

Abstract
An anthraquinone-linked duplex DNA oligomer containing 60 base pairs was synthesized by PCR. The strand complementary to the quinone-containing strand has four isolated GG steps, which serve as traps for a migrating radical cation. Irradiation of the quinone leads to electron transfer from the DNA to the quinone forming the anthraquinone radical anion and a base radical cation. The radical cation migrates through the DNA, causing reaction at GG steps revealed as strand breaks. The efficiency of strand cleavage falls off exponentially with distance from the quinone (slope = −0.02 Å−1). This finding necessitates reinterpretation of mechanisms proposed for radical cation migration in DNA. We propose that radical cations form self-trapped polarons that migrate by thermally activated hopping.
Interaction of light, ionizing radiation, and active reagents (1) with DNA can result in loss of an electron with formation of a radical cation (hole). A clear understanding of radical cation chemistry in duplex DNA is important because the products from its reaction can lead to mutations (2, 3). The key issues for this topic can be divided into three parts: radical cation injection, radical cation transport, and radical cation reaction.
Charge injection (oxidation) can be accomplished both by physical and chemical means. For example, exposure to γ radiation results in oxidation of DNA (4, 5), as do certain light-driven processes (6–9). Similarly, specially constructed DNA analogs have been prepared that contain functional groups that lead to radical cation formation on activation (10, 11). The reactions of radical cations in DNA are a very complex matter that is currently under active investigation (12, 13). Reaction products have been identified, and they indicate a dominant role for guanines. This is not unexpected because guanine is the most easily oxidized of the DNA bases (14, 15).
Radical cation transport in DNA is a controversial matter that has been probed by diverse methods (16–23). Recent results demonstrate that a radical cation injected into duplex DNA may migrate through several base pairs until it is irreversibly trapped, typically, at a GG step (24, 25). However, the mechanism and distance dependence for radical cation migration are still being debated (26).
The idea that duplex DNA may provide a special pathway for electron transport through the ordered π-electron system of the bases has been contemplated for more than 35 years (27). Interest intensified when Barton and coworkers reported rapid photoinduced electron transfer over a distance >40 Å (28). This result led them to conclude that DNA is a “molecular wire” and that charge transport occurs through a “π-way” of well stacked DNA bases (17). The mechanism proposed to account for this rapid, long-distance charge transport was based on the electron transfer theory of superexchange (29). A key parameter in this approach is the extent to which the bridge orbitals of the intervening DNA bases couple the electron donor and electron acceptor. This coupling is symbolized by β, and Barton’s first report of its magnitude indicated that β ≥ 0.2 Å−1 for duplex DNA (28). This remarkable finding triggered an extensive investigation of the mechanism of charge transport through DNA. Experiments on diverse systems yielded β values in the range of 0.6–1.3 Å−1 (8, 10, 20–22, 25). There is currently no general agreement about the magnitude of β in duplex DNA. And, more critically, there is no certainty that it is appropriate to view charge transport in DNA as superexchange through bridge orbitals.
In earlier work on this topic, it was suggested that charge transport in DNA occurs by “hopping” of a radical cation localized on one base to a neighboring base (30, 31). This mechanism does not require extensive coupling through a π-way of bridge orbitals, and thus the characterization of the distance dependence with β is misleading. Very recently, Giese and coworkers reported an examination of sequence-dependent long- and short-range charge transfer in DNA (10). They conclude that superexchange occurs but that the particular sequence of bases determines when this mechanism operates. In particular, their data suggest that superexchange occurs between nearby guanines but that three or four intervening AT sequences stop this process. Ratner comments that this suggestion might explain the wide range of results from previous experiments (32). Jortner et al. (26) come to a similar conclusion from a theoretical analysis that shows that both superexchange and hopping can occur in certain circumstances. They suggest a parallel superexchange-sequential charge-hopping mechanism, including both unistep transfer and multistep transport processes (33).
In a very recent report, Kelley and Barton (34) used DNA duplexes containing analogs of adenine to examine electron transfer. They report values for β ranging from 0.1 to 1.0 Å−1 determined by base stacking and reaction energetics. These findings encouraged them to propose that new paradigms need to be developed to account for both the insulator and wire-like DNA properties. Finally, Barton and coworkers (25) describe an examination of the long-range (≈200 Å) oxidative damage to guanines initiated by irradiation of metal complexes linked to duplex DNA. They report a shallow distance dependence that appears to be controlled, in part, both by the base sequence separating the metal from the oxidized guanine and by the identity of the metal. These very latest findings lead them to conclude that the movement of charge through duplex DNA may be described by a hole hopping mechanism.
Herein we report the photochemistry of an anthraquinone derivative (AQ) covalently linked to a 60-base pair DNA oligomer [DNA(1)] (Fig. 1) whose complementary strand, DNA(2), contains four isolated GG steps. Reactions leading to strand cleavage of DNA(2) at the GG steps are triggered by irradiation of the remote AQ. Quantitative analysis of the strand cleavage efficiency reveals an exponential distance dependence for charge transport over 55 base pairs by a mechanism that we identify as phonon-assisted polaron-like hopping.
