

Abstract
A 5-(2′-Deoxyuridinyl)methyl radical (1) was independently generated from three photochemical precursors and is the first example of a DNA radical that forms interstrand cross-links. Oxygen labeling experiments support generation of 1 by all precursors. Interstrand cross-links are produced upon irradiation of DNA containing any of the precursors. Cross-linking occurs via reaction with the opposing 2′-deoxyadenosine and is independent of O2. The independence of cross-link formation on O2 is explained by kinetic analysis, which shows that the radical reacts reversibly with O2. Examination of the effects of glutathione on cross-link formation under anaerobic conditions suggests that adoption of the syn-conformation by 1 is the rate-limiting step in the process. Interstrand cross-link formation is reversible in the presence of a good nucleophile. The stability of the interstrand cross-link suggests that the isolated molecule is a rearrangement product of that formed in solution. The rearrangement is a consequence of the isolation procedure but also occurs slowly in solution. Oxygen independent cross-link formation may be useful for the purposeful damage of DNA in hypoxic tumor cells, where O2 is deficient.
The biological importance of DNA damage is evident in many ways. The double sword nature of these chemical transformations is illustrated by their involvement in the etiology and treatment of cancer.1-3 In addition, there is a growing appreciation of the importance of DNA damage in aging.4-6 DNA radicals are an important family of reactive intermediates that give rise to modified intact polymers and also lead to single-and double-strand breaks.7-9 Recently, we reported that an independently generated 5-(2′-deoxyuridinyl)methyl radical (1) produced interstrand cross-links (ISCs) with an opposing 2′-deoxyadenosine (dA) (Scheme 1).10 The radical is derived from formal hydrogen atom abstraction from the methyl group of thymidine and is not believed to be formed in large amounts by ionizing radiation despite the low bond dissociation energy (BDE) of the carbon-hydrogen bonds and their accessibility in the major groove. The preliminary report concerning 1 was the first explicit example of DNA interstrand cross-link (ISC) formation through a nucleic acid radical. ISCs are often responsible for the cytotoxic effects of DNA damaging agents.11,12 Hence, ISC formation by a radical produced in even small amounts (e.g., 1) by agents such as γ-radiolysis could be biologically significant. Our interest in the radical-mediated cross-link formation is enhanced by the observation that the process involving 1 is independent of O2. This suggests that, in addition to being chemically novel, the formation of interstrand cross-links from 1 may be useful in hypoxic cells, such as those present in tumors.13 DNA damaging agents, such as γ-radiolysis, are less efficient in hypoxic cells because many of the chemical pathways require O2. Herein we describe experiments that further elucidate and substantiate this novel mechanism for DNA interstrand cross-link formation.
