

Abstract
In smooth muscle cells, localized intracellular Ca2+ transients, termed “Ca2+ sparks,” activate several large-conductance Ca2+-activated K+ (KCa) channels, resulting in a transient KCa current. In some smooth muscle cell types, a significant proportion of Ca2+ sparks do not activate KCa channels. The goal of this study was to explore mechanisms that underlie fractional Ca2+ spark-KCa channel coupling. We investigated whether membrane depolarization or ryanodine-sensitive Ca2+ release (RyR) channel activation modulates coupling in newborn (1- to 3-day-old) porcine cerebral artery myocytes. At steady membrane potentials of −40, 0, and +40 mV, mean transient KCa current frequency was ∼0.18, 0.43, and 0.26 Hz and KCa channel activity [number of KCa channels activated by Ca2+ sparks × open probability of KCa channels at peak of Ca2+ sparks (NPo)] at the transient KCa current peak was ∼4, 12, and 24, respectively. Depolarization between −40 and +40 mV increased KCa channel sensitivity to Ca2+ sparks and elevated the percentage of Ca2+ sparks that activated a transient KCa current from 59 to 86%. In a Ca2+-free bath solution or in diltiazem, a voltage-dependent Ca2+ channel blocker, steady membrane depolarization between −40 and +40 mV increased transient KCa current frequency up to ∼1.6-fold. In contrast, caffeine (10 μM), an RyR channel activator, increased mean transient KCa current frequency but did not alter Ca2+ spark-KCa channel coupling. These data indicate that coupling is increased by mechanisms that elevate KCa channel sensitivity to Ca2+ sparks, but not by RyR channel activation. Overall, KCa channel insensitivity to Ca2+ sparks is a prominent factor underlying fractional Ca2+ spark uncoupling in newborn cerebral artery myocytes.
Keywords: ryanodine-sensitive calcium release channel, calcium-activated potassium channel, membrane potential
Arterial smooth muscle cell contractility is differentially regulated by local and global elevations in intracellular Ca2+ concentration ([Ca2+]i) (16). Global nanomolar [Ca2+]i elevations, caused by Ca2+ influx from the extracellular space and release from intracellular stores, stimulate contraction via the activation of Ca2+/calmodulin-dependent myosin light chain kinase (8, 16). In contrast, localized micromolar [Ca2+]i transients, termed “Ca2+ sparks,” oppose contraction (16, 21).
Ca2+ sparks are induced by activation of several ryanodine-sensitive Ca2+ release (RyR) channels on the sarcoplasmic reticulum (SR) (5, 16). In smooth muscle cells, a Ca2+ spark can activate multiple large-conductance Ca2+-activated K+ (KCa) channels, resulting in a transient KCa current (3, 16, 21). In the arterial wall, transient KCa currents induce membrane hyperpolarization, which reduces voltage-dependent Ca2+ channel activity and, thus, global [Ca2+]i and contractility (21). The differential regulation of arterial smooth muscle contractility by local and global Ca2+ signals exemplifies how a single signaling element can control opposing cellular functions in the same cell.
Several important features facilitate differential regulation of arterial smooth muscle contractility by local and global [Ca2+]i elevations, including the spatial and temporal nature of the Ca2+ signals and the proximity and Ca2+ sensitivity of downstream target proteins (16). One important feature of Ca2+ sparks that allows specificity of signaling is that these events do not contribute significantly to global [Ca2+]i because of their rapid and localized properties (16, 21). Another important aspect is that KCa channels are sensitive to micromolar [Ca2+]i elevations, such as those generated by Ca2+ sparks (22, 33). As such, KCa channels are relatively insensitive to global nano-molar [Ca2+]i elevations that signal contraction (22, 33).
In adult rat cerebral artery smooth muscle cells, the effective coupling of Ca2+ sparks to KCa channels is strong, and at a physiological membrane potential of −40 mV, essentially all Ca2+ sparks activate a transient KCa current (6, 23). However, in adult human and newborn porcine cerebral arterial, adult feline esophageal, and Bufo marinus stomach smooth muscle cells, the effective coupling of Ca2+ sparks to KCa channels is considerably weaker (14, 18, 27, 32). In these smooth cell types, a significant proportion of Ca2+ sparks do not activate a transient KCa current (∼20–40%), and the amplitude correlation between these events is less robust than in rat cerebral artery smooth muscle cells (14, 18, 27, 32). However, underlying causes of weak coupling and mechanisms that enhance coupling in these smooth muscle cell types are unclear.
The goal of this study was to investigate mechanisms that underlie fractional Ca2+ spark-KCa channel coupling in smooth muscle cells. Ca2+ spark-KCa channel coupling was studied in newborn porcine cerebral artery smooth muscle cells, which exhibit a weak coupling phenotype similar to that observed in other smooth muscle cell types, including human cerebral artery smooth muscle cells (27). We investigated whether an increase in KCa channel Ca2+ sensitivity or RyR channel activation enhances coupling. Data suggest that coupling is determined primarily by KCa channel sensitivity to Ca2+ sparks and indicate that RyR channel activation alone does not influence coupling.
MATERIALS AND METHODS
Tissue preparation
All procedures used were approved by the University of Tennessee Animal Care and Use Committee. Newborn pigs (1–3 days old, 1–2.5 kg body wt) were anesthetized with ketamine hydrochloride (33 mg/kg im) and acepromazine (3.3 mg/kg im). The brain was removed and maintained in ice-cold HEPES-buffered physiological saline solution (PSS) containing (in mM) 134 NaCl, 6 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, with pH adjusted to 7.4 with NaOH. Isolated arteries (50–200 μm) were dissected from the brain and cleaned to remove basolateral connective tissue. Individual smooth muscle cells were dissociated from cerebral arteries by a procedure described previously (13).