Figure 1.

Oligonucleotide sequences DNA(1) and DNA(2) and the structure of the anthraquinone conjugate, AQ, which in AQ-DNA(1) is linked to its 5′ terminus.
MATERIALS AND METHODS
General.
Oligonucleotides were prepared and purified by using standard methods. The anthraquinone phosphoramidite was synthesized by previously published methods (24, 35). β detection of PAGE products was carried out with a Scanalytics (Bellerica, MA) AMBIS imaging system.
Preparation of Radiolabeled DNA.
5′-end-labeling was performed by using T4 polynucleotide kinase and [γ-32P]dATP. A 250-pmol sample of duplex DNA was incubated with 5.0 μl of [γ-32P]ATP (6,000 Ci/mmol) and 1.0 μl (8 units) of T4 polynucleotide kinase in a total volume of 20 μl at 50°C for 60 min. After incubation, the DNA sample was suspended in denaturing loading buffer and was purified on a 20% nondenaturing polyacrylamide gel. After autoradiography, the band corresponding to AQ-DNA was excised from the gel, was eluted in 350 μl of elution buffer [0.5 M NH4OAc/10 mM Mg(OAc)2/1.0 mM EDTA/0.1% SDS) at 37°C for 4 h, and was centrifuged at 12,000 × g for 5 min. The DNA was precipitated from the supernatant by addition of 3.0 μl of 10 mM MgSO4, 5.0 μl of 3M NaOAc (pH 5.2), and 700 μl of cold ethanol. The mixture was vortexed, was placed on dry ice for 30 min, and was centrifuged at 12,000 × g for 30 min, and the supernatant was removed. The resulting pellets were washed three times with 80% ethanol. A+G and T sequence markers were produced according to the Maxam–Gilbert sequencing protocol (36).
Photocleavage.
Samples were prepared by diluting labeled DNA (5,000 cpm) to 20 μl with water buffered with 10 mM sodium phosphate (pH 7.0). Samples were irradiated in 1.5-ml microcentrifuge tubes by using a Rayonet (Barnsford, CT) photoreactor (λ = 350 nm). The tubes were suspended from a rotating sample holder and were cooled from below. After irradiation, 5.0 μl of 3M NaOAc (pH 5.3), 0.5 μl of glycogen, and 100 μl of cold ethanol were added in the order indicated. The DNA was precipitated on dry ice for 30 min followed by centrifugation. The pellets were washed twice with 100 μl of 80% ethanol, were dried for 5 min at low heat by Speedvac, were treated with piperidine if required, and were dissolved in 5.0 μl of denaturing formamide loading buffer. The photocleavage products were separated by electrophoresis on a 20% gel and were detected by autoradiography.
Formamidopyrimidine Glycosolase (Fpg) Enzymatic Digestion.
The standard reaction mixture (10 μl) 50 mM Tris⋅HCl (pH 7.5), 2 mM EDTA, 70 mM NaCl, and 10 μg of Fpg was incubated with 5 μM DNA at 37°C for 5 min. Reactions were terminated by heating at 70°C followed by ethanol precipitation at −20°C. The reaction mixture was analyzed by 20% PAGE containing 7 M urea.
Preparation of AQ-DNA by PCR.
Duplexes were prepared by PCR using 19-mer primers and DNA(2) as the template (37). PCR reactions contained 0.2 mM dNTPs, 10 mM Tris⋅HCl (pH 9.0), 50 mM KCl, 0.1% Trition X-100, 1 mM MgCl2, 10% DMSO, and 2.5 units of Taq polymerase per 100 μl reaction. Primer concentrations were 2.5 μM with 0.75 pmol of template. The primer sequences were 5′-CTT TGG TTC CTT GGT CAG C-3′ (with or without AQ at the 5′-end) and 5′-TAC GTG GCT TTT CGG TCA C-3′. The DNA was initially heated to 94°C for 60 s and underwent 32 cycles of denaturation, annealing, and extension of 94°C for 60 s, 45°C for 60 s, and 72°C for 45 s, respectively. The final extension was at 72°C for 5 min. After dialysis with Centricon 10 spin devices (Amicon), the duplexes were purified by nondenaturing agarose gel electrophoresis. Their analysis by capillary zone gel electrophoresis reveals two peaks, and matrix-assisted laser desorption ionization spectroscopy of AQ-DNA(1)/DNA(2) shows a peak with m/z = 37,316, corresponding to the duplex (m/z calculated = 37,306).
RESULTS
It was previously shown that UV irradiation of intercalated or covalently bound AQ leads to electron transfer giving the quinone radical anion and a base radical cation (24, 30, 38, 39). The radical anion is quenched by O2, and the radical cation migrates through duplex DNA. Trapping of the migrating radical cation with water or O2 occurs at the 5′-G of GG steps of the duplex DNA. Reaction is revealed as strand cleavage by treatment of the irradiated DNA with piperidine or with Fpg. These results demonstrate that a radical cation injected into DNA by irradiation of the AQ can be transported through the DNA and will cause a reaction at a remote site. Our previous work did not probe the distance dependence of the charge transport and did not provide evidence to distinguish between the various mechanistic postulates.