Scheme 1.
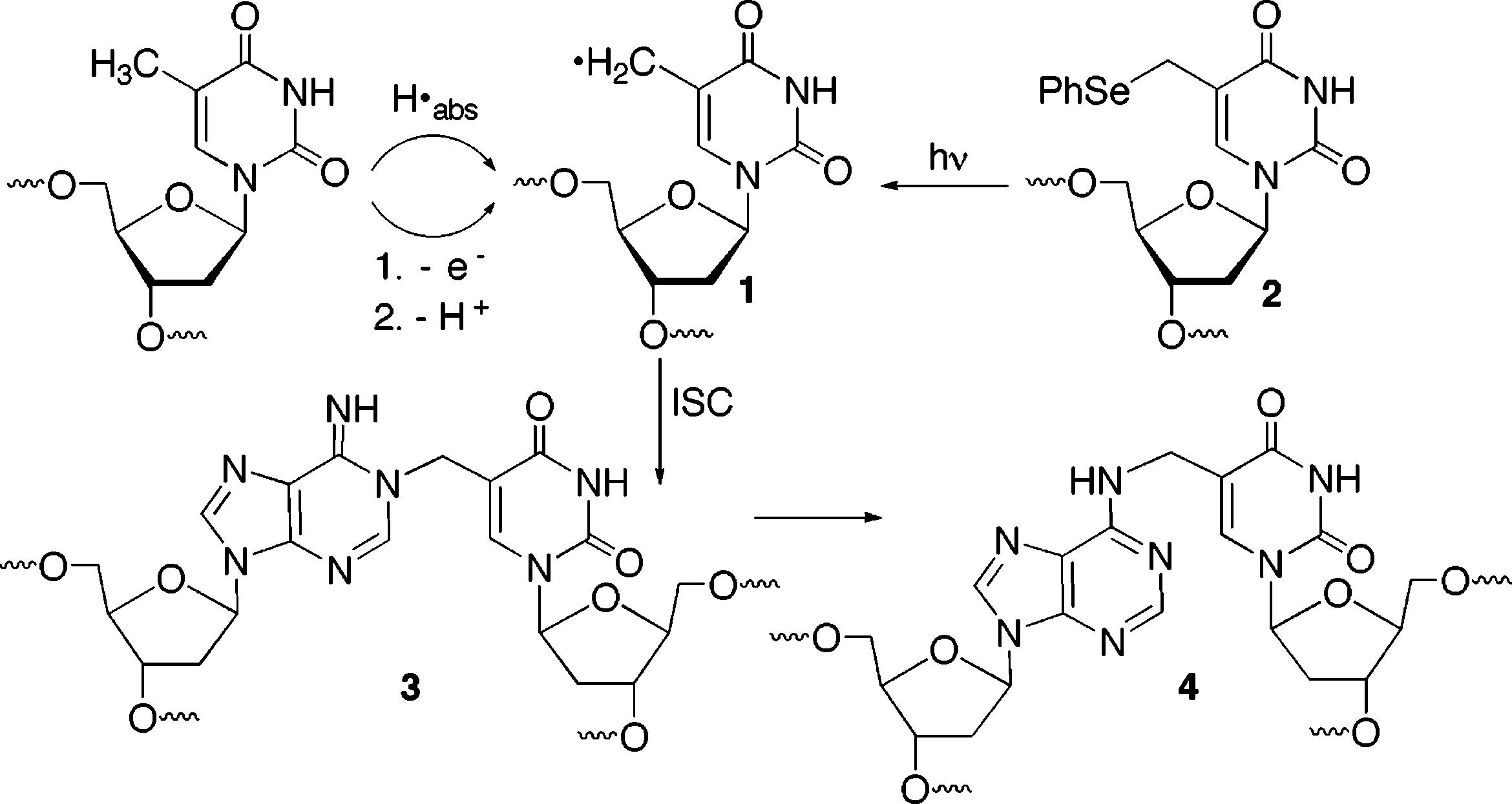
Generation of, and Interstrand Cross-Link Formation by the 5-(2′-Deoxyuridinyl)methyl Radical (1)
In addition to arising from direct hydrogen atom abstraction, the 5-(2′-deoxyuridinyl)methyl radical (1) can be formed from a two-step process, such as that induced by the direct effect of γ-radiolysis (Scheme 1). In accordance with these mechanistic possibilities, the radical has been implicated as an intermediate in γ-radiolysis through product studies and in experiments in which photoinduced electron transfer is used to oxidize thymidine.7,14,15 Oxygenated products for which 1 can serve as a precursor, most notably 5-formyl-2′-deoxyuridine (9) and 5-hydroxymethyl-2′-deoxyuridine (8), have been detected in studies on the reactivity of thymidine and DNA.16 Prior to the report by Hong, 1 was independently generated from other photolabile precursors in a nucleoside, dinucleotide, and single-stranded oligonucleotides.17,18 Generation of 1 in dinucleotides and single-stranded oligonucleotides led to the formation of tandem lesions.18 Tandem lesions are defined as two contiguously damaged nucleotides. Cadet characterized a tandem lesion derived from 1, involving formation of a covalent between the radical and C8 of the adjacent 2′-deoxyguanosine. Structurally similar tandem lesions were also detected when the analogous radical (5) derived from 5-methyl-2′-deoxycytidine was produced in a dinucleotide.19 However, in contrast to other  nucleobase radicals, alkali-labile tandem lesions were not detected by gel electrophoresis when 1 was produced from 2 in duplex DNA.20-22 Interstrand cross-links were the sole products detected when 1 was produced from 2 in duplex DNA under aerobic or anaerobic conditions.10 Given this surprising result, we investigated the mechanism for ISC formation further. Herein we provide further support that the 5-(2′-deoxyuridinyl)-methyl radical (1) is responsible for cross-linking. An explanation for why ISC formation is independent of O2 and other issues are also addressed.
nucleobase radicals, alkali-labile tandem lesions were not detected by gel electrophoresis when 1 was produced from 2 in duplex DNA.20-22 Interstrand cross-links were the sole products detected when 1 was produced from 2 in duplex DNA under aerobic or anaerobic conditions.10 Given this surprising result, we investigated the mechanism for ISC formation further. Herein we provide further support that the 5-(2′-deoxyuridinyl)-methyl radical (1) is responsible for cross-linking. An explanation for why ISC formation is independent of O2 and other issues are also addressed.
