

Abstract
N-Methyltryptophan oxidase (MTOX), a flavoenzyme from Escherichia coli, catalyzes the oxidative demethylation of secondary amino acids such as N-methyltryptophan or N-methylglycine (sarcosine). MTOX is one of several flavin-dependent amine oxidases whose chemical mechanism is still debated. The kinetic properties of MTOX with the slow substrate sarcosine were determined. Initial rate data are well-described by the equation for a ping-pong kinetic mechanism, in that the V/KO2 value is independent of the sarcosine concentration at all accessible concentrations of oxygen. The kcat/Ksarc pH profile is bell-shaped, with pKa values of 8.8 and about 10; the latter value matches the pKa value of the substrate nitrogen. The kcat pH profile exhibits a single pKa value of 9.1 for a group that must be unprotonated for catalysis. There is no significant solvent isotope effect on the kcat/Ksarc value. With N-methyl-2H3-glycine as the substrate, there is a pH-independent kinetic isotope effect on kcat, kcat/Ksarc, and the rate constant for flavin reduction, with an average value of 7.2. Stopped-flow spectroscopy with both the protiated and deuterated substrate failed to detect any intermediates between the enzyme–substrate complex and the fully reduced enzyme. These results are used to evaluate proposed chemical mechanisms.
N-Methyltryptophan oxidase (MTOX)1 catalyzes the oxidative demethylation of N-methyl amino acids (Scheme 1). The earliest studies of the enzyme, dating back to the 1940s, showed that MTOX is commonly found in both prokaryotes and eukaryotes, including humans (6-10). It was rediscovered in Escherichia coli in 1996 because of its 43% amino acid identity to the more thoroughly studied monomeric sarcosine oxidase (MSOX) (11). Like MSOX, MTOX is a monomeric enzyme with a covalently attached flavin cofactor (12). Although MTOX can catalyze demethylation of sarcosine (N-methylglycine), it has a preference for bulky hydrophobic substrates, such as N-methyl-l-tryptophan (11).
The flavoproteins, which catalyze amine oxidation, have thus far fallen into two structural groups. d-Amino acid oxidase (13), MSOX (3) and glycine oxidase (14) all have a similar overall fold, although the relative orientation of the substrate and flavin differs in MSOX from that in the other two. Sarcosine is a substrate for d-amino acid oxidase (15), although the products are glycolate and methylamine instead of glycine and formaldehyde, the products with MSOX and MTOX. Monoamine oxidase B (16), polyamine oxidase (17), and l-amino acid oxidase (18) form a separate structural class. While a number of these enzymes have been the subject of kinetic, spectroscopic, and structural studies, only in the case of d-amino acid oxidase have mechanistic (19) and structural (13) studies led to a consensus that the reaction involves hydride transfer. In contrast, the chemical mechanism of amine oxidation by the remaining amine oxidases is still debated.
Scheme 1.
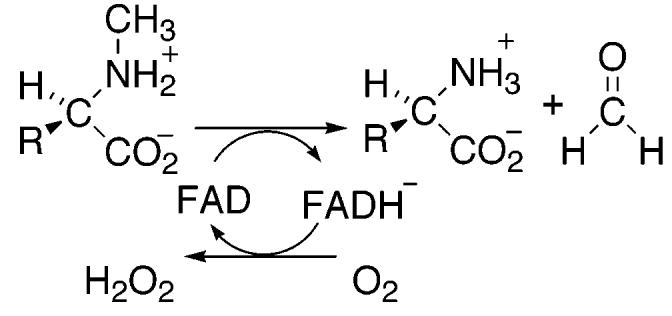
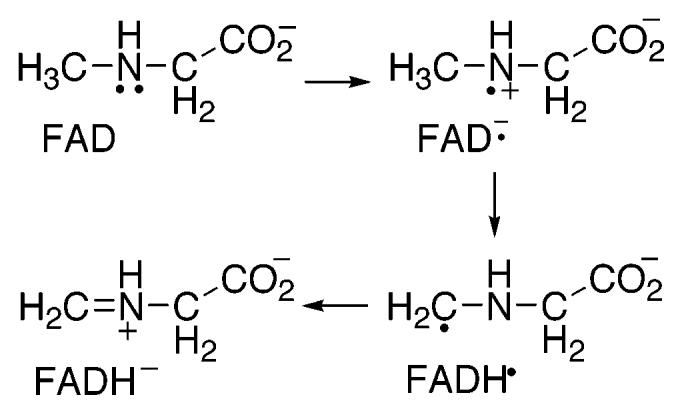
A variety of mechanisms have been proposed for amino acid oxidation by sarcosine oxidase and MTOX (2, 12, 20-22). The mechanism shown in Scheme 2 involves two separate single electron transfers. The initial transfer of an electron from the substrate nitrogen to the flavin forms a flavin semiquinone, which is then further reduced by either separate proton and electron transfers as shown or by a hydrogen atom transfer. The mechanism shown in Scheme 3 involves a concerted electron and proton transfer in the form of a hydride transfer from the substrate methyl group. In the mechanism of Scheme 4, the substrate nitrogen attacks the flavin cofactor to form a covalently bound flavin intermediate; this is followed by the loss of a proton from the substrate methyl carbon and electron rearrangement to give the final products. Whereas the hydride transfer mechanism (Scheme 3) involves no intermediates between fully oxidized and fully reduced flavin, the mechanisms of Schemes 2 and 4 involve a third, intermediate flavin species. The absorbance changes associated with the reduction of the flavin in MSOX or MTOX by a number of substrates have previously been monitored at 25 and 4 °C (1, 22, 23). In all cases, flavin reduction could be described as an isosbestic conversion from fully oxidized flavin to fully reduced flavin with no detectable intermediates. However, these observations are insufficient to conclusively assign the hydride transfer mechanism as the mechanism of amine oxidation. As previously noted (21, 23), formation of the postulated intermediates may occur during a rate-limiting step. In such a scenario, the rate of decay of the flavin intermediate would be greater than the rate of its formation, thereby preventing any detectable concentration of the intermediate species. In the present study, we utilize pH and isotope effects to identify the rate-limiting step of sarcosine turnover by MTOX and to probe for intermediates prior to carbon–hydrogen bond cleavage.
Scheme 2.
Scheme 3.
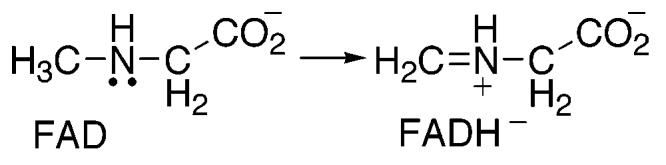
Scheme 4.

EXPERIMENTAL PROCEDURES
Materials
N-Methyl-l-tryptophan was purchased from Aldrich Chemical Co. (Milwaukee, WI). Sarcosine (N-methylglycine) was purchased from Sigma Chemical Company (St. Louis, MO). N-Methyl-2H3-glycine was purchased from CDN Isotopes (Quebec, Canada). DNA vectors and E. coli cells were from Novagen (Madison, WI). The HiTrap Chelating HP column was from Amersham Pharmacia Biotech (Uppsala, Sweden).
