed] [Google Scholar]
- MOTULSKY H.J, CHRISTOPOULOS A.Analyzing kinetic binding data Fitting Models to Biological Data Using Linear and Nonlinear Regression 2003New-York: Oxford University Press; 245–251.eds. Motulsky, H.J. & Christopoulos, A., pp [Google Scholar]
- MOTULSKY H.J., MAHAN L.C. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol. Pharmacol. 1984;25:1–9. [PubMed] [Google Scholar]
- RANG H.P. The kinetics of action of acetylcholine antagonists in smooth muscle. Proc. R. Soc. Lond. B. Biol. Sci. 1966;164:488–510. doi: 10.1098/rspb.1966.0045. [DOI] [PubMed] [Google Scholar]
- SCHREIBER G., HENIS Y.I., SOKOLOVSKY M. Analysis of ligand binding to receptors by competition kinetics. Application to muscarinic antagonists in rat brain cortex. J. Biol. Chem. 1985a;260:8789–8794. [PubMed] [Google Scholar]
- SCHREIBER G., HENIS Y.I., SOKOLOVSKY M. Rate constants of agonist binding to muscarinic receptors in rat brain medulla. Evaluation by competition kinetics. J. Biol. Chem. 1985b;260:8795–8802. [PubMed] [Google Scholar]
- SCHREIBER G., HENIS Y.I., SOKOLOVSKY M. Application of competition kinetics to investigate rat brain muscarinic receptors. Isr. J. Med. Sci. 1987;23:36–43. [PubMed] [Google Scholar]
- SMITH D.A., JONES B.C., WALKER D.K. Design of drugs involving the concepts and theories of drug metabolism and pharmacokinetics. Med. Res. Rev. 1996;16:243–266. doi: 10.1002/(SICI)1098-1128(199605)16:3<243::AID-MED2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI T., BELVISI M.G., PATEL H., WARD J.K., TADJKARIMI S., YACOUB M.H., BARNES P.J. Effect of Ba 679 BR, a novel long-acting anticholinergic agent, on cholinergic neurotransmission in guinea pig and human airways. Am. J. Respir. Crit. Care Med. 1994;150:1640–1645. doi: 10.1164/ajrccm.150.6.7952627. [DOI] [PubMed] [Google Scholar]
- TASHKIN D.P. Is a long-acting inhaled bronchodilator the first agent to use in stable chronic obstructive pulmonary disease. Curr. Opin. Pulmonary Med. 2005;11:21–128. doi: 10.1097/00063198-200503000-00004. [DOI] [PubMed] [Google Scholar]
- TAYEBATI S.K., VITALI D., SCORDELLA S., AMENTA F. Muscarinic cholinergic receptor subtypes in rat cerebellar cortex: light microscope autoradiography of age-related changes. Brain Res. 2001;889:256–259. doi: 10.1016/s0006-8993(00)03146-2. [DOI] [PubMed] [Google Scholar]
- TICE M.A., HASHEMI T., TAYLOR L.A., MCQUADE R.D. Distribution of muscarinic receptor subtypes in rat brain from postnatal to old age. Brain Res. Dev. Brain Res. 1996;92:70–76. doi: 10.1016/0165-3806(95)01515-9. [DOI] [PubMed] [Google Scholar]
- TRANKLE C., KOSTENIS E., MOHR K. Muscarinic allosteric modulation: M2/M3 subtype selectivity of gallamine is independent of G-protein coupling specificity. Naunyn. Schmiedebergs Arch. Pharmacol. 2001;364:172–178. doi: 10.1007/s002100100441. [DOI] [PubMed] [Google Scholar]
- VAN NOORD J.A., BANTJE T.A., ELAND M.E., KORDUCKI L., CORNELISSEN P.J. A randomised controlled comparison of tiotropium and ipratropium in the treatment of chronic obstructive pulmonary disease. The Dutch Tiotropium Study Group. Thorax. 2000;55:289–294. doi: 10.1136/thorax.55.4.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANNUCCHI M.G., PEPEU G. Muscarinic receptor modulation of acetylcholine release from rat cerebral cortex and hippocampus. Neurosci. Lett. 1995;190:53–56. doi: 10.1016/0304-3940(95)11498-l. [DOI] [PubMed] [Google Scholar]
- VINCKEN W., VAN NOORD J.A., GREEFHORST A.P., BANTJE T.A., KESTEN S., KORDUCKI L., CORNELISSEN P.J. Improved health outcomes in patients with COPD during 1 yr's treatment with tiotropium. Eur. Respir. J. 2002;19:209–216. doi: 10.1183/09031936.02.00238702. [DOI] [PubMed] [Google Scholar]