Results and Discussion
Independent Generation of 5-(2′-Deoxyuridinyl)methyl Radical (1) from Three Photolabile Precursors. Preliminary product studies were consistent with independent generation of 1 from the phenyl selenide (2, Scheme 2).23 More complete studies were undertaken because the radical was not directly observed and interstrand cross-link formation could be rationalized using other electrophilic species as reactants. For example, we recently showed that oxidation of 2 by 1O2 or NaIO4 also produces an ISC with the opposing dA following [2,3] sigmatropic rearrangement of the selenoxide.24 Formal solvolysis of 2 would produce a carbocation capable of alkylating dA. Although the formation of oxygenated thymidine derivatives was indicative of the intermediacy of 1, isotopic labeling was used to further develop this connection (Table 1). The source of the hydroxyl group oxygen atom in 5-hydroxymethyl-2′-deoxyuridine (8) was determined using 18O-labeled H2O and O2 in the presence of glutathione (50 mM). The ratio of the M+ + 2:M+ ions was insignificantly higher when H218O was used as solvent. However, bubbling 18O2 through the solution prior to and during photolysis resulted in an approximate 100-fold increase in the ratio of the M+ + 2:M+ ions, confirming that O2 is the source of the hydroxyl group oxygen in 8.
Scheme 2.
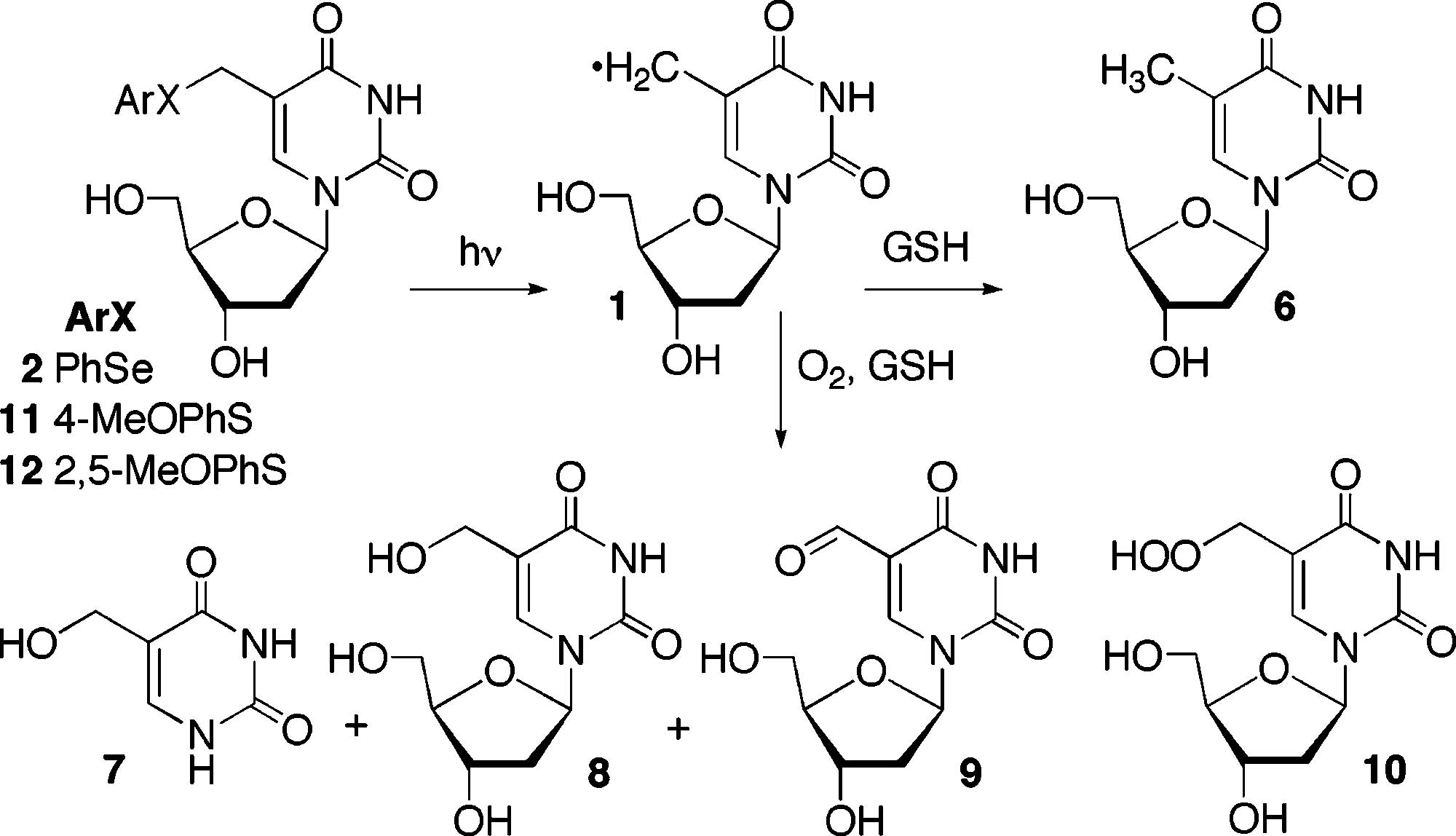
Trapping Products Produced from the 5-(2′-Deoxyuridinyl)methyl Radical (1)
Table 1.