DNA Manipulation
Genomic DNA was prepared from BL21DE3* E. coli cells using the Qiagen genomic DNA purification kit. The MTOX gene was amplified by PCR using the oligonucleotides 5′-CCATGAGAGCAGAGAGAATTCAATGAAATACGATCT (oligonucleotide 1) and 5′-AAAACTTTCTCATTCTAGAGGCCGTAATGATGATTATTG and then ligated into pUC18 between the EcoRI and XbaI sites. The gene was later amplified with oligonucleotide 1 and the oligonucleotide 5′-AGAGGACGAAATGACTCGAGTTGGAAGCGGG and subcloned into pET23d between the EcoRI and XhoI sites in frame with the C-terminal His6 tag to form the plasmid pETMTOX. The resulting recombinant protein sequence is lengthened by a leucine, a glutamate, and the His6 tag.
Protein Expression and Purification
A culture of Luria–Bertani broth (LB) containing 100 μg/mL ampicillin was inoculated from a single colony of BL21DE3* E. coli containing pETMTOX and grown overnight at 37 °C. A total of 6 L of LB containing 100 μg/mL ampicillin were inoculated from the overnight culture using 10 mL of culture/L. The cells were grown at 37 °C to an absorbance at 600 nm of 0.6–0.8 and induced with 120 mg of IPTG/L. After an additional 5 h, the cells were harvested by centrifugation at 5000g for 30 min at 4 °C. The cell pellets (approximately 20 g) were resuspended in 100 mL of 10 mM sodium pyrophosphate, 100 μg/mL phenylmethylsulfonyl fluoride, 1 μM leupeptin, and 1 μM pepstatin at pH 8.0. Cells were lysed using a French Press, and the cell debris was removed by centrifugation at 29000g for 20 min at 4 °C. The clarified lysate was filtered through a 0.45 μm filter and loaded onto a 5 mL HiTrap Chelating HP column charged with nickel sulfate. Using an Amersham-Pharmacia Biotech AKTA FPLC, the column was washed first with 15 mL of 10 mM pyrophosphate at pH 8.0 and then with a 10 mL gradient of 0–6 mM imidazole in 10 mM pyrophosphate at pH 8.0. The protein was eluted with a 25 mL linear gradient of 6–120 mM imidazole. Fractions that had A280/A460 values less than 15 were pooled and precipitated at 75% saturated ammonium sulfate. The protein was resuspended in a minimal volume and dialyzed against 10 mM sodium pyrophosphate and 10% glycerol at pH 8.0. The resulting protein exhibited a single band in polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate. Approximately 130 mg of holoenzyme could be purified from a 6 L cell growth. Enzyme samples were stored at −80 °C at a concentration of 250–300 μM. The flavin content of purified enzyme samples was determined as described by Wagner et al. (12)
Enzyme Assays
The rate of oxygen consumption was measured using a Hansatech oxygen-monitoring system (Norfolk, U.K.) with a computer-interfaced graphical mode or a Yellow Springs Instrument model 5300 oxygen electrode (Yellow Springs, OH). Steady-state kinetic parameters were routinely determined at 25 °C under atmospheric oxygen concentrations in the constant ionic strength buffer 0.1 M ACES, 52 mM Tris, 52 mM ethanolamine-HCl, and 1 M KCl, unless stated otherwise. When necessary, the oxygen concentration was varied by bubbling a reaction sample with the desired oxygen/argon mixture in the oxygen electrode cuvette until a constant oxygen concentration was observed. The reaction was then started by the immediate addition of enzyme. To measure the solvent isotope effect, the above constant ionic strength buffer was prepared in D2O. The pD was calculated by adding 0.4 unit to the reading on the pH meter. Deuterium kinetic isotope effect values were calculated from direct comparison of the rates of oxidation of methyl-deuterated and methyl-protiated sarcosine under identical conditions. The rate of flavin reduction was determined by anaerobic stopped-flow spectroscopy using an Applied Photophysics SX.18MV stopped-flow spectrophotometer. Buffers used for anaerobic experiments included 5 mM glucose and were cycled 10 times between vacuum and argon prior to the addition of glucose oxidase (10 μg/mL). The active enzyme concentration was calculated from the absorbance at 456 nm using an extinction coefficient of 13.3 mM−1 cm−1 (12).
Data Analysis
The kinetic data were analyzed using Kaleidagraph (Synergy Software, Reading, PA), Igor (Wave-metrics, Lake Oswego, OR), and SPECFIT (Spectrum Software Associates, Marlborough, MA). When the concentration of only one substrate was varied, initial rate data were fit to either the Michaelis–Menten equation or a straight line. When the concentrations of both substrates were varied, initial rate data were fit to different models described by eqs 1-3; eq 1 describes a sequential reaction, eq 2 describes a ping-pong reaction, and eq 3 describes an ordered rapid equilibrium reaction. The kcat/Ksarc and kcat values at different pH or pD values were fit to eqs 4 and 5, where C is the pH-independent value of kcat/Ksarc, L is the concentration of the lyonium ion, and K1 and K2 are the dissociation constants for the ionizable groups. Deuterium kinetic isotope effect values were obtained from fits of the data to eq 6. This equation describes equal isotope effects on kcat and kcat/Ksarc; Fi is the fraction of the heavy atom, and EKIE is the isotope effect minus 1. Stopped-flow traces were fit to eq 7, which describes a monophasic exponential decay; k is the first-order rate constant, At is the absorbance at time t, and A∞ is the final absorbance. The rate constants for flavin reduction at different concentrations of sarcosine were analyzed using eq 8; kred is the rate constant for flavin reduction at saturating sarcosine concentrations, and Kd is the apparent dissociation constant.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
RESULTS
Steady-State Kinetics
The C terminus of the recombinant protein encoded by the pETMTOX vector has two extra amino acids followed by a His6 tag. Analysis of the percent flavin content of purified enzyme indicated approximately 35% of the enzyme-contained FAD. The concentration of the holoenzyme determined from the flavin spectrum was used in all calculations of kinetic parameters. The steady-state kinetic parameters for this form of MTOX were determined using atmospheric oxygen concentrations in 0.1 M potassium pyrophosphate and 1 mM EDTA at pH 8.0 with both N-methyltryptophan and sarcosine as the substrates and are in good agreement with previously published values (22) (Table 1). Thus, the His6 tag does not affect the enzymatic activity of this enzyme.
Table 1.
Apparent Steady-State Kinetic Parameters for His6-Tagged MTOX at pH 8.0a
| N-methyltryptophan | sarcosine | |
|---|---|---|
| kcat (min−1) | 1190 ± 50 | 23 ± 7 |
| Km (mM) | 0.25 ± 0.02 | 44 ± 5 |
| kcat/Ksarc (mM−1 min−1) | 4700 ± 200 | 0.51 ± 0.04 |
Air-saturated 0.1 M potassium phosphate and 1 mM EDTA at pH 8.0 and 25 °C.
Slow substrates, such as sarcosine in the case of MTOX, are of great utility in the study of enzyme mechanisms, in that chemical steps are typically rate-limiting in such cases, allowing intrinsic rate constants, isotope effects, and pKa values to be measured using steady-state kinetics (24). Consequently, sarcosine was characterized as a substrate for MTOX to probe the chemical mechanism. To determine the oxygen dependence of sarcosine turnover, initial rates of oxygen consumption were measured in a constant ionic strength buffer at pH 9.4 and 25 °C, while varying both oxygen and sarcosine concentrations. The resulting data (Figure 1) were fit to eqs 1-3, which describe sequential, ping-pong, and ordered rapid equilibrium mechanisms, respectively. The resulting kinetic parameters are shown in Table 2. Fitting the data to eq 3 required assignment of oxygen as the first substrate. The lines shown in Figure 1 are from the fit of the data by eq 2, which describes a ping-pong mechanism. While the fit to eq 1 is statistically better than the fit to eq 2, the value of Ki,sarc is not significantly different from zero. More importantly, either fit predicts a sufficiently low Km for oxygen that kcat/Ksarc values can be treated as independent of oxygen concentrations under atmospheric oxygen concentrations and kcat values can be determined at ambient concentrations.