Confocal Ca2+ imaging
Arterial smooth muscle cells were placed in HEPES-buffered PSS containing 10 μM fluo 4-AM for 20 min at room temperature. The cells were then washed with HEPES-buffered PSS for 30 min to allow indicator deesterification. Fluo 4 was imaged using a laser scanning confocal microscope (Oz, Noran Instruments, Middleton, WI) and a ×60 water immersion objective (1.2 NA) attached to a microscope (model TE300, Nikon). Fluo 4 was illuminated at 488 nm with use of a krypton-argon laser, and emitted light >500 nm was captured. Images (56.3 × 52.8 μm) were recorded every 8.3 ms (i.e., 120 images per second). When a slit width of 100 μm was used, the z resolution (full width at half-maximal amplitude) of the imaging system was 7 μm, as determined by subresolution (100-nm-diameter) fluorescent beads. Electrophysiological and fluorescence measurements were synchronized using a light-emitting diode placed above the recording chamber that was triggered during acquisition. Each isolated smooth muscle cell was imaged for 10 s under each condition. Custom analysis software (kindly provided by Dr. M. T. Nelson, University of Vermont) was used to detect Ca2+ sparks in smooth muscle cells. For detection of Ca2+ sparks, an area 1.54 × 1.54 μm (7 × 7 pixels, i.e., 2.37 μm2) in each image (F) was divided by a baseline (F0) that was determined by averaging 10 images without Ca2+ spark activity. The entire image area was analyzed to detect Ca2+ sparks. A Ca2+ spark was identified as a local increase in F/F0 that was >1.2. Mean Ca2+ spark frequency and standard error of the mean under each condition were calculated by averaging individual cellular frequencies. Spatial spread of the Ca2+ spark was calculated at half-maximal amplitude. Changes in local or global [Ca2+]i were calculated using the pseudoratio method (5)
where K is the apparent affinity of fluo 4 for Ca2+ [770 nM (28)], R is the fractional fluorescence increase (F/F0), and [Ca2+]rest is [Ca2+]i at F0. Global Ca2+ fluorescence was calculated from the same images used for Ca2+ spark analysis and was the mean pixel value of 100 different images acquired over 10 s. Global [Ca2+]i at 0 and +40 mV were calculated from the cellular change in F/F0 from −40 mV (determined with fura 2; see Intracellular Ca2+ measurements using fura 2).
Patch-clamp electrophysiology
Isolated cells were allowed to adhere to a glass coverslip in the bottom of a chamber for 10 min before experimentation. K+ currents were measured using the perforated-patch configuration of the patch-clamp technique with an Axo-patch 200B amplifier (Axon Instruments, Union City, CA). The bath solution was HEPES-buffered PSS. Where appropriate, Ca2+-free bath solution was prepared by substitution of equimolar CaCl2 with NaCl and addition of 1 mM EGTA. The pipette solution contained (in mM) 110 potassium aspartate, 30 KCl, 10 NaCl, 1 MgCl2, 10 HEPES, and 0.05 EGTA, with pH adjusted to 7.2 with KOH. Membrane currents were filtered at 1 kHz and digitized at 4 kHz. In each patch under each condition, transient KCa current frequency and amplitude were calculated from ≥5 min of continuous gap-free data. At −40, 0, and +40 mV, in the presence of thapsigargin (500 nM), an SR Ca2+-ATPase blocker that inhibits Ca2+ sparks (16), a maximum of two, three, and six KCa channel openings, respectively, were observed (n = 5 cells). Therefore, at −40, 0, and +40 mV in control, a transient KCa current was defined as the simultaneous opening of three, four, or seven KCa channels, respectively. Single KCa channel current amplitude at each voltage was calculated using amplitude histograms.
Intracellular Ca2+ measurements using fura 2
Cerebral arteries were incubated in HEPES-buffered PSS containing 5 μM fura 2-AM and 0.05% Pluronic F-127 for 45 min at room temperature. After they were washed, the arteries were allowed 15 min for indicator deesterification. Fura 2 was alternately excited with 340- or 380-nm light with use of a xenon arc lamp and a personal computer-driven hyperswitch (Ionoptix, Milton, MA). Background corrected ratios were collected every 1 s at 510 nm with use of a photomultiplier tube (Ionoptix). For calibration of confocal Ca2+ imaging data, the extracellular K+ concentration was elevated from 6 to 30 mM by substitution of equimolar K+ for Na+; 30 mM K+ depolarizes arterial smooth muscle cells to ∼−40 mV (10), a voltage applied in transient KCa current measurements. [Ca2+]i values were calculated from fura 2 fluorescence measurements using the following equation (9)
where R is the ratio of fluorescence at 340 nm to fluorescence at 380 nm, Rmin and Rmax are the minimum and maximum fluorescence ratios determined in Ca2+-free and saturating Ca2+ solutions, respectively, Sf2/Sb2 is the ratio of Ca2+-free to Ca2+-replete emissions at 380-nm excitation, and Kd is the dissociation constant for fura 2 [282 nM (19)]. For determination of Rmin, Rmax, Sf2, and Sb2 at the end of the experiments and in separate experiments, the Ca2+ permeability of smooth muscle cells was increased with 10 μM ionomycin and the cells were perfused with a high-Ca2+ (10 mM) or Ca2+-free (no added Ca2+, 5 mM EGTA) solution. Elevation of extracellular K+ from 6 to 30 mM or from approximately −60 to −40 mV increased arterial wall Ca2+ from 104 ± 17 to 244 ± 29 nM (n = 7 arteries, P < 0.05).
Statistical analysis
Values are means ± SE; n refers to the number of events analyzed, unless otherwise specified. Student's t-tests were used for comparison of paired or unpaired data and Student-Newman-Keuls test for comparison of multiple data sets. When data sets were not normally distributed, the Kruskal-Wallis test with Dunn's multiple comparisons test was used for statistical comparison. Linear regression was used to calculate statistical correlation between the amplitude of Ca2+ sparks and evoked transient KCa currents (Origin, OriginLab, Northampton, MA). Analysis of covariance of linear regression was used to compare amplitude correlation data sets (Graphpad Prism, San Diego, CA). P < 0.05 was considered significant.