DNA(1) consists of 60 bases and an AQ functional group linked at its 5′ terminus through a four-atom tether. This tether limits the number of possible interactions of the AQ with the duplex DNA. Molecular modeling and spectroscopic experiments reveal that the tether is too short to allow the AQ to intercalate (24). However, intimate π-electron overlap between the AQ and the DNA bases does occur. This is attributed to end-capping of the DNA by the AQ caused by hydrophobic (40) and donor–acceptor interactions. The end-capped AQ is closely associated with its neighboring, terminal base pair in the DNA duplex. Importantly, end-capping should not distort the structure of DNA, as can occur when an electron acceptor is intercalated (41).
DNA(2) is complementary to AQ-DNA(1). It contains four GG steps located at various distances from its 3′ terminus. G10 on DNA(2) is the 5′-G of the GG step that is closest to the AQ linked to the 5′ terminus of DNA(1). There are nine base pairs between the AQ and G10, including a 3-base pair AT sequence. Similarly, G28 is the 5′-G of a GG step located 28 base pairs from the AQ. The next 5′-G of a GG step in DNA(2) occurs at G46. A five-base run of AT sequences occurs between G28 and G46. The farthest 5′-G in a GG step of DNA(2) occurs at G55, which is 185 Å from the AQ.
A sample of AQ-DNA(1)/DNA(2) that had been 5′-end-labeled on the DNA(2) strand was irradiated at 350 nm. Fig. 2 shows an autoradiogram from the PAGE analysis of this sample after treatment with piperidine (Fig. 2, lane 6) or with Fpg (lane 7) (42). Clearly, selective cleavage of DNA(2) is seen at each 5′-G of the four GG steps. Further, the relative reactivity of 5′- and 3′-guanines of the GG steps (5′-G:3′-G, ≈3:1) is characteristic of radical cation reactions in duplex DNA (30). Control experiments showed that this reaction requires both light and AQ. Most importantly, Fig. 2 shows that the amount of strand cleavage depends on the distance between the 5′-G and the AQ. The farther the guanine from the quinone, the less likely it is that strand cleavage will occur at that base.
Figure 2.
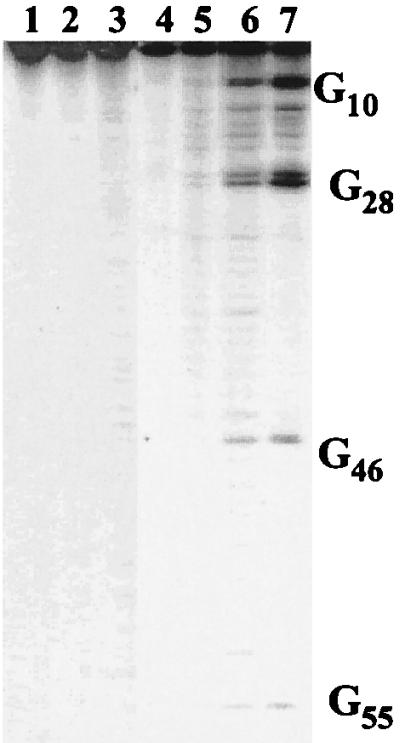
Autoradiogram of DNA(1) or AQ-DNA(1) hybridized with 32P-5′-labeled DNA(2) irradiated at 350 nm in a Rayonet photoreactor (16 lamps, 4 h, 25°C). Lanes: 1, DNA(1)/DNA(2), no irradiation; 2, same as lane 1, with irradiation; 3, same as lane 2, with piperidine treatment (30 min, 90°C); 4, AQ-DNA(1)/DNA(2), no irradiation; 5, same as lane 4, with irradiation; 6, same as lane 5, with piperidine treatment; 7, same as lane 5, with Fpg treatment (10 μg/ml, 30 min, 37°C).
Guanine damage may be caused by intramolecular or intermolecular reactions. We carried out control experiments to distinguish these possibilities. Triplet anthraquinone is capable of generating singlet oxygen (1O2), which adds indiscriminately to guanines and causes strand cleavage (43–45). It is unlikely that 1O2 is the cause of the cleavage seen in DNA(2) because isolated guanines do not react. Nevertheless, we probed for participation of 1O2 by changing the reaction solvent to D2O, where the 1O2 lifetime is lengthened (46, 47). There is no measurable isotope effect on the cleavage yield; consequently, 1O2 can have no more than a minor role in the long-range reaction. Irradiation of a mixture of AQ-DNA(1)/5′-32P-DNA(2) and noncomplementary GG-containing duplex DNA (0.3 and 5 μM, respectively) does not reduce the cleavage yield measured in DNA(2). Further, irradiation of AQ-DNA(1)/DNA(2) causes no detectable cleavage of an added radiolabeled, noncomplementary duplex. These experiments show that the long-distance reaction caused by irradiation of AQ-DNA(1)/DNA(2) must be intramolecular.