Source of Oxygen in 5-(Hydroxymethyl)-2′-deoxyuridine (8) Determined by Mass Spectrometry
| M++2:M+ |
|||
|---|---|---|---|
| substrate | H2O/O2 | H218O/O2 | H2O/18O2 |
| 2 | 0.05 | 0.08 | 6.1 |
| 11 | 0.01 | 0.03 | |
| 12 | 0.01 | 0.06 | |
Although the O2 trapping data from photolysis of 2 are consistent with the intermediacy of 1, we sought to strengthen the argument by developing other precursors to the 5-(2′-deoxyuridinyl)methyl radical (1). Irradiation of the respective phenyl sulfides at 254 nm are reported to produce the 5-(2′-deoxyuridinyl)methyl radical (1) and 5-(2′-deoxycytidinyl)-methyl radical (5).18,19 Using these molecules as a guide, the methoxy-substituted derivatives of thymidine (11, 12) were
| (1) |
| (2) |
synthesized with the intent of red-shifting the absorption so that 1 could be generated from them upon irradiation with lamps that emit maximally at 350 nm. The mono- and dimethoxy-substituted molecules had λmax (ε, M−1 cm−1) at 258 (6800) and 267 (5640) nm, respectively. However, the absorbance of solutions (<100 μM) of each tailed into the ≥320 nm region, and the photochemical conversion efficiency of 11 and 12 correlated with their relative absorption. Approximately 50% of 11 or 12 (50 μM) was converted upon irradiation in a Rayonet photoreactor for 5 or 30 min, respectively. Product trapping studies in the presence of GSH and O2 produced the mixture of oxygenated products and thymidine expected from the formation of 1. The mass balances (65–76%) were comparable to those measured for 350 nm photolysis of 2.23 Additional support for the intermediacy of 1 from the aryl sulfides was acquired by carrying out the photolyses in H218O and GSH (50 mM), whereupon no incorporation of 18O in 8 was detected (Table 1).
Reversible Trapping of 5-(2′-Deoxyuridinyl)methyl Radical (1) by O2. Product and isotope labeling studies support the formation of 1 and its trapping by O2 under the 350 nm photolysis conditions used to generate the radical in DNA. Oxygenated product formation from monomeric 1 was reconciled with the observation that interstrand cross-linking is independent of dioxygen by examining the possibility that O2 trapping is reversible. We probed for reversible formation of monomeric 13 (Scheme 3) by measuring the distribution of radical trapping products as a function of GSH concentration. The product dependence on GSH concentration was established by assuming that thymidine (6) is formed via reaction between 1 and GSH with bimolecular rate constant kGSH. Oxygenated products were assumed to emanate from 13 upon its reaction with GSH (kOO•) or by other processes (kOx, Scheme 3, eq 1). If O2 trapping (kO2) is reversible and one assumes that the peroxyl radical (13) is formed under steady-state conditions, the ratio of thymidine (6) to oxygenated products (7–10, TOx) should vary nonlinearly with GSH concentration (Scheme 3, eq 2). Under conditions where peroxyl radical trapping (kOO•[GSH]) is much greater than the rate constant describing loss of O2 from 13 (k−O2), the product ratio is expected to exhibit a linear dependence on GSH concentration typical of irreversible O2 trapping. Indeed, the nonlinear dependence of the ratio of thymidine to oxygenated products is evident at low thiol concentrations when the dimethoxy-substituted aryl sulfide (12) is photolyzed in the presence of GSH between 0.2 mM and 20 mM (Figure 1). Nonlinear regression analysis (R2 = 0.979) was used to solve for k−O2 (3.4 s−1), kGSH (6.9 × 106 M−1 s−1), and kOx (0.08 s−1). This analysis assumed a rate constant for the trapping of 1 by O2 (2 × 109 M−1 s−1) and that 13 is trapped by GSH with a rate constant commensurate with that reported for DNA peroxyl radicals (2 × 102 M−1 s−1).25 The magnitude of k−O2 is slightly greater than the respective rate constant determined for the peroxyl radical derived from the C4′-DNA radical but is within the range expected for such reactions.26,27 The value of the rate constant determined for reaction between 1 and GSH (kGSH) is within the realm of what one would expect for reaction between a thiol and an alkyl radical.28 This calibration suggests that the value determined for k−O2 is a reasonable estimate.