Figure 1.
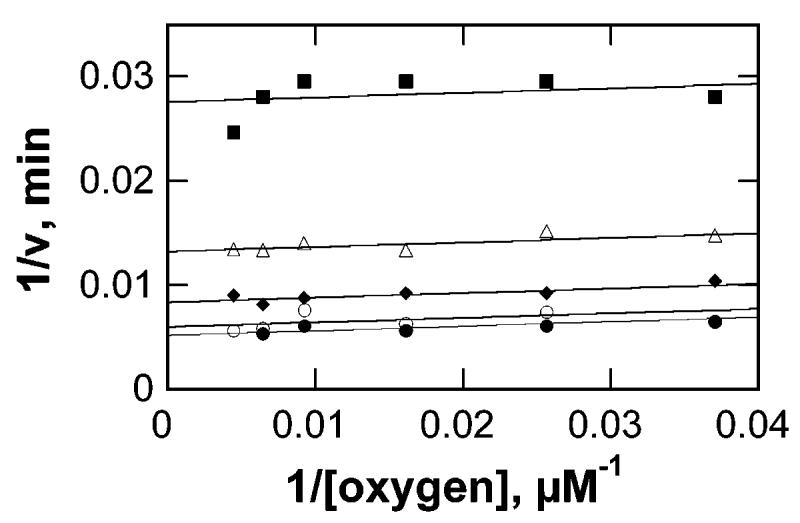
Initial rates of oxygen consumption by MTOX measured at various oxygen and sarcosine concentrations in 0.1 M ACES, 52 mM Tris, 52 mM ethanolamine-HCl, and 1 M KCl at pH 9.4 and 25 °C. The lines are from fits of the data to eq 2. The sarcosine concentrations were 18.5 mM (■), 46.3 mM (△), 92.6 mM (●), 185 mM (○), and 278 mM (●).
Table 2.
Kinetic Parameters for Sarcosine Oxidation by MTOX at pH 9.4a
| model (equation) | kcat (min−1) | KO2 (μM) | kcat/KO2 (min−1 μM−1) | Ksarc (mM) | kcat/Ksarc (min−1 μM−1) | Ki,sarc (mM) | χ 2 |
|---|---|---|---|---|---|---|---|
| sequential (1) | 251 ± 13 | 5 ± 3 | 50 ± 30 | 100 ± 14 | 2.5 ± 0.4 | 130 ± 160 | 1724 |
| ping-pong (2) | 262 ± 12 | 8 ± 2 | 33 ± 8 | 114 ± 10 | 2.3 ± 0.2 | NA | 1839 |
| rapid equilibrium (3) | 233 ± 8 | 16 ± 5 | 15 ± 5 | 83 ± 9 | 2.8 ± 0.8 | NA | 1964 |
0.1 M ACES, 52 mM Tris, 52 mM ethanolamine-HCl, and 1 M KCl at pH 9.4.
pH Effects
The kcat/Ksarc and kcat values were determined over the pH range 7.5–10.2, using a constant ionic strength buffer to minimize ionic strength effects. The kcat/Ksarc data were best fit by a bell-shaped profile as described by eq 4 (Figure 2A), with pKa values of 8.8 ± 0.1 and 10.0 ± 0.1. The pH dependence of the kcat value (Figure 2B) could be described by eq 5, with a pKa value of 9.1 ± 0.2 for a group, which must be unprotonated for catalysis.
Figure 2.
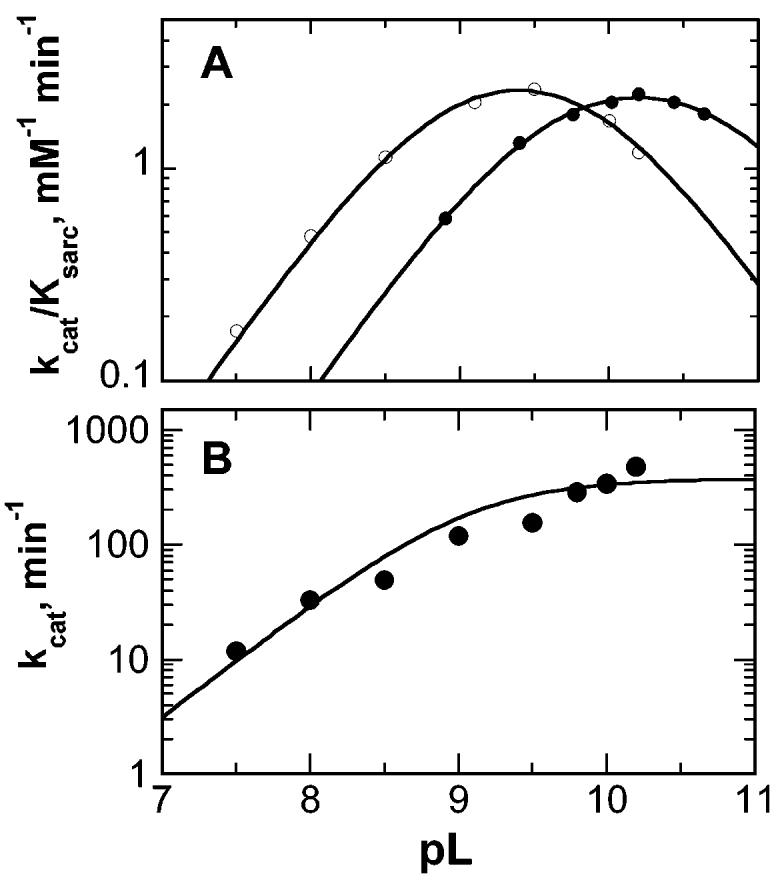
pH profiles with sarcosine as a substrate for MTOX. (A) kcat/Ksarc values in H2O (○) and D2O (●). The lines are from fits of the data to eq 4. (B) kcat pH profile. The line is from a fit of the data to eq 5. Conditions (A and B): 0.1 M ACES, 52 mM Tris, 52 mM ethanolamine-HCl, and 1 M KCl at 25 °C. The average error in the data for each profile is indicated by the size of the circles.
Isotope Effects
kcat/Ksarc values were also determined in D2O over the pD range 8.9–10.6. The kcat/Ksarc values again fit to a bell-shaped profile (eq 4) with the pKa values shifted to 9.5 ± 0.1 and 10.9 ± 0.1 (Figure 2A). A comparison of the pH independent kcat/Ksarc values calculated from the complete pL profiles using eq 4 gives a solvent isotope effect of 1.14 ± 0.10. Direct comparison of kcat/Ksarc values measured at the pH and pD optima (9.4 and 10.2, respectively) gave a solvent isotope effect of 1.04 ± 0.03. Because the high Km value for sarcosine above pH 9.5 limited the precision of the kcat value at high pH to ∼30%, the solvent isotope effect on the kcat value was not determined.
Deuterium kinetic isotope effects on steady-state kinetic parameters were measured using methyl-deuterated sarcosine (N-methyl-2H3-glycine). The isotope effect was measured at, above, and below the pH optimum of 9.4. In each case, the data fit well to eq 6, which assumes equal isotope effects on kcat and kcat/Ksarc. The magnitude of the isotope effect is pH-independent, with an average value of 6.9 ± 0.8 (Table 3).
Table 3.