The cleavage yield from irradiation of AQ-DNA(1)/5′-32P-DNA(2) was quantified by measuring the radioactivity in each band. Fig. 3 is a semilog plot of cleavage yield against distance between the 5′-G and the AQ. A clear linear relationship with slope of −0.02 Å−1 is revealed. This is a remarkable result that indicates an extraordinary ability for the radical cation to travel through DNA.
Figure 3.

Semi-log plot of cleavage intensity after piperidine treatment of irradiated AQ-DNA(1)/5′-32P-DNA(2) determined by counting the radioactivity with a β-detector. The measured counts in each band were normalized so that G10 ≡ 1.0. The distance scale was calculated assuming an average distance of 3.4 Å between base pairs. The error bars represent standard deviations calculated from four independent experiments.
DISCUSSION
Two extreme mechanisms of radical cation transport in DNA can be considered. In the first, DNA behaves like a molecular wire, and the radical cation is instantaneously delocalized through a continuous molecular orbital of the B-form helix. In this orbital, each base pair is in electronic contact with every other. In this model, the radical cation can “travel” instantaneously through the DNA independent of base sequence and distance (17). The second model is hole hopping, which presumes a discrete molecular orbital localized at each base with no significant electronic overlap between adjacent base pairs. The localized radical cation migrates (hops) by a thermally activated process requiring local structural distortion of the DNA (30). The results of our experiments, and those recently reported by others (10, 34), suggest a complex reality containing features from both of these limiting models (26, 32).
The key findings of this work are revealed in Fig. 3. Three facts are evident from inspection of this figure. First, radical cation-caused strand cleavage of DNA(2) occurs at each GG step. It is inconceivable that a single delocalized molecular orbital could extend over these many base pairs because DNA is a dynamic structure and the likelihood of all 55 base pairs being simultaneously well stacked is vanishingly small (48).
Second, Fig. 3 reveals a linear relationship between the log of cleavage efficiency and the distance between the site of charge injection and radical cation reaction. This is significant because AQ-DNA(1)/DNA(2) has different sequences between each GG step. For example, the segment between G28 and G46 contains, on its own strand, the base sequence 3′-AAATT-5′ whereas, between the AQ and G10, the longest AT stretch between a base pair containing a guanine is 2 bases long. We interpret the linear relationship independent of sequence to indicate that structural averaging of DNA occurs. Importantly, Giese and coworkers (10) report related measurements of radical cation migration that show an ≈10-fold decrease in rate for each AT base pair between guanines. There are systemic differences between our experiments and those of Giese that may account for these apparently contrasting results. We discuss structural averaging and these systemic differences below.
Third, Fig. 3 reveals that the distance dependence for charge migration is shallow. This is quantified by the slope of −0.02 Å−1. Because the mechanism for radical cation transport cannot be superexchange through bridge orbitals, it is misleading to refer to this slope as β. Following recent usage, we refer to the magnitude of the slope as γ without, at this point, attributing a mechanistic meaning to that designation (34). Any mechanism of charge transport in DNA must accommodate both the linear relationship and the magnitude of γ that is revealed in Fig. 3.
A Kinetic Model for γ.
Irradiation of AQ-DNA(1)/DNA(2) injects a radical cation at one end of the DNA duplex. We suppose that the cation may undergo one of three processes (see Scheme S1). First, it can migrate away from its site of generation. The initial step can only take it away from the point of charge injection. We designate the first-order rate constant kh to symbolize this process. In principle, kh could have a unique value that depends on the actual base sequence. For simplicity, and because averaging over base sequence is postulated to accommodate our findings, we assume that kh is independent of sequence. This implies that, after the first step, migration in either direction is equally likely. Averaging processes that yield a nearly constant value for the hopping probability are proposed below.
Scheme 1.

A second process that the radical cation may undergo is annihilation. By this we mean that it is consumed but does not generate strand cleavage. For example, irradiation of the AQ generates superoxide anion (O2⨪) (38). An encounter between O2⨪ and the radical cation will lead to annihilation and no net chemical reaction of the DNA. Similarly, any reagent (a metal ion, for example) capable of transferring an electron to the radical cation can cause its consumption. We designate the rate constant ka to symbolize consumption of the radical cation not leading to strand cleavage. It, too, must be an average, and, as a matter of fact, ka designates a process that is second order, or higher, kinetically, but will be treated here as a pseudo first-order rate constant by assuming that the concentration of reagent is constant and incorporated into the value of ka.
Finally, the third process is trapping of the radical cation that we designate with the rate constant kt. By “trapping,” we mean the consumption of the radical cation by a reaction that yields a chemical product that is revealed as strand cleavage (13). Trapping occurs primarily on the 5′-G of GG steps, which may be a consequence of either kinetic or thermodynamic considerations (41). As with ka, kt is treated as a pseudo first-order process because the reagent concentration is assumed to be constant.