Chemicals
Unless otherwise stated, all chemicals were obtained from Sigma Chemical (St. Louis, MO). Papain was purchased from Worthington Biochemical (Lakewood, NJ) and fluo 4-AM from Molecular Probes (Eugene, OR).
RESULTS
Membrane depolarization elevates transient KCa current frequency and activity in newborn cerebral artery smooth muscle cells
Steady membrane depolarization between −40 and 0 mV increased mean transient KCa current frequency from ∼0.18 to 0.43 Hz (Fig. 1, A and B). Further depolarization to +40 mV reduced transient KCa current frequency to ∼0.26 Hz (Fig. 1, A and B). In contrast, depolarization between −40 and +40 mV continually increased mean transient KCa current amplitude (Fig. 1, A and C). Transient KCa current amplitude (I) is dependent on the number of KCa channels activated by a Ca2+ spark (N), the open probability of KCa channels at the Ca2+ spark peak (Po), and single KCa channel amplitude (i), giving iNPo. Membrane depolarization increases the driving force for K+ and, thus, i. Therefore, transient KCa current amplitude data were normalized for voltage-dependent changes in driving force as follows: NPo = I/i. In the same patches used for transient KCa current analysis, single KCa channel amplitudes at −40, 0, and +40 mV were 2.8 ± 0.1, 4.8 ± 0.1, and 9.0 ± 0.1 pA, respectively (n = 13). Over the voltage range of −40 to +40 mV, transient KCa channel activity (i.e., NPo) increased from 4 to 23 (Fig. 1D). These data indicate that membrane depolarization elevates transient KCa current frequency and Ca2+ spark-induced KCa channel activity in newborn porcine cerebral artery smooth muscle cells.
Fig. 1.

Voltage dependence of transient Ca2+-activated K+ (KCa) current frequency, amplitude, and activity. A: original recordings obtained in the same newborn arterial smooth muscle cell illustrating effect of steady membrane potentials between −40 and +40 mV on transient KCa current frequency and amplitude. B: voltage dependence of transient KCa current frequency (n = 13 cells). C: depolarization-induced elevation in transient KCa current amplitude (n = 13 cells). D: depolarization-induced elevation in transient KCa current activity (n = 13 cells). NPo, number of KCa channels activated by Ca2+ sparks × open probability of KCa channels at peak of Ca2+ sparks. *P < 0.05 vs. −40 mV.
Membrane depolarization activates Ca2+ sparks and augments Ca2+ spark-induced KCa channel activation
To examine the mechanisms by which membrane depolarization elevates transient KCa current frequency and activity in newborn arterial smooth muscle cells, simultaneous measurements of Ca2+ sparks and transient KCa currents were acquired using confocal Ca2+ imaging in combination with patch-clamp electrophysiology.
At −40 mV, ∼59% of Ca2+ sparks activated a transient KCa current (Fig. 2, Table 1). Steady membrane depolarization from −40 to 0 mV elevated global F/F0 1.33-fold, which translates to an increase in global [Ca2+]i from 224 ± 29 nM (see materials and methods) to 363 nM. Depolarization from −40 to 0 mV elevated the amplitude of coupled and uncoupled Ca2+ sparks, with the mean amplitude of all Ca2+ sparks increasing from ∼874 to 1,424 nM. In contrast, mean Ca2+ spark spread was smaller and decay was faster at 0 mV that at −40 mV. Depolarization from −40 to 0 mV increased the percentage of Ca2+ sparks that activated a transient KCa current to ∼77%. Further depolarization from 0 to +40 mV reduced global [Ca2+]i to 271 nM, which is expected because of a reduction in the driving force for Ca2+ influx, decreased mean Ca2+ spark amplitude to ∼1,121 nM and reduced coupled and uncoupled Ca2+ spark amplitude. However, depolarization to +40 mV increased the percentage of Ca2+ sparks that activated a transient KCa current to ∼86%. Taken together, membrane depolarization between −40 and +40 mV is estimated to increase KCa channel sensitivity to Ca2+ sparks from ∼0.015 to 0.026 NPo/nM Ca2+ when the [Ca2+]i detected by fluo 4 is taken as an indicator of Ca2+ spark amplitude.
Fig. 2.

Membrane depolarization elevates KCa channel sensitivity to Ca2+ sparks. A: simultaneous recordings of whole cell current (top traces) and Ca2+ sparks (bottom traces) at −40, 0, and +40 mV. Fluo 4 fluorescence changes (F/F0) were measured in 2 different 1.54 × 1.54 μm (i.e., 2.37 μm2) areas of the cell in which Ca2+ sparks occurred. At −40 mV, 1 Ca2+ spark occurred at 1 location; at 0 mV, 4 Ca2+ sparks were observed at 2 locations; at +40 mV, 3 Ca2+ sparks occurred at 1 location. *, Ca2+ spark at its peak in the pseudocolored inset image. B: voltage dependence of relation between peak Ca2+ spark amplitude and activity (NPo) of evoked transient KCa currents (n = 14, 13, and 10 cells for −40, 0, and +40 mV, respectively). Linear regression with 95% confidence bands is illustrated with slopes of 0.005, 0.010, and 0.016 for −40, 0, and +40 mV, respectively. Membrane depolarization elevated linear correlation coefficient as follows: 0.04 for −40 mV, 0.28 for 0 mV, and 0.51 for +40 mV. Amplitudes of Ca2+ sparks and evoked transient KCa currents were significantly correlated at each voltage (P < 0.05 for each).
Table 1.