Deuterium Kinetic Isotope Effects on Sarcosine Oxidation by MTOX
| pH | kinetic isotope effect | pH | kinetic isotope effect |
|---|---|---|---|
| 8.0a,b | 6.8 ± 0.6 | 8.0d,e | 7.4 ± 0.5 |
| 9.4a,c | 6.6 ± 0.4 | 9.4c,d | 7.7 ± 0.3 |
| 10.0a,c | 7.4 ± 0.4 |
From steady-state kinetic analyses.
0.25 M potassium phosphate.
0.1 M ACES, 52 mM Tris, 52 mM ethanolamine-HCl, and 1 M KCl.
From anaerobic stopped-flow analyses with 250 mM sarcosine.
0.1 M Tris and 0.5 M KCl.
Rapid Reaction Kinetics
The reduction of the MTOX flavin by sarcosine under anaerobic conditions was monitored using single wavelength stopped-flow spectroscopy at various sarcosine concentrations. In all cases, the absorbance change upon flavin reduction could be described by a single-exponential decay (eq 7) (Figure 3). The observed pseudo-first-order rate constants obtained at different concentrations of sarcosine were fit to eq 8 to give kred and Kd values of 220 ± 17 min−1 and 160 ± 40 mM, respectively, for the protiated substrate at pH 9.4. These values are in good agreement with the steady-state kcat and Ksarc values, consistent with the reductive half-reaction being rate-limiting under steady-state conditions. At all sarcosine concentrations, reduction yielded the same spectrum of the fully reduced enzyme, consistent with the reduction being effectively irreversible. This conclusion is also supported by the fit of the data to eq 8, which applies to an irreversible step following substrate binding (25).
Figure 3.
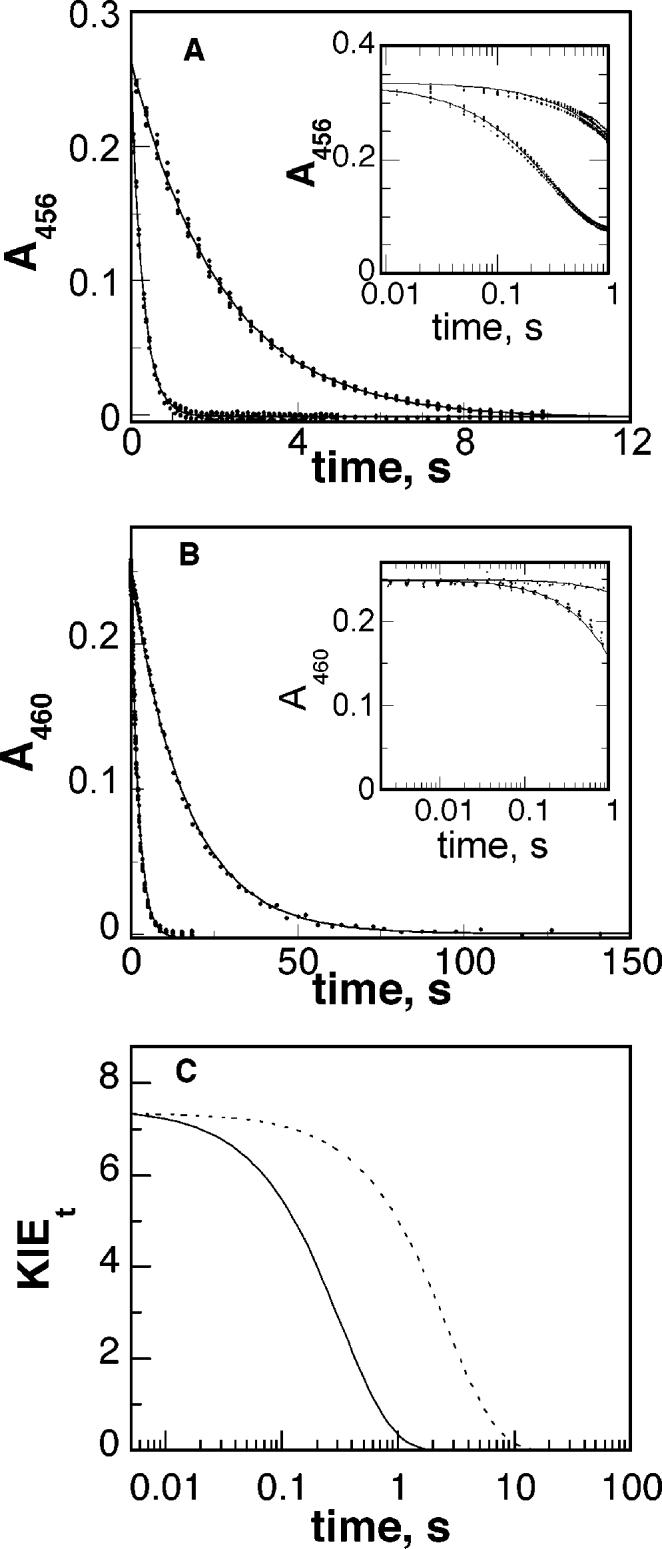
Representative time courses of flavin reduction by protiated and deuterated sarcosine at pH 9.4 (A) and 8.0 (B). Reactions contained 250 mM sarcosine (bottom curve in each panel) or deuterated sarcosine (top curve in each panel). The inset shows the first second on a logarithmic time scale. The lines are from the fits of the data to eq 7. Conditions were as described in Table 3. (C) Transient isotope effect for flavin reduction at pH 9.4 (⋯) and 8.0 (—).
The equal values for Dkcat and D(kcat/Ksarc) establish that the observed deuterium kinetic isotope effect is independent of the sarcosine concentration and therefore allows for the determination of deuterium kinetic isotope effect on the rate of flavin reduction (Dkred) using a single sarcosine concentration. The Dkred value was determined using 250 mM sarcosine at pH 8.0 and 9.4. Direct comparison of the rate constants for flavin reduction yielded isotope effects of 7.4 ± 0.5 and 7.7 ± 0.3, at pH 8.0 and 9.4, respectively. These values agree well with the kinetic isotope effects determined under steady-state conditions. Analysis of the transient isotope effects as described by Fisher (26, 27) is shown in Figure 3C. In both cases, the transient isotope effect starts at a value equal to the observed isotope effect and decreases exponentially over time. This is consistent with a single isotope-sensitive step.
Reduction of the enzyme by deuterated sarcosine was also followed with a photodiode array detector, allowing the entire flavin spectrum to be monitored simultaneously. No intermediates were seen between the spectra of the oxidized and the fully reduced enzyme, either at the pH optimum (data not shown) or at pH 8.0 (Figure 4A). The complete spectral changes could be modeled with a single-exponential decay with rate constants of 0.068 ± 0.001 and 0.43 ± 0.02 s−1 at pH 8.0 and 9.4, respectively. These numbers are in good agreement with the single wavelength data obtained under the same conditions (0.061 and 0.46 s−1). Figure 4B shows the spectra at zero and infinite time, which were extrapolated from the global analysis of flavin reduction at pH 8.0. Similar results were obtained at pH 9.4 (data not shown). The spectrum of the oxidized enzyme in the absence of the substrate is shown for comparison. The extrapolated spectra are those expected for fully oxidized and fully reduced flavins, consistent with a single-step, two-electron flavin reduction. The starting spectrum obtained from the global analysis is clearly different from that of the free enzyme. Differences in the spectra of the free enzyme and the extrapolated oxidized flavin spectra can be attributed to rapid substrate binding during the dead time of the stopped-flow instrument.2 The observed spectrum of the enzyme–substrate complex is similar to that seen upon binding d-α-aminobutyrate to d-amino acid oxidase at cryogenic temperatures (28) and to the spectrum of MTOX with N,N-dimethylglycine bound (29).