Application of probability theory to the kinetic model outlined above yields Eq. 1 as a description of the radical cation migration process. In Eq. 1, Pc is the amount of observed strand cleavage at a particular base and is directly proportional to the experimental value I in Fig. 3, n (≥2) is the number of steps the radical cation takes between the cleavage site and the point of charge injection at AQ, Pt is the probability of trapping the radical cation that is given by Eq. 2, Pf is the probability that the first, unique, step will occur and is given in Eq. 3, and Ph is the probability that the radical cation will hop before it is annihilated or trapped and is given in Eq. 4.
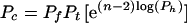 |
1 |
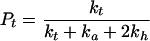 |
2 |
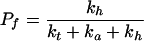 |
3 |
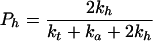 |
4 |
Estimates for Ph can be obtained within the proposed kinetic model from the slope (γ) in Fig. 3 and an assumption of the step length. If we assign the step length to be 1 base pair (3.4 Å), then Ph = 0.86. However, it is important to emphasize that there is no evidence that requires the step length to be 1 base pair or, more importantly, that it be a constant value. In fact, we suggest below that the step length will depend on the particular base pair sequence the radical cation encounters for each step. Variable step length is a key component of the mechanism we propose to average the value of kh.
A Model Mechanism: Phonon-Assisted Polaron-Like Hopping.
DNA is a dynamic structure in which bases move rapidly. Recent experiments indicate a liquid-like internal structure for DNA in which small amplitude motions occur on short time scales (49). And NMR spectral analyses provide evidence for sugar and base motions that occur with periods of 30–300 ps (50). We suggest that dynamical structural variation of DNA mixes the hole-hopping and molecular wire mechanisms for radical cation transport. This concept forms the key part of the mechanism we propose for radical cation migration in duplex DNA that we designate as phonon-assisted polaron-like hopping.
A polaron is a radical ion self-trapped by structural distortion of its containing medium (51). The polaron may migrate by tunneling or by phonon-assisted (thermally activated) hopping (52). Baverstock (53) proposed that formation of a one-dimensional polaron in DNA provides a means for energy translation. The key concepts of the polaron-like hopping model are depicted schematically in Fig. 4.
Figure 4.

Symbolic representation of a polaron-like distortion in duplex DNA. Vertical lines represent any DNA base (solid for purines, dashed for pyrimidines). The polaron-like distortion is contained within the rectangle symbolized by a decrease in base pair distance. The radical cation charge and free radical density of the polaron is portrayed as delocalized throughout its structure. Hopping of the polaron, in this case with a step size of 1 base pair, to the right is illustrated by one base pair joining the structure (on the right) and one leaving (on the left).
It seems certain that injection of a charge into DNA will cause its structure to change (54). Base radical cations are electron-deficient, and it is naïve to expect that the DNA will not rapidly adjust its local structure to relieve this deficiency. One likely structural change is a reduction of the intrabase distance caused by a change in inclination (η) between adjacent base pairs (55). The ability of adjacent bases to stabilize the radical cation by electron donation will increase as the distance between them decreases. A second structural change is a decrease in the twist angle (Ω) by rotation around the z axis of the DNA. This unwinding will increase the π-electron overlap between bases and will stabilize the radical cation. Another possible distortion is a shift in proton donation of the hydrogen bonds that form the base pairs. The pKa of a localized base radical cation is different from that of the neutral base (56, 57). Obviously, the pKa of bases carrying delocalized (partial) positive charge also will change, and hydrogen bonds in duplex DNA will shift to accommodate this change. It is the total of these structural changes that we depict in Fig. 4 as the polaron-like distortion. Simply put, the polaron-like distortion is a section of duplex DNA surrounding a base radical cation with a structure modified so that stabilization of the radical cation results. The detailed properties of the polaron distortion (lifetime, reactivity, and number of base pairs involved) will depend on the specific base sequence, and for this reason it is not strictly a polaron.
The structural distortion caused by introduction of the radical cation is not expected to extend indefinitely through the DNA. The cause of the distortion is minimization of free energy. As the size of the polaron increases, the energy required to distort the DNA structure will be balanced by decreasing additional stabilization of the radical cation gained by the incorporation of an additional base. Thus, we characterize the polaron as a “self-trapped” local distortion of the DNA. We postulate further that electronic overlap between bases within the polaron is greater than in standard DNA because it is formed in response to a need for electron donation. As a consequence, the radical cation becomes delocalized within the base pairs in the polaron-like distortion, and its transport therein might appear to occur by a superexchange mechanism.