Regulation of Ca2+ spark and transient KCa current properties by membrane potential
| −40 mV | 0 mV | +40 mV | |
|---|---|---|---|
| Ca2+ sparks | |||
| Amplitude, All nM | 874 ± 89 (65) | 1,424 ± 100*(74) | 1,121 ± 147 (62) |
| Coupled | 927 ± 123 (45) | 1,377 ± 114*(58) | 1,187 ± 180 (50) |
| Uncoupled | 755 ± 77 (20) | 1283 ± 97*(16) | 849 ± 99 (12) |
| Spread, μm2 | 3.56 ± 0.35 (17) | 3.08 ± 0.39*(17) | 3.58 ± 0.30 (17) |
| Decay (t1/2), ms | 61.3 ± 5.3 (17) | 52.6 ± 3.0*(17) | 59.2 ± 3.6 (17) |
| Coupling, % | 58.6 ± 6.4 | 76.5 ± 4.2* | 86.2 ± 5.1* |
| Global Ca2+, nM | 224 ± 29 | 363 | 271 |
| KCa transients | |||
| Amplitude, pA | 34.7 ± 5.9 (39) | 88.4 ± 11.6*(52) | 251.5 ± 24.3*(41) |
| NPo | 13.9 ± 2.4 (39) | 22.1 ± 2.9*(52) | 31.4 ± 3.0*(41) |
Values are means±SE of number of events in parentheses. t1/2, half time; KCa, Ca2+-activated K+; NPo, number of KCa channels activated by Ca2+ spark × open probability of KCa channels at peak of Ca2+ spark.
P < 0.05 vs. −40 mV.
Diltiazem or removal of extracellular Ca2+ blocks depolarization-induced elevations in transient KCa current frequency, but not activity
To investigate the contribution of Ca2+ influx to the depolarization-induced increase in Ca2+ spark-KCa channel coupling, voltage-dependent transient KCa current regulation was measured in a Ca2+-free bath solution or in the presence of diltiazem (50 μM), a voltage-dependent Ca2+ channel blocker.
At −40 mV, removal of extracellular Ca2+ reduced transient KCa current frequency to 0.41 ± 0.10 of control (P < 0.05) but did not change transient KCa current amplitude (0.99 ± 0.05 of control, P > 0.05, n = 5 cells). At −40, 0, and +40 mV, 50 μM diltiazem reduced transient KCa current frequency to 0.43 ± 0.06, 0.24 ± 0.03, and 0.46 ± 0.03 of control, respectively (P < 0.05 for each), but did not alter transient KCa current amplitude (1.00 ± 0.10, 1.17 ± 0.10, and 1.07 ± 0.05 of control, respectively, P > 0.05 for each, n = 6 cells). More importantly, in the absence of extracellular Ca2+ or in the continued presence of diltiazem, steady membrane depolarization between −40 and +40 mV increased mean transient KCa current frequency up to 1.6-fold (Fig. 3A). Over the same voltage range, transient KCa current activity (NPo) increased up to approximately fourfold (Fig. 3B). These data indicate that steady membrane depolarization elevates transient KCa current frequency and activity in the absence of extracellular Ca2+ entry or voltage-dependent Ca2+ channel activation.
Fig. 3.
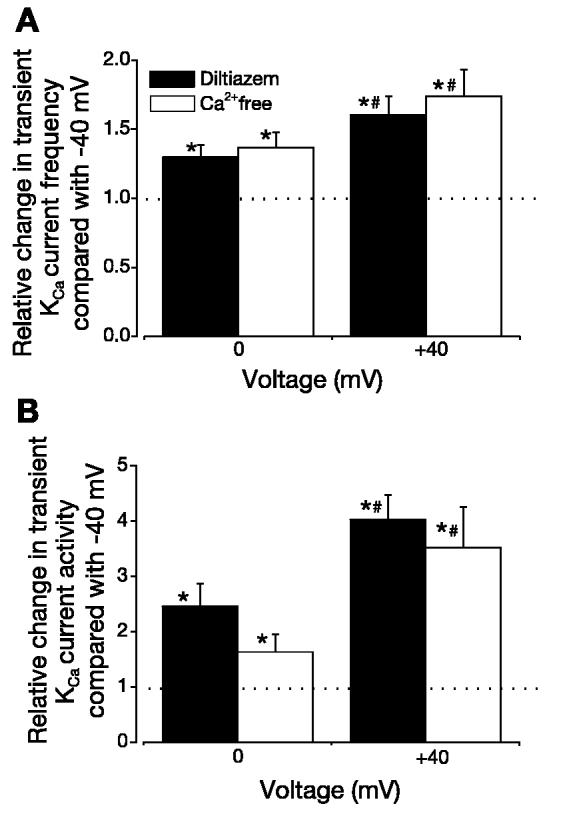
In the absence of Ca2+ influx, membrane depolarization elevates transient KCa current frequency and amplitude. In a Ca2+-free bath solution (n = 5 cells) or in the continued presence of 50 μM diltiazem (n = 6 cells), steady membrane depolarization elevates transient KCa current frequency (A) and activity (B). Dotted lines indicate control level. *P < 0.05 vs. −40 mV. #P < 0.05 vs. 0 mV.
Caffeine activates Ca2+ sparks and transient KCa currents
To determine whether RyR channel activation alters Ca2+ spark-KCa channel coupling in arterial smooth muscle cells, we studied transient KCa current regulation by caffeine, an RyR channel activator.