Scheme 3.
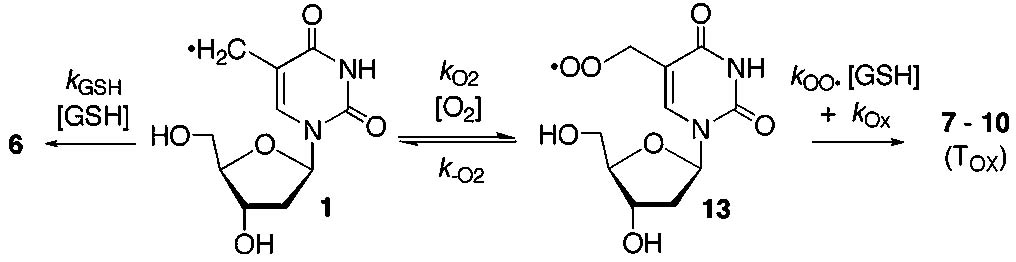
Reversible Trapping of the 5-(2′-Deoxyuridinyl)methyl Radical (1) by O2
Scheme 4.
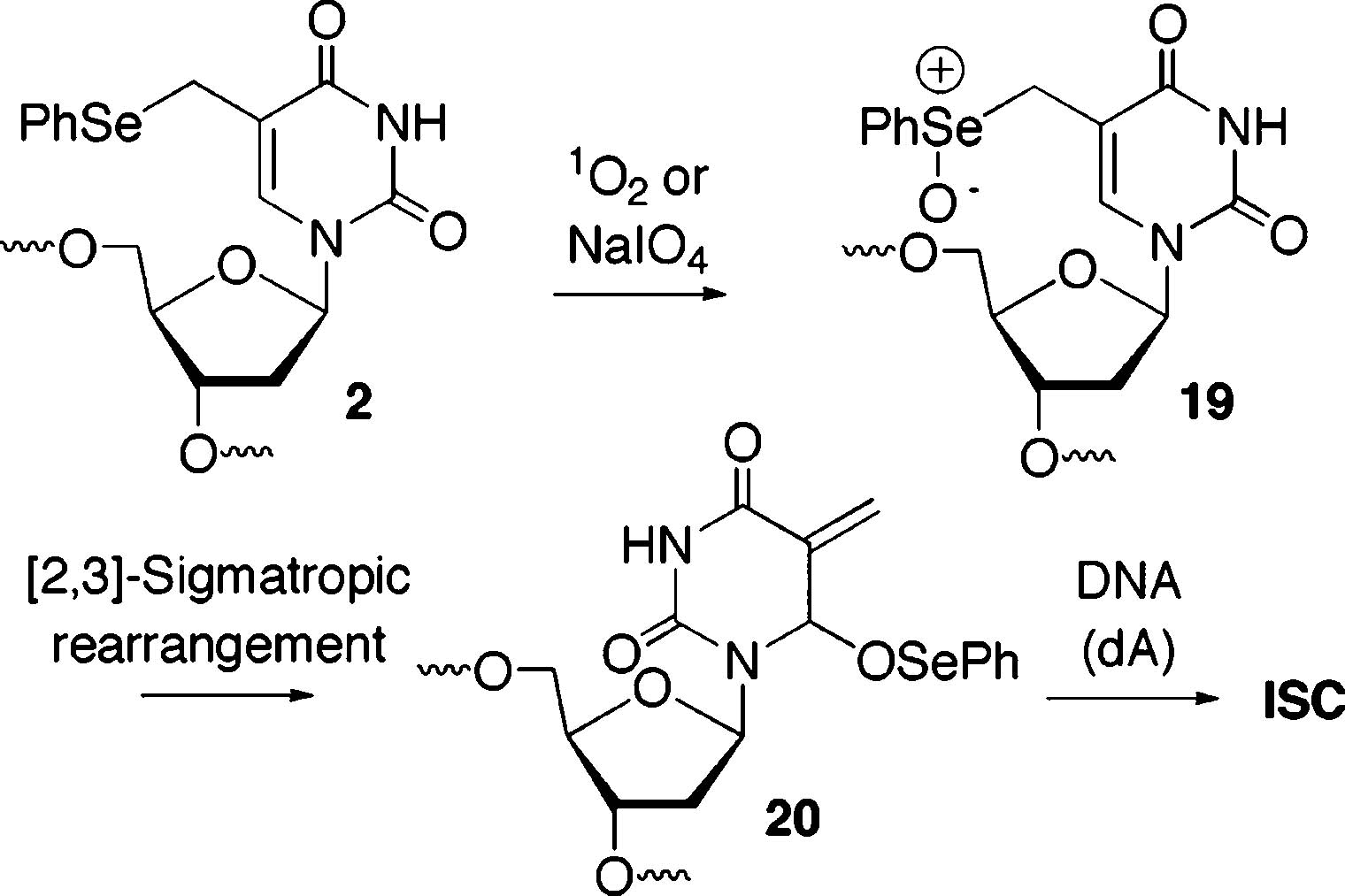
Interstrand Cross-Link Formation via Methide (20) Formation
Interstrand Cross-Linking by a Common Intermediate Produced from Three Photolabile Molecules. The above experiments demonstrate that the phenyl selenide (2) and aryl sulfides (11, 12) produce the nucleoside radical upon 350 nm photolysis. Further evidence was sought to support that the 5-(2′-deoxyuridinyl)methyl radical (1) was responsible for DNA interstrand cross-links. Concern that ISC formation was the result of an excited-state reaction between the phenyl selenide (2) and a neighboring nucleotide was minimized by showing that duplexes containing 2 (14–16) produce cross-links (Table 2).29 The ISC yield was significantly lower when the radical precursor was flanked by dG. However, lower yields were not accompanied by other products such as direct strand breaks or alkali-labile lesions. The lower ISC yield was not a result of  less efficient photochemical conversion of 2. ISC yield does not increase when photolysis of 15 or 16 is extended to 60 or 90 min, indicating that radical generation is complete within 30 min (data not shown). The lower ISC yield obtained from the sequence in which 1 is produced when flanked by dGs and the absence of direct strand breaks or alkali-labile lesions is consistent with the formation of tandem lesions as described by Cadet.18 Further support for radical generation from 2 in DNA is gleaned from ESI-MS analysis following anaerobic photolysis of 14 in the presence of GSH (50 mM), which showed that an oligonucleotide containing thymidine is formed at the expense of ISC.30 Duplexes (17, 18) containing 11 or 12 also produce an ISC upon photolysis in the presence or absence of O2 (Table 