In this model, part of the averaging required for kh is accomplished by delocalization of the radical cation through the bases of the polaron. This would be evidenced by sequence-dependent oxidation potentials (Eox) for the DNA bases. Reliable values of Eox for the DNA bases are available (14). We are aware of no oxidation potential measurements for bases in duplex DNA. However, some insight into polaron averaging is available from recent ab initio calculations for guanine-containing hot-spots by Saito et al. (15). They report calculated ionization potentials (Ip) for 16 standard B-form duplex sequences. For example, the 5-mer fragments TAGAT and TTGTT have Ip values of 6.73 and 6.96 eV, respectively. The calculated Ip for an isolated GC base pair is 7.34 eV (58, 59). Clearly, even in the undistorted duplex DNA structure, neighboring bases contribute to radical cation stabilization. This effect can only become more significant in the polaron in which the structure of the DNA has been distorted to accommodate the radical cation. There seems little doubt that the differences between the Eox of isolated bases will be reduced when they are incorporated into DNA. This is an important component of the averaging we propose that generates a nearly constant value for kh.
Long-range radical cation migration cannot occur by superexchange. We suggest instead that the internal structural dynamical motions of DNA cause the polaron to migrate. By this we mean that thermal motions of the base pairs in and near the structural distortion lead to their leaving or joining the polaron. We designate this as phonon-assisted hopping. The number of base pairs leaving and joining the polaron distortion in a hop will depend on the local sequence. For example, Saito et al. (15) calculate Ip values equal to 6.96 eV for both the CGC 3-mer and for the TTGTT 5-mer. Thus, with the assumption that radical cation stabilization in the distorted structures parallels the change in Ip, the polaron can hop from a CGC to an adjacent TTGTT sequence with no change in free energy. This hop has a step size of 4 base pairs (C1 → G4) in the sequence CGC1 T2T3G4 TT. In this regard, polaron-like hopping is an example of the familiar Marcus mechanism for electron transfer (60). In this instance, the solvent reorganization (λ) is dominated by the motions of the bases in and adjacent to the polaron.
Within this kinetic model, the step size for each polaron hop will be determined by the number of bases required to minimize the free energy change. For example, the calculated Ip for the 5-mers TAGTT and TTGTT are 6.93 and 6.96 eV, respectively. Thus, the step size for a polaron in the sequence TAG TTG TT will be 3 base pairs. Similarly, the Ip of the 3-mers AGC and CGC are calculated to be 7.01 and 6.96 eV, respectively. The step size in the sequence AGC GC then would be 2 base pairs. Combining these three examples, a polaron can migrate through the 21-mer sequence CGC TTG TTA GCG CTA GTT GTT in five hops with step sizes of 4, 4, 2, 4, and 3 base pairs and a maximum change in Ip of only 0.07 eV. Variable step size is a second sequence averaging that can lead to a nearly constant value for kh.
This model can be applied to the results observed for AQ-DNA(1)/DNA(2). Consider the sequence between G28 and G46, where previous proposals require radical migration to be retarded by the 5-base pair AT run (10). We divide the sequence arbitrarily into four polarons: 3′-AAGG28AAA-5′; 3′-TTGATT-5′; 3′-ACGTC-5′; and 3′-ACTGG46C-5′. In this construction, three hops each with step lengths of 6 base pairs are required for the radical cation to migrate from G28 to G46. The Ip of these sequences have not been calculated. However, based on the available results (15), the Ip difference between them will be a small. Without experimental measurements of Eox for relevant duplex DNA sequences, assignment of polaron composition and step length is arbitrary. Nevertheless, it is clear that the concept of phonon-assisted polaron hopping permits the averaging of sequence differences required to accommodate our findings.
Implicit in the discussion thus far is the assumption that radical cation density is confined primarily to one strand of duplex DNA. However, we have evidence that a radical cation injected on one strand can hop to its complementary strand (D. Ly and G.B.S., unpublished work). Interstrand migration plays a critical part in the proposal of Giese and coworkers (10). Inclusion of interstrand hopping provides another mechanism for the averaging of sequence differences. Of course, there is no necessary requirement that every possible DNA sequence will give the same value for Ph. This accommodates the recent observation that radical cation migration through multiple 5′-TA-3′ steps is diminished faster than for other sequences (25). And it reconciles our findings with those reported by Giese coworkers. (10) Also, in their experiments, the radical cation is formed in a chemical reaction that introduces a strand break. It is possible that ka and kt will have different values near a frayed end than in an internal DNA sequence.
The phonon-assisted polaron hopping model described for radical cation migration can be generalized to accommodate results for other systems. For example, the quenching of fluorescence by a remote electron donor can occur instantaneously if the fluorescer and donor are part of an appropriate distortion in the DNA structure in the instant when the electronically excited state is created (34). In this view, the distance dependence reported for instantaneous quenching is not a measure of superexchange (β) but describes the probability that a structural distortion extends as far as the quencher when the excited state is formed. In a like manner, dynamic distance-dependent quenching will depend on the probability that a distortion extends to the quencher during the lifetime of the excited state (22). Thus, time-resolved emission of a fluorescer linked to DNA may not be a measure of superexchange but may gauge the probability that a structural distortion extending to the quencher will be formed within the lifetime of the emitter.