At −40, 0, and +40 mV, 10 μM caffeine increased transient KCa current frequency ∼1.5-, 1.6-, and 1.5-fold, respectively (Fig. 4A). In contrast, over the same voltage range, caffeine did not alter transient KCa channel activity (NPo; Fig. 4B). To investigate the effects of caffeine on Ca2+ spark properties and Ca2+ spark-KCa channel coupling, we used simultaneous patch-clamp electrophysiology and confocal Ca2+ imaging. Experiments were performed at 0 mV, because caffeine was most effective at activating transient KCa currents at this voltage. Caffeine increased mean global Ca2+ from ∼363 to 419 nM but reduced mean peak Ca2+ spark amplitude from ∼1,956 to ∼1,375 nM (Table 2). Caffeine also increased mean Ca2+ spark spatial spread from ∼2.9 to 3.6 μm2. Caffeine did not alter Ca2+ spark decay, the percentage of Ca2+ sparks that activated a transient KCa current, the amplitude relation between sparks and transient KCa currents, or transient KCa channel activity (NPo; Table 2, Fig. 5). These data indicate that RyR channel activation decreases Ca2+ spark amplitude (i.e., the local subsarcolemmal [Ca2+]i activating KCa channels) and elevates spatial spread of Ca2+ sparks, which would increase the number of KCa channels impacted by the spark. The combination of these changes in Ca2+ spark properties results in no net change in Ca2+ spark-KCa channel coupling.
Fig. 4.
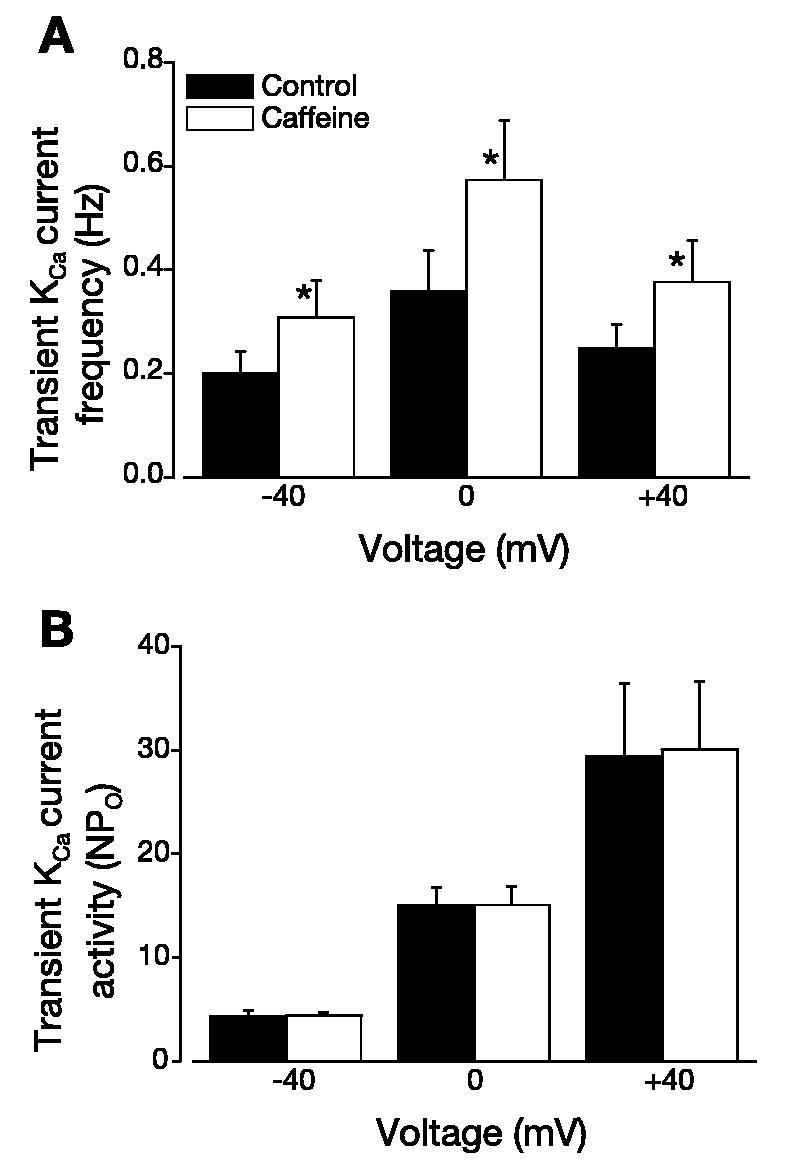
Caffeine elevates transient KCa current frequency, but not activity. A: 10 μM caffeine increased transient KCa current frequency at steady membrane potentials of −40, 0, and +40 mV (n = 6 cells). B: caffeine did not alter transient KCa current activity at −40, 0, and +40 mV (n = 6 cells). *P < 0.05 vs. control at the same voltage.
Table 2.
Regulation of Ca2+ spark and transient KCa current properties by caffeine
| Control | Caffeine (10 μM) | |
|---|---|---|
| Ca2+ sparks | ||
| Amplitude: all, nM | 1,915 ± 213 (33) | 1,375 ± 81*(42) |
| Spread, μm2 | 2.92 ± 0.29 (20) | 3.61 ± 0.31*(20) |
| Decay (t½), ms | 54.5 ± 3.9 (16) | 50.7 ± 5.7 (16) |
| Coupling, % | 75.4 ± 4.9 | 75.8 ± 5.1 |
| Global Ca2+, nM | 363 | 419 ± 9* |
| KCa transient activity (NPo) | 15.5 ± 1.4 (25) | 15.7 ± 1.0 (32) |
Values are means ± SE of number of events in parentheses.
P < 0.05 vs. control.
Fig. 5.