Figure 4.
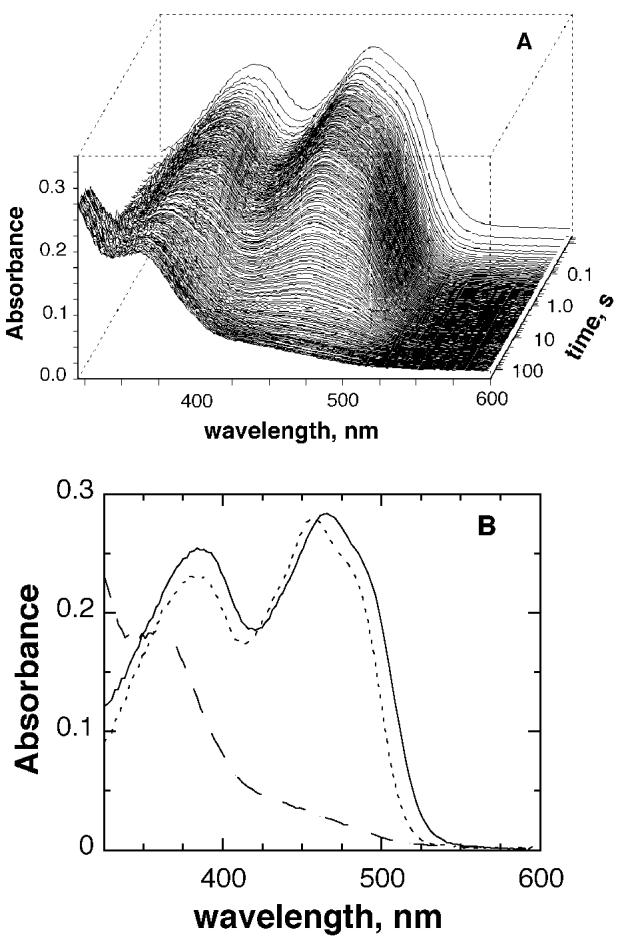
(A) Changes in the absorbance spectrum of the enzyme-bound flavin during anaerobic reduction of MTOX by deuterated sarcosine at pH 8.0. The conditions were as described for Figure 3. (B) Extrapolated starting (—) and final (- - -) spectra, obtained by global analysis of the spectra using a single-step kinetic model, and the spectrum of the free enzyme (⋯).
Scheme 5.
DISCUSSION
The mechanism of substrate oxidation by flavin-dependent amine oxidases is still widely debated. The chemical mechanisms proposed for methyltryptophan oxidase and sarcosine oxidase are shown in Schemes 2-4. Although the mechanisms shown in Schemes 2 and 4 involve the formation of a flavin intermediate between the fully oxidized and fully reduced flavin species, no spectroscopic evidence of such intermediates has been reported. However, formation of the postulated intermediates may occur during a rate-limiting step, preventing the accumulation of detectable intermediate concentrations. The slow substrate sarcosine was chosen for the study with the expectation that chemical steps rather than substrate binding or product release would be rate-limiting, allowing intrinsic isotope effects and pKa values to be determined. All of the kinetic data presented here are consistent with cleavage of the sarcosine carbon–hydrogen bond being rate-limiting, in line with this prediction.
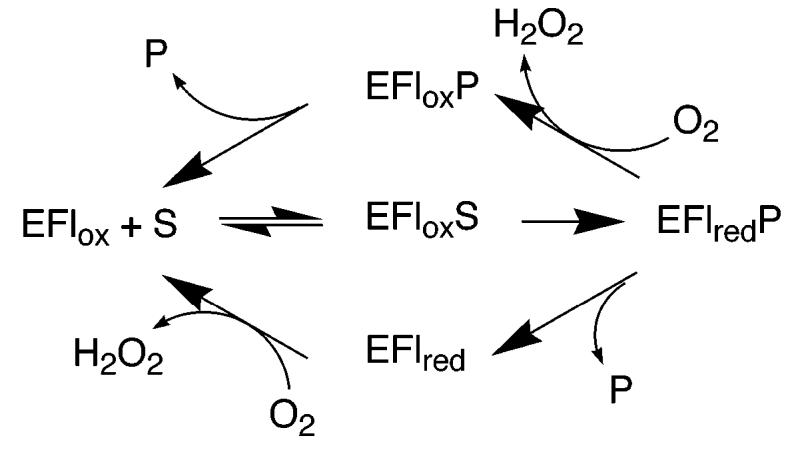
With virtually all flavoprotein oxidases studied to date, the steady-state kinetic mechanism can be described by a variation of the mechanism of Scheme 5 (30). Here, the enzyme-containing oxidized flavin (EFlox) binds and oxidizes the substrate (S), producing the reduced enzyme–product complex (EFlredP); this species reacts with molecular oxygen or an alternative electron acceptor in the case of the related dehydrogenases. Whether the oxidized product (P) dissociates before or after reoxidation of the flavin varies with the specific enzyme and the substrate. This mechanism results in steady-state kinetic patterns, which fit to either the equation for a ping-pong mechanism or that for a sequential mechanism; the reversibility of the flavin reduction determines whether the rate equation contains the extra term in Kia, which distinguishes ping-pong and sequential mechanisms (31). Khanna and Jorns (22) previously reported that the initial rate data for N-methyltryptophan turnover by MTOX are consistent with the upper loop of Scheme 5. In contrast, the data with sarcosine as the substrate were fit to a rapid equilibrium kinetic model with oxygen binding as the first substrate. This was the first report of oxygen binding by an oxidized flavoprotein, although H269Q3 MSOX was subsequently reported to have a similar kinetic pattern, in contrast to the wild-type enzyme (1). However, as shown here (Figure 3), MTOX is readily reduced by sarcosine in the absence of oxygen, and the kinetic parameters for that reduction establish the free oxidized enzyme as kinetically competent for reduction; this rules out any mechanism in which oxygen must bind before sarcosine. The initial rate data described here for sarcosine turnover are fit better by the equations for ping-pong and sequential kinetics, in line with the common kinetic mechanisms for flavoprotein oxidases of Scheme 5, than by the equation for a rapid equilibrium mechanism with oxygen binding first. The reason for the discrepancy with the earlier data is not clear, but the data of Figure 3 show that the kcat/KO2 value is effectively independent of the sarcosine at accessible concentrations of oxygen. For an intersecting line pattern to be seen in double-reciprocal plots for flavoprotein oxidases requires that the reduction step must be significantly reversible, so that there are reversible steps connecting the binding of the oxidized substrate and oxygen (31). The rapid reaction kinetics described here are most consistent with this reduction being effectively irreversible, consistent with eq 2 being appropriate. Most critically for the present work, whether the data are fit to eq 1 or 2, the KO2 value is sufficiently low that kcat and kcat/Ksarc values can be treated as independent of oxygen concentrations under atmospheric oxygen concentrations.