CONCLUSIONS
The results described above demonstrate that long-range charge transfer in DNA falls off exponentially with distance and can extend to ≈200 Å. We propose a kinetic model to accommodate this. The mechanism is based on the fact that DNA is a dynamic structure on the time scale of charge transport experiments. The structure of DNA responds to the radical cation by creating the self-trapped distortion we characterize as a polaron. The migration of the polaron through DNA occurs as a consequence of normal vibrational fluctuations (phonons). This mechanism bears a close resemblance to classical electron transfer processes described by Marcus theory. Averaging of differences in the local sequence occurs through variation of the number of bases that comprise the polaron and by variability in the step size the polaron takes when it hops. An appealing aspect of this mechanistic proposal is that it can be generalized to reconcile experimental and theoretical findings that previously seemed contradictory.
Acknowledgments
We are thankful to Professors Laren Tolbert and Loren Williams of this department for insightful advice on polaron transport in DNA and help in the design of the DNA sequence. We are grateful to Professor Christian Houdre, of the School of Mathematics, Georgia Institute of Technology, for assistance in the analysis of the hopping model. This work was supported by grants from the National Institutes of Health and the National Science Foundation, for which we are grateful.
ABBREVIATIONS
- Fpg
formamidopyrimidine glycosolase
- Ip
ionization potential
References
- 1.Tuppurainen K, Lotjonen S, Laatikainen R, Vartiainen T, Maran U, Strandberg M, Tamm T. Mutat Res. 1991;247:97. doi: 10.1016/0027-5107(91)90037-o. [DOI] [PubMed] [Google Scholar]
- 2.Demple B, Harrison L. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 3.Loft S, Poulsen H E. J Mol Med. 1996;74:297–312. doi: 10.1007/BF00207507. [DOI] [PubMed] [Google Scholar]
- 4.Melvin T, Botchway S W, Parker A W, O’Neill P. J Am Chem Soc. 1996;118:10031–10036. [Google Scholar]
- 5.Steenken S. Chem Rev. 1989;89:503–520. [Google Scholar]
- 6.Kasai H, Yamaizumi Z, Berger M, Cadet J. J Am Chem Soc. 1992;114:9692–9694. [Google Scholar]
- 7.Saito I, Takayama M, Sugiyama H, Nakatani K, Tsuchida A, Yamamoto M. J Am Chem Soc. 1995;117:6406–6407. [Google Scholar]
- 8.Hall D B, Holmlin R E, Barton J K. Nature (London) 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 9.Stemp E D A, Holmlin R E, Barton J K. Nature (London) 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 10.Meggers E, Michel-Beyerle M E, Giese B. J Am Chem Soc. 1998;120:12950–12955. [Google Scholar]
- 11.Tronche C, Goodman B K, Greenberg M M. Chem Biol. 1998;5:263–271. [PubMed] [Google Scholar]
- 12.Pogozelski W K, Tullius T D. Chem Rev. 1998;98:1089–1108. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 13.Burrows C J, Muller J G. Chem Rev. 1998;98:1109–1154. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 14.Steenken S, Jovanovic S V. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 15.Saito I, Nakamura T, Nakatani K, Yoshioka Y, Yamaguchi K, Sugiyama H. J Am Chem Soc. 1998;120:12686–12687. [Google Scholar]
- 16.Arkin M R, Stemp E D A, Holmlin R E, Barton J K, Horman A, Olson E J C, Barbara P F. Science. 1996;273:475–480. doi: 10.1126/science.273.5274.475. [DOI] [PubMed] [Google Scholar]
- 17.Turro N J, Barton J K. J Biol Inorg Chem. 1998;3:201–209. [Google Scholar]
- 18.Priyadarshy S, Risser S M, Beratan D N. J Phys Chem. 1996;100:17678–17682. [Google Scholar]
- 19.Beratan D N, Priyadarshy S, Risser S M. Chem Biol. 1997;4:3–8. doi: 10.1016/s1074-5521(97)90230-1. [DOI] [PubMed] [Google Scholar]
- 20.Brun A M, Harriman A. J Am Chem Soc. 1992;114:3656–3660. [Google Scholar]
- 21.Meade T J, Kayyem J F. Angew Chem Int Ed Engl. 1995;34:352–354. [Google Scholar]
- 22.Lewis F D, Wu T, Zhang Y, Letsinger R L, Greenfield S R, Wasielewski M R. Science. 1997;277:673–676. doi: 10.1126/science.277.5326.673. [DOI] [PubMed] [Google Scholar]
- 23.Fukui K, Tanaka K. Angew Chem Int Ed Engl. 1998;37:158–161. [Google Scholar]
- 24.Gasper S M, Schuster G B. J Am Chem Soc. 1997;119:12762–12771. [Google Scholar]