2). Cross-linking generated by the aryl sulfide is also quenched by GSH.30 Finally, the assumption that cross-linking occurred exclusively between the radical center and the opposing dA was verified by examining the hydroxyl radical cleavage of the cross-link product that was produced from photolysis of 18.30,31 All of these observations are consistent with the proposal that photolysis of the respective duplexes containing 2, 11, and 12 produces interstrand cross-links via a common intermediate, 5-(2′-deoxyuridinyl)methyl radical (1).
less efficient photochemical conversion of 2. ISC yield does not increase when photolysis of 15 or 16 is extended to 60 or 90 min, indicating that radical generation is complete within 30 min (data not shown). The lower ISC yield obtained from the sequence in which 1 is produced when flanked by dGs and the absence of direct strand breaks or alkali-labile lesions is consistent with the formation of tandem lesions as described by Cadet.18 Further support for radical generation from 2 in DNA is gleaned from ESI-MS analysis following anaerobic photolysis of 14 in the presence of GSH (50 mM), which showed that an oligonucleotide containing thymidine is formed at the expense of ISC.30 Duplexes (17, 18) containing 11 or 12 also produce an ISC upon photolysis in the presence or absence of O2 (Table 2). Cross-linking generated by the aryl sulfide is also quenched by GSH.30 Finally, the assumption that cross-linking occurred exclusively between the radical center and the opposing dA was verified by examining the hydroxyl radical cleavage of the cross-link product that was produced from photolysis of 18.30,31 All of these observations are consistent with the proposal that photolysis of the respective duplexes containing 2, 11, and 12 produces interstrand cross-links via a common intermediate, 5-(2′-deoxyuridinyl)methyl radical (1).
Table 2.