With sarcosine as the substrate for MTOX, the reductive half-reaction is clearly rate-limiting. The rate constant for flavin reduction at saturating sarcosine concentrations and the Kd value for sarcosine determined from the analysis of the reduction kinetics match the kcat and Ksarc values from steady-state kinetics. The agreement between the Kd value and the Km value for sarcosine is also consistent with a lack of an external forward commitment to catalysis for this substrate.4 This conclusion is supported by the pH independence of the deuterium isotope effect on the kcat/Ksarc value (32). The identity of Dkcat/Ksarc, Dkcat, and the isotope effect measured from the reduction kinetics is consistent with a lack of any significant internal or reverse commitment, so that the average isotope effect of 7.2 obtained from both steady-state and rapid reaction analyses is likely to be the intrinsic deuterium isotope effect. The magnitude of this value is consistent with the expectation for the intrinsic isotope effect upon cleavage of a carbon–hydrogen bond in a trideuterated methyl group. The value of 7.2 agrees well with the deuterium isotope effect of 7.3 for the heterotetrameric sarcosine oxidase from Arthrobacter sp. (2), suggesting that the extent of carbon–hydrogen bond cleavage in the transition state is similar for the two enzymes, even though sarcosine is a slow substrate for MTOX.
The bell-shaped kcat/Ksarc pH profile is consistent with two groups in the free enzyme or substrate whose protonation state is critical for productive binding of substrates, with pKa values of 8.8 ± 0.1 and approximately 10. While the inability to measure initial rates at high pH because enzyme instability greatly limits the precision of the upper pKa value, a pKa value of 10 would match the pKa value of the substrate amino group (33), suggesting that the zwitterionic form of the substrate is preferred and that the lower pKa is due to a group on the enzyme which must be unprotonated. This interpretation is consistent with the MTOX kcat profile, which shows only the lower pKa value for a group, which must be unprotonated, suggesting that the acidic limbs of both profiles reflect the same event, the deprotonation of the substrate amino group in the enzyme–substrate complex. Although pH studies have not been reported with sarcosine as a substrate for MSOX, Zhao and Jorns (20) reported that the limiting rate of flavin reduction by proline decreases below a pKa of 8.0, while the Kd value for proline is pH-independent over the pH range 6.5–9.0, similarly suggesting that MSOX binds zwitterionic proline and deprotonates it prior to flavin reduction.
Oxidation of the carbon–nitrogen bond in zwitterionic sarcosine by MTOX necessarily requires the loss of a hydrogen from the substrate nitrogen. The lack of a solvent isotope effect on the kcat/Ksarc value limits the possibilities for the timing of the cleavage of this nitrogen–hydrogen bond. If MTOX prefers the zwitterionic form of sarcosine, as suggested by the above pH profiles, deprotonation of the amino group must occur after the substrate binds. If this deprotonation occurred in the same step as carbon–hydrogen bond cleavage, there would be a significant solvent isotope effect, because carbon–hydrogen bond cleavage is rate-limiting. Because there is no significant solvent isotope effect on the kcat/Ksarc value, deprotonation of the enzyme-bound sarcosine most likely occurs in a rapid and reversible step preceding carbon–hydrogen bond cleavage5. Such a rapid deprotonation of the heteroatom of the enzyme-bound substrate prior to flavin reduction has recently been demonstrated for both flavocytochrome b2 (34) and choline oxidase (35). In both enzymes, an active-site histidine (36, 37) removes the hydroxyl proton to form the alkoxide prior to carbon–hydrogen bond cleavage. In the crystal structure of MSOX with N,N-dimethylglycine bound, the only residue within hydrogen-bonding distance (2.8 Å) of the amino nitrogen of the inhibitor is the carbonyl oxygen of Gly344 (4); formation of a hydrogen bond in this case would require that the nitrogen be protonated, consistent with the conclusion drawn from the binding affinities. The nearest residue capable of acting as a base in accepting a proton from the nitrogen is His270, the N1 of which is 4.3 Å away; this residue is conserved in MTOX (His263) (11). Mutation of His270 to asparagine results in a decrease in catalytic activity but does not significantly change the observed pKa value of 8, suggesting that it is not the residue responsible for this pKa in the pH profile, although it still may be the active-site base (1). An alternative possibility for this pKa is the flavin itself, for which a pKa value of 8.4 was determined from the effect of pH on the flavin spectrum at 4 °C (29).
The changes in the flavin spectrum, which occur upon reduction of MTOX by sarcosine, are fully consistent with a minimal kinetic model in which binding of the amino acid is very rapid and is followed by a single step in which a hydride equivalent is transferred to the flavin. While these results do not rule out additional intermediates, they place limits on the extent to which they accumulate and thus on their energetics. The kinetic isotope effects establish that carbon–hydrogen bond cleavage occurs in the rate-limiting transition state for flavin reduction. This rules out rate-limiting formation of an intermediate in which the carbon–hydrogen bond is intact. Furthermore, for an intermediate between the oxidized enzyme–substrate complex and the reduced enzyme, the rate constant for productive decay necessarily equals the rate constant for flavin reduction. In the case of deuterated sarcosine, in which cleavage of the carbon–hydrogen bond has been slowed by 7-fold, the large decrease in the rate constant for reduction should result in a comparable increase in the accumulation of any such intermediate. If one assumes that any intermediate that accumulates to 35% would be detected, the lack of such a detectable intermediate with the deuterated substrate places an upper limit of 5% on the maximum amount of the intermediate, which builds up prior to carbon–hydrogen bond cleavage. The present data do not rule out a mechanism in which an intermediate is formed at lower concentrations than this because it is at a free energy well above that of the enzyme–substrate complex. Still, the lack of detectable intermediates is most simply explained by a lack of intermediates, as is the case for a hydride transfer mechanism.6
This conclusion is supported by structural data. Although structural information is not available for MTOX, a crystal structure is available for MSOX (4). In several crystal structures with product analogues bound, the carbon corresponding to the reactive carbon of the substrate is within 4 Å of the flavin N5, an arrangement which is common in flavoproteins catalyzing dehydrogenation reactions (38). This distance is compatible with transfer of the methyl hydrogen to the flavin N5, as in a hydride transfer mechanism.
The anionic flavin semiquinone and flavin adducts proposed as intermediates in Schemes 2 and 4, respectively, should be readily detectable if they accumulate significantly. The spectrum of the flavin semiquinone in MTOX has a visible absorbance maximum at 393 nm with an ε393 of 28 800 M−1 cm−1 (29), compared to the maximum of the free oxidized enzyme at 456 nm of 13 300 M−1 cm−1 (Figure 4) (12), and would be readily distinguished. Flavin N(5) and C(4a) adducts have spectra that resemble that of fully reduced flavin, with an additional shoulder or peak at 300–400 nm (39). While such an intermediate might be difficult to distinguish from a reduced enzyme–product complex in an analysis such as that shown in Figure 4A, its presence would result in a decrease in the apparent absorbance of the enzyme–substrate complex; this is ruled out by comparison of the spectra of the free oxidized enzyme and of the intermediate formed upon binding of sarcosine (Figure 4B).7
Some evidence for radical mechanisms for MTOX and related enzymes has come from studies of MSOX with substrate analogues. N-Cyclopropylglycine will slowly inactivate MSOX; the reaction simultaneously forms a species with a partially bleached visible absorbance spectrum, which very slowly decomposes to fully reduced enzyme (21). While the structure of the modified cofactor was not determined, the inactivation was attributed to one-electron oxidation of the inhibitor to the cyclopropyl radical, which then reacted with the anionic flavin semiquinone. However, cyclopropyl compounds have been found to inactivate other flavoprotein oxidases by mechanisms that are not analogous to oxidation of more normal substrates. Thus, a number of inhibitors, which would be expected to utilize radical intermediates for inactivation, have been described for monoamine oxidase (40). Recent elucidation of the three-dimensional structure of monoamine oxidase B shows that the amino acid residue, which is labeled by some of these inhibitors, is well outside the active site (16), suggesting that inactivation and normal catalysis utilize different mechanisms. Methanol oxidase (41) and cholesterol oxidase (42), two members of a family of homologous flavoprotein alcohol oxidases (43), are both inactivated by cyclopropyl compounds; in this case, the available evidence is most consistent with hydride transfer mechanisms for these enzymes (35, 44, 45).