- 25.Nunez M, Hall D B, Barton J K. Chem Biol. 1999;6:85–97. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- 26.Jortner J, Bixon M, Langenbacher T, Michel-Beyerle M E. Proc Natl Acad Sci USA. 1998;95:12759–12765. doi: 10.1073/pnas.95.22.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eley D D, Spivey D I. Trans Faraday Soc. 1962;58:411–415. [Google Scholar]
- 28.Murphy C J, Arkin M R, Jenkins Y, Ghatlia N D, Bossman S H, Turro N J, Barton J K. Science. 1993;262:1025–1029. doi: 10.1126/science.7802858. [DOI] [PubMed] [Google Scholar]
- 29.McConnell H M. J Chem Phys. 1961;35:508–513. [Google Scholar]
- 30.Ly D, Kan Y, Armitage B, Schuster G B. J Am Chem Soc. 1996;118:8747–8748. [Google Scholar]
- 31.Armitage B, Ly D, Koch T, Frydenlund H, Orum H, Batz H G, Schuster G B. Proc Natl Acad Sci USA. 1997;94:12320–12325. doi: 10.1073/pnas.94.23.12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ratner M. Nature (London) 1999;397:480–481. doi: 10.1038/17232. [DOI] [PubMed] [Google Scholar]
- 33.Holstein W L, Boudart M. J Phys Chem B. 1997;101:9991–9994. [Google Scholar]
- 34.Kelley S O, Barton K K. Science. 1999;283:375–381. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- 35.Breslin D T, Yu C, Ly D, Schuster G B. Biochemistry. 1997;36:10463–10471. doi: 10.1021/bi9702750. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 37.Mori K, Subasinghe C, Cohen J S. FEBS Lett. 1989;249:213–218. doi: 10.1016/0014-5793(89)80626-x. [DOI] [PubMed] [Google Scholar]
- 38.Armitage B A, Yu C, Devadoss C, Schuster G B. J Am Chem Soc. 1994;116:9847–9859. [Google Scholar]
- 39.Breslin D T, Schuster G B. J Am Chem Soc. 1996;118:2311–2319. [Google Scholar]
- 40.Guckian K M, Schweitzer B A, Ren R X-F, Sheils C J, Paris P L, Tahmassebi D C, Kool E T. J Am Chem Soc. 1996;118:8182–8183. doi: 10.1021/ja961733f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gasper S M, Armitage B, Hu G G, Shui X, Yu C, Williams L D, Schuster G B. J Am Chem Soc. 1998;120:12402–12409. [Google Scholar]
- 42.David S S, Williams S D. Chem Rev. 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 43.Blazek E R, Peak J G, Peak M J. Photochem Photobiol. 1989;49:607–613. doi: 10.1111/j.1751-1097.1989.tb08431.x. [DOI] [PubMed] [Google Scholar]
- 44.Floyd R A, West M S, Eneff K L, Schneider J E. Arch Biochem Biophys. 1989;273:106–111. doi: 10.1016/0003-9861(89)90167-7. [DOI] [PubMed] [Google Scholar]
- 45.Devasagayam T P A, Steenken S, Obendorf M S W, Schulz W A, Sies H. Biochemistry. 1991;30:6283–6289. doi: 10.1021/bi00239a029. [DOI] [PubMed] [Google Scholar]
- 46.Rogers M A J, Snowden P T. J Am Chem Soc. 1982;104:5541–5543. [Google Scholar]
- 47.Showen K B, Showen R L. Solvent Isotope Effects on Enzyme Systems. New York: Academic; 1982. [PubMed] [Google Scholar]
- 48.Packer K J, Hunter C A. J Mol Biol. 1998;280:407–420. doi: 10.1006/jmbi.1998.1865. [DOI] [PubMed] [Google Scholar]
- 49.Brauns E B, Murphy C J, Berg M A. J Am Chem Soc. 1998;120:2449–2456. [Google Scholar]
- 50.Borer P N, Pante S R, Kumar A, Zanatta N, Martin A, Hakkinen A, Levy G C. Biochemistry. 1994;33:2441–2450. doi: 10.1021/bi00175a012. [DOI] [PubMed] [Google Scholar]
- 51.Sewell G L. Polarons and Excitions. New York: Plenum; 1962. [Google Scholar]
- 52.Emin D. Handbook of Conducting Polymers. New York: Dekker; 1986. [Google Scholar]
- 53.Baverstock K F, Cundall R B. Nature (London) 1988;332:312–313. doi: 10.1038/332312b0. [DOI] [PubMed] [Google Scholar]
- 54.Colson A-O, Besler B, Sevilla M D. J Phys Chem. 1992;96:9787. [Google Scholar]
- 55.Sriram M, Wang A H-J. In: Bioorganic Chemistry: Nucleic Acids. Hecht S M, editor. New York: Oxford Univ. Press; 1996. pp. 105–143. [Google Scholar]
- 56.Englander S W, Kallenbach N R, Heeger A J, Krumhansl J A, Litwin S. Proc Natl Acad Sci USA. 1980;77:7222–7226. doi: 10.1073/pnas.77.12.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steenken S. Biol Chem. 1997;378:1293–1297. [PubMed] [Google Scholar]
- 58.Sugiyama H, Saito I. J Am Chem Soc. 1996;118:7063–7068. [Google Scholar]
- 59.Prat F, Houk K N, Foote C S. J Am Chem Soc. 1998;120:845–846. [Google Scholar]
- 60.Marcus R A. Annu Rev Phys Chem. 1964;15:155–196. [Google Scholar]