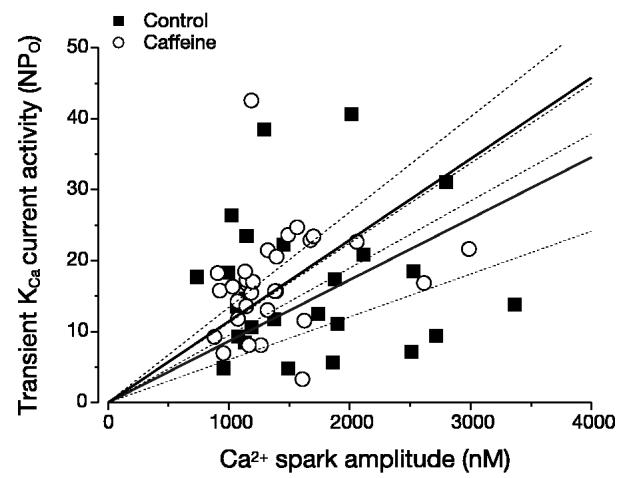
Caffeine does not alter Ca2+ spark-KCa channel coupling. Caffeine did not alter relation between peak Ca2+ spark amplitude and transient KCa current activity (NPo). Control and caffeine data were obtained from the same 7 cells. Linear regression with 95% confidence bands are illustrated with slopes of 0.009 and 0.011 for control and caffeine, respectively. Correlation coefficients were 0.11 and 0.53 for control and caffeine data, respectively. Amplitudes of Ca2+ sparks and evoked transient KCa currents were significantly correlated for control and caffeine (P < 0.0001 for each).
DISCUSSION
The regulation of Ca2+ spark-KCa channel coupling by mechanisms that activate KCa and RyR channels was studied in newborn cerebral artery smooth muscle cells, in which a significant proportion of Ca2+ sparks do not activate a transient KCa current. Membrane depolarization between −40 and +40 mV increased 1) transient KCa current frequency and activity (NPo), 2) the percentage of Ca2+ sparks that activated a transient KCa current from 59 to 86%, and 3) the sensitivity of KCa channels to Ca2+ sparks. Ca2+ influx or voltage-dependent Ca2+ channel activation was not obligatory for membrane depolarization to elevate transient KCa current frequency and activity. In contrast, RyR channel activation elevated transient KCa current frequency solely by causing an increase in Ca2+ spark frequency. RyR channel activation did not change Ca2+ spark-KCa channel coupling or transient KCa current activity. These data indicate that KCa channel Ca2+ sensitivity, rather than RyR channel activity, is a principal factor that underlies fractional Ca2+ spark coupling in newborn cerebral artery smooth muscle cells.
Membrane depolarization between −40 and 0 mV increased global [Ca2+]i, Ca2+ spark amplitude, KCa channel sensitivity to Ca2+ sparks, and the percentage of Ca2+ sparks that activated a transient KCa current. Further depolarization to +40 mV decreased Ca2+ spark amplitude and reduced global [Ca2+]i, which was expected because of a reduction in driving force for Ca2+ influx. However, depolarization from 0 to +40 mV further increased the percentage of Ca2+ sparks that activated a transient KCa current and elevated KCa channel sensitivity to Ca2+ sparks. These data suggest that, in newborn arterial smooth muscle cells, effective coupling and percent coupling of Ca2+ sparks to KCa channels are modulated primarily by KCa channel sensitivity to Ca2+ sparks, rather than by RyR channel activity. An explanation for these findings is that membrane depolarization increases KCa channel apparent Ca2+ sensitivity, which would increase the impact of sparks on KCa channel Po (4, 12, 20). The depolarization-induced elevation in transient KCa current frequency most likely occurs through an increase in the percentage of Ca2+ sparks that activate KCa channels. In support of this conclusion, in the presence of diltiazem or in the absence of extracellular Ca2+, both of which would block depolarization-induced Ca2+ spark activation (13, 17), depolarization elevated transient KCa current frequency and activity. In murine colonic myocytes, a reduction in extracellular Ca2+ reduced local intracellular Ca2+ transients but elevated transient KCa current frequency and amplitude by removing protein kinase C-mediated KCa channel inhibition (2). In contrast, in the present study, removal of extracellular Ca2+ or diltiazem reduced transient KCa current frequency but did not alter amplitude. These data suggest Ca2+ sparks are activated by Ca2+ influx through voltage-dependent Ca2+ channels, as previously reported (13, 17), and illustrate differences in the mechanisms by which Ca2+ spark-KCa channel coupling is modulated by Ca2+ influx pathways in colonic and arterial smooth muscle cells.
KCa channel “Ca2+ sensitivity” has previously been used to describe 1) the Ca2+ concentration that induces half-maximal activation at a given voltage, 2) the slope of the Ca2+-activity relation at a defined voltage, and 3) a shift in half-maximal potential for a given Ca2+ concentration change (4). Depolarization shifts the Ca2+ concentration-KCa channel activity relation leftward (4) and increases the percentage of Ca2+ sparks that activate KCa channels. The present data dispute the possibility that uncoupling occurs because KCa channels within the vicinity of Ca2+ spark sites are absent or incapable of activation. The Kd for Ca2+ of newborn porcine arteriole smooth muscle cell KCa channels is 31 μM at 0 mV, which is high compared with that of KCa channels in other smooth muscle cell preparations, including human coronary artery and rat cerebral artery (22, 26, 30). Conceivably, uncoupling may occur because KCa channel Ca2+ sensitivity is lower in uncoupled than in strongly coupled cell types. Other likely explanations are that uncoupled Ca2+ sparks are of lower amplitude (present study and Ref. 14) and/or the distance between uncoupled spark release sites and the sarcolemma is greater, both of which would result in lower spark-induced subsarcolemmal Ca2+ elevations. In B. marinus stomach smooth muscle cells, some Ca2+ spark sites generate sparks that reliably activate transient KCa currents, whereas other locations consistently generate uncoupled sparks (31). In the amphibian preparation, sites that generate uncoupled Ca2+ sparks may be located near sarcolemma that is devoid of KCa channels or populated by inactivatable KCa channels (31). However, in newborn cerebral artery smooth muscle cells, Ca2+ spark-KCa channel coupling is increased by membrane depolarization and carbon monoxide, which elevate KCa channel apparent Ca2+ sensitivity (14, 15, 30). Similarly, in guinea pig bladder smooth muscle cells, membrane depolarization between −50 and −20 mV elevated Ca2+ spark coupling (11). Thus KCa channel localization near Ca2+ spark sites and regulation by Ca2+ sparks appear to differ in mammalian and amphibian smooth muscle cells.