In summary, the present results are most simply explained by a hydride transfer mechanism for MTOX, in that no intermediates are detected prior to rate-limiting carbon–hydrogen bond cleavage. The structural similarities among the other known and predicted members of the MTOX/MSOX family suggest that these enzymes also utilize a similar mechanism. These results add to the growing body of evidence for hydride transfer mechanisms in flavoenzymes that oxidize amines, alcohols, α-amino acids, and α-hydroxy acids (for a recent review, see ref 46).
Footnotes
This work was supported in part by Grants to P.F.F. from the NIH (R01 GM58698) and The Welch Foundation (A-1245) and by NIH Grant T32 GM08523 (to E.C.R.).
Abbreviations: MTOX, N-methyltryptophan oxidase; MSOX, monomeric sarcosine oxidase; LB, Luria-Bertani broth.
The increase in absorbance intensity in the 350 nm region is not seen in fresh enzyme samples and is therefore attributed to slight enzyme denaturation after several hours at 25 °C.
H270 was previously described as H269 (1, 2) based on the crystal structure of MSOX (3), which is missing the initial methionine residue.
The external commitment is defined as the ratio of the rate constant for cleavage of the CH bond to the rate constant for substrate dissociation from the enzyme–substrate complex. A significant commitment can yield a Km value significantly different from the Kd value, perturbed pKa values, and isotope effects less than the intrinsic values.
A simpler explanation for the lack of a solvent isotope effect is that the enzyme binds the anionic form of the substrate, so that the only effect of D2O is on the concentration of the active form of the substrate, which is present at a given pH; this would result in a shift in the pH optimum but no decrease in the activity at the pH optimum, as is indeed the case. However, such a model is difficult to rationalize with the kcat profile and the published data with proline.
The present data do not rule out a carbanion mechanism in which transfer of electrons from the carbanion to the flavin is much faster than carbon–hydrogen bond cleavage. However, because d-amino acid oxidase, which is homologous to MSOX and MTOX, is generally accepted not to involve a carbanion intermediate and the pKa of the methyl protons of sarcosine would be expected to be significantly higher than the α proton of an amino acid, a carbanion mechanism would seem the least likely possibility.
An alternative possibility that is not generally proposed for this family of enzymes is hydrogen atom abstraction from the substrate methyl group. While a hydrogen atom abstraction would be much more energetically unfavorable than electron transfer, it would be isotope-sensitive. This would be expected to form the neutral flavin radical; however, the pKa for the neutral flavin radical in MSOX has been estimated to be below pH 6 (4), compared to the pH optimum above 9. Moreover, neutral flavin radicals have reduced absorbance around 460 nm compared to oxidized flavin and broad absorbance bands from around 500 to 650 nm with extinction coefficients of 3000 to 10 000 M−1 cm−1 (5). No absorbance changes are seen in this longer wavelength region upon MTOX flavin reduction with sarcosine. Still, because radical formation would necessarily occur in the rate-limiting step, the flavin radical would decay much more rapidly than it would form, making its detection difficult. Such a slow formation of an unstable neutral radical followed by a rapid electron transfer is effectively indistinguishable from a hydride transfer.
REFERENCES
- 1.Zhao G, Song H, Chen Z, Mathews S, Jorns MS. Monomeric sarcosine oxidase: Role of histidine 269 in catalysis. Biochemistry. 2002;41:9751–9764. doi: 10.1021/bi020286f. [DOI] [PubMed] [Google Scholar]
- 2.Harris RJ, Meskys R, Sutcliffe MJ, Scrutton NS. Kinetic studies of the mechanism of carbon–hydrogen bond breakage by the heterotetrameric sarcosine oxidase of Arthrobacter sp. 1–IN. Biochemistry. 2000;39:1189–1198. doi: 10.1021/bi991941v. [DOI] [PubMed] [Google Scholar]
- 3.Trickey P, Wagner MA, Jorns MS, Mathews FS. Monomeric sarcosine oxidase: Structure of a covalently flavinylated amine oxidizing enzyme. Structure. 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- 4.Wagner MA, Trickey P, Chen Z.-w., Mathews FS, Jorns MS. Monomeric sarcosine oxidase: 1. Flavin reactivity and active site binding determinants. Biochemistry. 2000;39:8813–8824. doi: 10.1021/bi000349z. [DOI] [PubMed] [Google Scholar]
- 5.Massey V, Palmer G. On the existence of spectrally distinct classes of flavoprotein semiquinones. A new method for the quantitative production of flavoprotein semiquinones. Biochemistry. 1966;5:3181–3189. doi: 10.1021/bi00874a016. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida T, Fukuyama S. Metabolic changes in nitrogen-substituted amino acids. VIII. Demethylase. 2. Distribution. J. Biochem. 1944;36:349–353. [Google Scholar]
- 7.Moritani M. Demethylase. IV. Kinetics and reaction mechanism. Hukuoka Acta. Med. 1952;43:651–658. [Google Scholar]
- 8.Magaki I. Abrine demethylase of Escherichia coli. Osaka Daigaku Igaku Zasshi. 1955;7:359–367. [Google Scholar]
- 9.Wang S-P. Demethylase of bacteria. Taiwan Yixuehui Zazhi. 1957;56:362–369. [Google Scholar]
- 10.Ting S-M, Tung T. Some properties of the particulate and soluble oxidative de-methylases from rabbit kidney. Taiwan Yixuehui Zazhi. 1966;65:169–177. [Google Scholar]
- 11.Koyama Y, Ohmori H. Nucleotide sequence of the Escherichia coli solA gene encoding a sarcosine oxidase-like protein and characterization of its product. Gene. 1996;181:179–183. doi: 10.1016/s0378-1119(96)00500-8. [DOI] [PubMed] [Google Scholar]
- 12.Wagner MA, Khanna P, Jorns MS. Structure of the flavocoenzyme of two homologous amine oxidases: Monomeric sarcosine oxidase and N-methyltryptophan oxidase. Biochemistry. 1999;38:5588–5595. doi: 10.1021/bi982955o. [DOI] [PubMed] [Google Scholar]
- 13.Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B. Crystal structure of D-amino acid oxidase: A case of active site mirror-image convergent evolution with flavocytochrome b2. Proc. Natl. Acad. Sci. U.S.A. 1996;93:7496–7501. doi: 10.1073/pnas.93.15.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Settembre EC, Dorrestein PC, Park J, Augustine AM, Begley TP, Ealick SE. Structural and mechanistic studies on thiO, a glycine oxidase essential for thiamin biosynthesis in Bacillus subtilis. Biochemistry. 2003;42:2971–2981. doi: 10.1021/bi026916v. [DOI] [PubMed] [Google Scholar]