Interstrand Cross-Link Formation as a Function of Sequence and Radical Precursor
| local sequence (substrate) | % cross-link |
|
|---|---|---|
| aerobic | anaerobic | |
| 5′-C2C (14) | 25.7 ± 0.7 | 28.3 ± 0.6 |
| 5′-G2G (15) | 12.5 ± 0.5 | 12.6 ± 0.6 |
| 5′-T2T (16) | 20.0 ± 0.8 | 20.1 ± 0.7 |
| 5′-C11C (17) | 20.7 ± 0.8 | 19.8 ± 0.4 |
| 5′-C12C (18) | 20.6 ± 0.2 | 20.6 ± 0.6 |
A Methide is Not Responsible for Interstrand Cross-Links Produced upon Photolysis of Phenyl Selenide (2) or Aryl Sulfide (11, 12) Containing Duplex DNA. Oxidation of 2 by NaIO4 or 1O2 produces the methide (20), which reacts with nucleophiles, including the opposing dA in 14 (Scheme 4).24 In contrast, treatment of monomeric 12 with NaIO4 produces a diastereomeric mixture of sulfoxide 21, which does not rearrange even upon heating to 90 °C. This observation is consistent with  the expected higher barrier for a [2,3]-sigmatropic rearrangement faced by an allylic sulfoxide compared to that for a similar selenoxide.32 As expected, based upon this result, ISC is not observed upon treatment of 17 or 18 with NaIO4. This provides further indirect support that photochemical-mediated ISC formation from 17 and 18 proceeds via the 5-(2′-deoxyuridinyl)methyl radical (1). Because the phenyl selenide (2) produces cross-linked DNA upon NaIO4 oxidation, additional measures were taken to rule this process out. Cross-linking by the methide was envisioned to involve nucleophilic attack by N1 of dA (Scheme 4) on 20 (although we cannot rule out an SN1 process). An exogenous nucleophile could compete with dA for 20, but certainly not with 5-(2′-deoxyuridinyl)methyl radical (1). Reaction of monomeric 2 with NaIO4 (5 mM) in the presence of NaN3 gave rise to the expected alkyl azide (22), indicating that azide is compatible with the conditions in which cross-linking was carried out using this reagent.30 Addition of NaN3 (300 mM)
the expected higher barrier for a [2,3]-sigmatropic rearrangement faced by an allylic sulfoxide compared to that for a similar selenoxide.32 As expected, based upon this result, ISC is not observed upon treatment of 17 or 18 with NaIO4. This provides further indirect support that photochemical-mediated ISC formation from 17 and 18 proceeds via the 5-(2′-deoxyuridinyl)methyl radical (1). Because the phenyl selenide (2) produces cross-linked DNA upon NaIO4 oxidation, additional measures were taken to rule this process out. Cross-linking by the methide was envisioned to involve nucleophilic attack by N1 of dA (Scheme 4) on 20 (although we cannot rule out an SN1 process). An exogenous nucleophile could compete with dA for 20, but certainly not with 5-(2′-deoxyuridinyl)methyl radical (1). Reaction of monomeric 2 with NaIO4 (5 mM) in the presence of NaN3 gave rise to the expected alkyl azide (22), indicating that azide is compatible with the conditions in which cross-linking was carried out using this reagent.30 Addition of NaN3 (300 mM)  to a mixture of 14 and NaIO4 (5 mM) reduced the amount of cross-linked product by ∼60% regardless of whether the nucleophile is added before or after the phenyl selenide is oxidized (Table 3). However, the yield of ISC produced by photolysis of 14 or 18 was unaffected by this concentration of NaN3. These data suggest that ISC formation does not proceed via a methide when DNA containing 2 or 12 is photolyzed and that 5-(2′-deoxyuridinyl)methyl radical (1) remains the most viable species responsible for this product.