Regardless of voltage, caffeine, which elevates RyR channel Ca2+ sensitivity (24), induced a similar relative increase in transient KCa current frequency but did not change transient KCa channel activity. These data suggest that caffeine activates transient KCa currents by elevating Ca2+ spark frequency. Caffeine also elevated global [Ca2+]i and reduced Ca2+ spark amplitude, presumably by causing SR Ca2+ leak and a reduction in SR Ca2+ load, respectively (6). Caffeine also increased Ca2+ spark spread, presumably by elevating the number of RyR channels that contribute to sparks through localized Ca2+-induced Ca2+ release. In pulmonary artery smooth muscle cells, 500 μM caffeine did not change Ca2+ spark amplitude (calculated as F/F0) but elevated Ca2+ spark frequency, duration, and spread (25). In B. marinus stomach smooth muscle cells, caffeine increased the number of spark sites from ∼42 to 400 (31). Conceivably, caffeine may have also generated Ca2+ sparks at additional sites in newborn cerebral artery smooth muscle cells. However, the low Ca2+ spark frequency in newborn arterial smooth muscle cells and the 10-s time limit required for imaging to avoid laser-induced cell damage precluded systematic examination of this possibility. Nevertheless, the net effect of Ca2+ spark spatial and temporal changes was no net change in the mean percentage or effective Ca2+ spark-KCa channel coupling. Thus, in newborn porcine cerebral artery smooth muscle cells, RyR channel activation elevates transient KCa current frequency by elevating Ca2+ spark frequency, and not by altering Ca2+ spark-KCa channel coupling.
Caffeine, at low micromolar concentrations, induces a KCa channel-sensitive vasodilation in pressurized newborn cerebral arteries (1). Carbon monoxide increases KCa channel apparent Ca2+ sensitivity and Ca2+ spark-KCa channel coupling in smooth muscle cells and dilates newborn porcine cerebral arteries (14, 15, 30). These findings show that an elevation in Ca2+ spark frequency alone or an increase in Ca2+ spark-KCa channel coupling induces vasodilation through KCa channel activation. In the present study, membrane depolarization within the physiological range [i.e., ca. −60 to −20 mV (19)] would increase Ca2+ spark-Ca2+ channel coupling by only ∼10–15%. The increase in coupling alone would be predicted to have only a small effect on membrane potential. However, the combination of an increase in coupling and depolarization-induced transient KCa current frequency and amplitude elevation would increase K+ current through KCa channels, produce membrane hyperpolarization, and oppose pressure-induced constriction (16). Within the physiological range of voltages, Ca2+ spark-KCa channel coupling in newborn myocytes does not reach 100%, allowing additional mechanisms that enhance KCa channel Ca2+ sensitivity to augment coupling and further enhance KCa channel activity [e.g., carbon monoxide (14)]. As such, signaling elements that increase KCa channel Ca2+ sensitivity will be more effective vasodilators in myocytes that exhibit fractional coupling than in cells with 100% coupling. Furthermore, messengers that elevate Ca2+ spark frequency and coupling to KCa channels, including reactive oxygen species (7, 29) and carbon monoxide (14, 15), should produce the most significant KCa channel-dependent vasodilation.
In summary, the present data indicate that, in newborn porcine cerebral artery smooth muscle cells, fractional Ca2+ spark coupling occurs through KCa channel insensitivity to Ca2+ sparks. Uncoupled Ca2+ sparks can be coupled by mechanisms that elevate KCa channel Ca2+ sensitivity. In contrast, RyR channel activation alone reduces Ca2+ spark amplitude and increases Ca2+ spark spread, resulting in no net change in Ca2+ spark coupling.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
GRANTS
This research was supported by National Heart, Lung, and Blood Institute Grants HL-77678 and HL-67061 (to J. H. Jaggar) and HL-42851 and HL-34059 (to C. W. Leffler).