- 15.Naoi M, Yagi K. Oxidation of sarcosine and N-alkyl derivatives of glycine by d-amino acid oxidase. Biochim. Biophys. Acta. 1976;438:61–70. doi: 10.1016/0005-2744(76)90223-0. [DOI] [PubMed] [Google Scholar]
- 16.Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- 17.Binda C, Coda A, Angelini R, Federico R, Ascenzi P, Mattevi A. A 30 Å long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure. 1999;7:265–276. doi: 10.1016/s0969-2126(99)80037-9. [DOI] [PubMed] [Google Scholar]
- 18.Pawelek PD, Cheah J, Coulombe R, Macheroux P, Ghisla S, Vrielink A. The structure of l-amino acid oxidase reveals the substrate trajectory into an enantiomerically conserved active site. EMBO J. 2000;19:4204–4215. doi: 10.1093/emboj/19.16.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. Nitrogen isotope effects as probes of the mechanism of d-amino acid oxidase. J. Am. Chem. Soc. 2000;122:12896–12897. [Google Scholar]
- 20.Zhao G, Jorns MS. Monomeric sarcosine oxidase: Evidence for an ionizable group in the E•S complex. Biochemistry. 2002;41:9747–9750. doi: 10.1021/bi020285n. [DOI] [PubMed] [Google Scholar]
- 21.Zhao G, Qu J, Davis FA, Jorns MS. Inactivation of monomeric sarcosine oxidase by reaction with N-(cyclopropyl)-glycine. Biochemistry. 2000;39:14341–14347. doi: 10.1021/bi001421w. [DOI] [PubMed] [Google Scholar]
- 22.Khanna P, Schuman Jorns M. N-Methyltryptophan oxidase from Escherichia coli: Reaction kinetics with N-methyl amino acid and carbinolamine substrates. Biochemistry. 2001;40:1451–1459. doi: 10.1021/bi002442t. [DOI] [PubMed] [Google Scholar]
- 23.Wagner MA, Jorns MS. Monomeric sarcosine oxidase: 2. Kinetic studies with sarcosine, alternate substrates, and a substrate analogue. Biochemistry. 2000;39:8825–8829. doi: 10.1021/bi000350y. [DOI] [PubMed] [Google Scholar]
- 24.Cleland WW. Enzyme kinetics as a tool for determination of enzyme mechanisms. In: Bernasconi CF, editor. Investigation of Rates and Mechanism. Part I. 4th ed. Vol. 6. John Wiley and Sons; New York: 1986. pp. 791–870. [Google Scholar]
- 25.Strickland S, Palmer G, Massey V. Determination of dissociation constants and specific rate constants of enzyme–substrate (or protein–ligand) interactions from rapid reaction kinetic data. J. Biol. Chem. 1975;250:4048–4052. [PubMed] [Google Scholar]
- 26.Fisher HF, Saha SK. Interpretation of transient-state kinetic isotope effects. Biochemistry. 1996;35:83–88. doi: 10.1021/bi9509107. [DOI] [PubMed] [Google Scholar]
- 27.Maniscalco SJ, Tally JF, Fisher HF. The interpretation of multiple-step transient-state kinetic isotope effects. Arch. Biochem. Biophys. 2004;425:165–172. doi: 10.1016/j.abb.2004.02.041. [DOI] [PubMed] [Google Scholar]
- 28.Yagi K, Lange R, Douzou P. Spectroscopic demonstration of an initial stage of the complex of d-amino acid oxidase and its substrate d-α-aminobutyric acid. Biochem. Biophys. Res. Commun. 1980;97:370–374. doi: 10.1016/0006-291x(80)90274-0. [DOI] [PubMed] [Google Scholar]
- 29.Khanna P, Schuman Jorns M. Characterization of the FAD-containing N-methyltryptophan oxidase from Escherichia coli. Biochemistry. 2001;40:1441–1450. doi: 10.1021/bi0024411. [DOI] [PubMed] [Google Scholar]
- 30.Bright HJ, Porter DJT. Flavoprotein oxidases. In: Boyer P, editor. The Enzymes. 3rd ed. Vol. 12. Academic Press; New York: 1975. pp. 421–505. [Google Scholar]
- 31.Palmer G, Massey V. Mechanisms of flavoprotein catalysis. In: Singer TP, editor. Biological Oxidation. John Wiley and Sons; New York: 1968. pp. 263–300. [Google Scholar]
- 32.Cook PF, Cleland WW. pH variation of isotope effects in enzyme-catalyzed reactions. 1. Isotope- and pH-dependent steps the same. Biochemistry. 1981;20:1797–1805. doi: 10.1021/bi00510a014. [DOI] [PubMed] [Google Scholar]
- 33.Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for Biochemical Research. 3rd ed. Clarendon Press; Oxford, U.K.: 1986. [Google Scholar]
- 34.Sobrado P, Fitzpatrick PF. Solvent and primary deuterium isotope effects show that lactate CH and OH bond cleavages are concerted in Y254F flavocytochrome b2, consistent with a hydride transfer mechanism. Biochemistry. 2003;42:15208–15214. doi: 10.1021/bi035546n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan F, Gadda G. On the catalytic mechanism of choline oxidase. J. Am. Chem. Soc. 2005 doi: 10.1021/ja044541q. in press. [DOI] [PubMed] [Google Scholar]
- 36.Gaume B, Sharp RE, Manson FDC, Chapman SK, Reid GA, Lederer F. Mutation to glutamine of histidine 373, the catalytic base of flavocytochrome b2 (l-lactate dehydrogenase) Biochimie. 1995;77:621–630. doi: 10.1016/0300-9084(96)88177-6. [DOI] [PubMed] [Google Scholar]
- 37.Ghanem M, Gadda G. On the catalytic role of the conserved active site residue His466 of choline oxidase. Biochemistry. 2005 doi: 10.1021/bi048056j. in press. [DOI] [PubMed] [Google Scholar]
- 38.Fraaije MW, Mattevi A. Flavoenzymes: Diverse catalysts with recurrent features. Trends Biochem. Sci. 2000;25:126–132. doi: 10.1016/s0968-0004(99)01533-9. [DOI] [PubMed] [Google Scholar]
- 39.Ghisla S, Massey V, Choong YS. Covalent adducts of lactate oxidase. Photochemical formation and structure identification. J. Biol. Chem. 1979;254:10662–10669. [PubMed] [Google Scholar]
- 40.Silverman RB. Radical ideas about monoamine oxidase. Acc. Chem. Res. 1995;28:335–342. [Google Scholar]
- 41.Sherry B, Abeles RH. Mechanism of action of methanol oxidase, reconstitution of methanol oxidase with 5-deazaflavin, and inactivation of methanol oxidase by cyclopropanol. Biochemistry. 1985;24:2594–2605. doi: 10.1021/bi00332a002. [DOI] [PubMed] [Google Scholar]
- 42.McCann AE, Sampson NS. A C6–flavin adduct is the major product of irreversible inactivation of cholesterol oxidase by 2α,3α-cyclopropano-5α-cholestan-3β-ol. J. Am. Chem. Soc. 2000;122:35–39. [Google Scholar]
- 43.Cavener DR. GMC oxidoreductases a newly defined family of homologous proteins with diverse catalytic activities. J. Mol. Biol. 1992;223:811–814. doi: 10.1016/0022-2836(92)90992-s. [DOI] [PubMed] [Google Scholar]
- 44.Menon V, Hsieh C-T, Fitzpatrick PF. Substituted alcohols as mechanistic probes of alcohol oxidase. Bioorg. Chem. 1995;23:42–53. [Google Scholar]
- 45.Lario PI, Sampson N, Vrielink A. Sub-atomic resolution crystal structure of cholesterol oxidase: What atomic resolution crystallography reveals about enzyme mechanism and the role of the FAD cofactor in redox activity. J. Mol. Bio. 2003;326:1635–1650. doi: 10.1016/s0022-2836(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 46.Fitzpatrick PF. Carbanion versus hydride transfer mechanisms in flavoprotein–catalyzed dehydrogenations. Bioorg. Chem. 2004;32:125–139. doi: 10.1016/j.bioorg.2003.02.001. [DOI] [PubMed] [Google Scholar]