REFERENCES
- 1.Ahmed A, Waters CM, Leffler CW, Jaggar JH. Ionic mechanisms mediating the myogenic response in newborn porcine cerebral arteries. Am J Physiol Heart Circ Physiol. 2004;287:H2061–H2069. doi: 10.1152/ajpheart.00660.2004. [DOI] [PubMed] [Google Scholar]
- 2.Bayguinov O, Hagen B, Kenyon JL, Sanders KM. Coupling strength between localized Ca2+ transients and K+ channels is regulated by protein kinase C. Am J Physiol Cell Physiol. 2001;281:C1512–C1523. doi: 10.1152/ajpcell.2001.281.5.C1512. [DOI] [PubMed] [Google Scholar]
- 3.Benham CD, Bolton TB. Spontaneous transient outward currents in single visceral and vascular smooth muscle cells of the rabbit. J Physiol. 1986;381:385–406. doi: 10.1113/jphysiol.1986.sp016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carl A, Lee HK, Sanders KM. Regulation of ion channels in smooth muscles by calcium. Am J Physiol Cell Physiol. 1996;271:C9–C34. doi: 10.1152/ajpcell.1996.271.1.C9. [DOI] [PubMed] [Google Scholar]
- 5.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 6.Cheranov SY, Jaggar JH. Sarcoplasmic reticulum calcium load regulates rat arterial smooth muscle calcium sparks and transient KCa currents. J Physiol. 2002;544:71–84. doi: 10.1113/jphysiol.2002.025197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheranov SY, Jaggar JH. TNF-α dilates cerebral arteries via NAD(P)H oxidase-dependent Ca2+ spark activation. Am J Physiol Cell Physiol. 2006;290:C964–C971. doi: 10.1152/ajpcell.00499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 9.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 10.Harder DR. Comparison of electrical properties of middle cerebral and mesenteric artery in cat. Am J Physiol Cell Physiol. 1980;239:C23–C26. doi: 10.1152/ajpcell.1980.239.1.C23. [DOI] [PubMed] [Google Scholar]
- 11.Herrera GM, Heppner TJ, Nelson MT. Voltage dependence of the coupling of Ca2+ sparks to BKCa channels in urinary bladder smooth muscle. Am J Physiol Cell Physiol. 2001;280:C481–C490. doi: 10.1152/ajpcell.2001.280.3.C481. [DOI] [PubMed] [Google Scholar]
- 12.Jackson WF, Blair KL. Characterization and function of Ca2+-activated K+ channels in arteriolar muscle cells. Am J Physiol Heart Circ Physiol. 1998;274:H27–H34. doi: 10.1152/ajpheart.1998.274.1.H27. [DOI] [PubMed] [Google Scholar]
- 13.Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. 2001;281:C439–C448. doi: 10.1152/ajpcell.2001.281.2.C439. [DOI] [PubMed] [Google Scholar]
- 14.Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D,ES, Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res. 2002;91:610–617. doi: 10.1161/01.res.0000036900.76780.95. [DOI] [PubMed] [Google Scholar]
- 15.Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ Res. 2005;97:805–812. doi: 10.1161/01.RES.0000186180.47148.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- 17.Jaggar JH, Stevenson AS, Nelson MT. Voltage dependence of Ca2+ sparks in intact cerebral arteries. Am J Physiol Cell Physiol. 1998;274:C1755–C1761. doi: 10.1152/ajpcell.1998.274.6.C1755. [DOI] [PubMed] [Google Scholar]
- 18.Kirber MT, Etter EF, Bellve KA, Lifshitz LM, Tuft RA, Fay FS, Walsh JV, Fogarty KE. Relationship of Ca2+ sparks to STOCs studied with 2D and 3D imaging in feline oesophageal smooth muscle cells. J Physiol. 2001;531:315–327. doi: 10.1111/j.1469-7793.2001.0315i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magleby KL. Gating mechanism of BK (Slo1) channels: so near, yet so far. J Gen Physiol. 2003;121:81–96. doi: 10.1085/jgp.20028721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 22.Perez GJ, Bonev AD, Nelson MT. Micromolar Ca2+ from sparks activates Ca2+-sensitive K+ channels in rat cerebral artery smooth muscle. Am J Physiol Cell Physiol. 2001;281:C1769–C1775. doi: 10.1152/ajpcell.2001.281.6.C1769. [DOI] [PubMed] [Google Scholar]
- 23.Perez GJ, Bonev AD, Patlak JB, Nelson MT. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol. 1999;113:229–238. doi: 10.1085/jgp.113.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pessah IN, Stambuk RA, Casida JE. Ca2+-activated ryanodine binding: mechanisms of sensitivity and intensity modulation by Mg2+, caffeine, and adenine nucleotides. Mol Pharmacol. 1987;31:232–238. [PubMed] [Google Scholar]
- 25.Remillard CV, Zhang WM, Shimoda LA, Sham JS. Physiological properties and functions of Ca2+ sparks in rat intrapulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L433–L444. doi: 10.1152/ajplung.00468.2001. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka Y, Meera P, Song M, Knaus HG, Toro L. Molecular constituents of maxi KCa channels in human coronary smooth muscle: predominant α + β subunit complexes. J Physiol. 1997;502:545–557. doi: 10.1111/j.1469-7793.1997.545bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wellman GC, Nathan DJ, Saundry CM, Perez G, Bonev AD, Penar PL, Tranmer BI, Nelson MT. Ca2+ sparks and their function in human cerebral arteries. Stroke. 2002;33:802–808. doi: 10.1161/hs0302.104089. [DOI] [PubMed] [Google Scholar]
- 28.Woodruff ML, Sampath AP, Matthews HR, Krasnoperova NV, Lem J, Fain GL. Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J Physiol. 2002;542:843–854. doi: 10.1113/jphysiol.2001.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xi Q, Cheranov SY, Jaggar JH. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ Res. 2005;97:354–362. doi: 10.1161/01.RES.0000177669.29525.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activates KCa channels in newborn cerebral arteriole smooth muscle cells by increasing the apparent Ca2+ sensitivity of α-subunits. Am J Physiol Heart Circ Physiol. 2004;286:H610–H618. doi: 10.1152/ajpheart.00782.2003. [DOI] [PubMed] [Google Scholar]
- 31.ZhuGe R, Fogarty KE, Baker SP, McCarron JG, Tuft RA, Lifshitz LM, Walsh JV., Jr Ca2+ spark sites in smooth muscle cells are numerous and differ in number of ryanodine receptors, large-conductance K+ channels, and coupling ratio between them. Am J Physiol Cell Physiol. 2004;287:C1577–C1588. doi: 10.1152/ajpcell.00153.2004. [DOI] [PubMed] [Google Scholar]
- 32.ZhuGe R, Fogarty KE, Tuft RA, Lifshitz LM, Sayar K, Walsh JV., Jr Dynamics of signaling between Ca2+ sparks and Ca2+-activated K+ channels studied with a novel image-based method for direct intracellular measurement of ryanodine receptor Ca2+ current. J Gen Physiol. 2000;116:845–864. doi: 10.1085/jgp.116.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.ZhuGe R, Fogarty KE, Tuft RA, Walsh JV., Jr Spontaneous transient outward currents arise from microdomains where BK channels are exposed to a mean Ca2+ concentration on the order of 10 μM during a Ca2+ spark. J Gen Physiol. 2002;120:15–27. doi: 10.1085/jgp.20028571. [DOI] [PMC free article] [PubMed] [Google Scholar]