to a mixture of 14 and NaIO4 (5 mM) reduced the amount of cross-linked product by ∼60% regardless of whether the nucleophile is added before or after the phenyl selenide is oxidized (Table 3). However, the yield of ISC produced by photolysis of 14 or 18 was unaffected by this concentration of NaN3. These data suggest that ISC formation does not proceed via a methide when DNA containing 2 or 12 is photolyzed and that 5-(2′-deoxyuridinyl)methyl radical (1) remains the most viable species responsible for this product.
Scheme 5.
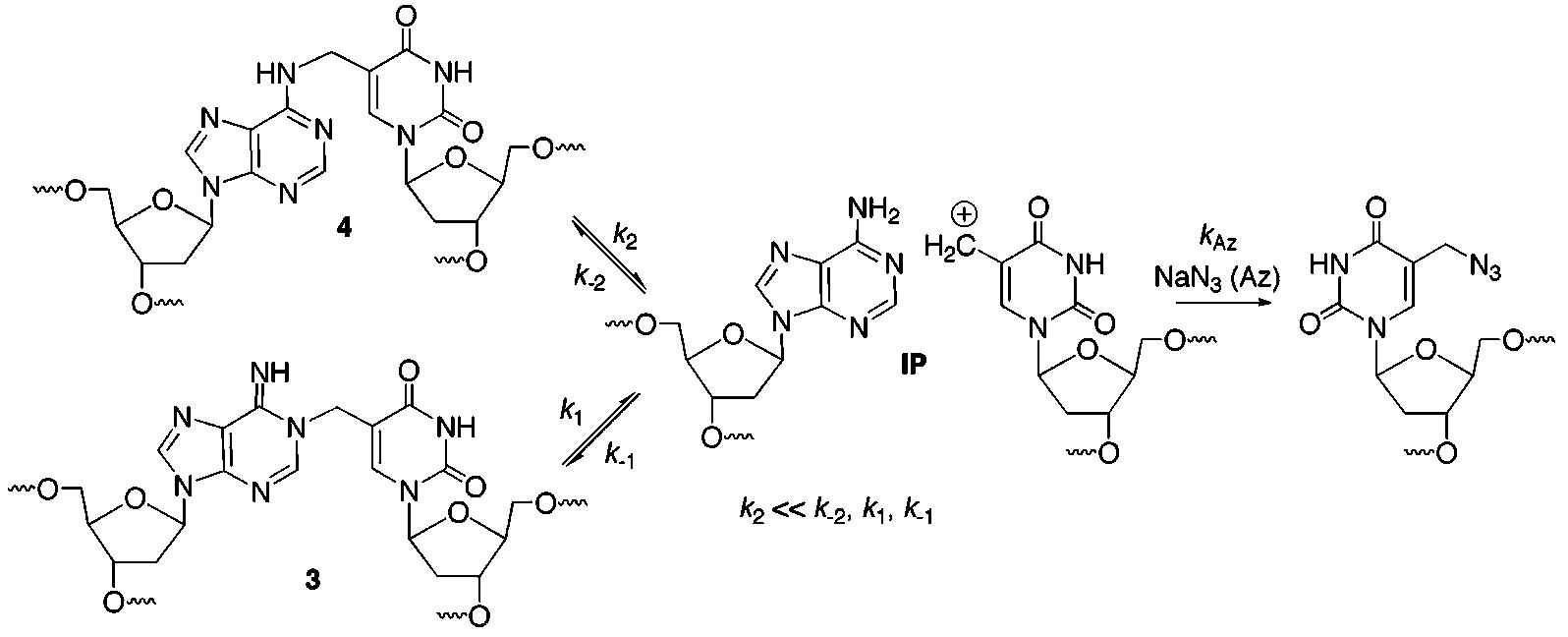
Detecting Reversibility of Interstrand Cross-Linking via Trapping with Azide
Table 3.
Effect of NaN3 on Interstrand Cross-Linking in Duplexes Subjected to Photolysis or NaIO4 Conditions
| % cross-linking |
||||
|---|---|---|---|---|
| duplex | method | no NaN3 | NaN3 (before oxid) | NaN3 (after oxid) |
| 14 | h ν | 29.9 ± 1.0 | 32.1 ± 1.5a | 29.3 ± 0.5a |
| 14 | NaIO4 | 49.2 ± 0.2 | 21.7 ± 0.7a | 18.6 ± 0.4a |
| 18 | h ν | 18.7 ± 1.4 | 17.5 ± 0.4b | |
[NaN3] = 0.3 M.
[NaN3] = 0.1 M.
What Is the Structure of the Cross-Linked Product? The N6-alkyl adduct (4) was isolated from photolyzed 14 following enzyme digestion.10 It was postulated that 4 resulted from rearrangement of an initially formed N1-adduct (3, Scheme 1). If 3 is the initially formed product, whether it rearranged as a result of the purification procedure is mechanistically relevant. The structure of the cross-linked product is also biologically important because the position in dA that is alkylated could
| (3) |
affect how the lesion is repaired.33,34 We sought to determine if the ISC product in solution is 3 or 4 by taking advantage of reversible alkylation of dA at N1.35 N6-Alkylation is irreversible under the mild incubation conditions used in this study. Duplex 14 was cross-linked under photochemical conditions and by reaction with NaIO4 in side-by-side reactions.24 After precipitation of the sodium periodate reaction, the cross-linked products were incubated at 37 °C under identical buffer conditions in the absence and presence of NaN3 (0.3 M) or GSH (5 mM). Aliquots were removed over the course of 4.6 days. In the absence of azide, ISC product(s) were stable regardless of how they were produced (Figure 2). The ISC product(s) were also stable in the presence of GSH (5 mM), which is present at a similar concentration in cells. However, in the presence of NaN3 the amount of ISC products decreased steadily (Figure 2) and followed first-order kinetics.30 The rate constant for decomposition of the cross-linked DNA produced via photolysis was (2.9 ± 0.1) × 10−6 s−1 (t1/2 = 66.0 h). The ISC generated by periodate decomposed at a rate that was within experimental error of the photochemically generated cross-linked DNA ((3.0 ± 0.1) × 10−6 s−1, t1/2 = 64.2 h). The similarity in the observed rate constants suggests that the cross-links produced by the two reaction conditions are the same. Moreover, the reversibility of the ISCs suggest that 4 is produced during the isolation procedure. Molecular modeling and the inherent nucleophilicity of N1 in dA lead us to suggest that 3 is the initial cross-linked product formed in duplex DNA from syn-5-(2′-deoxyuridinyl)-methyl radical (1) or the methide (20).10,35
Scheme 6.
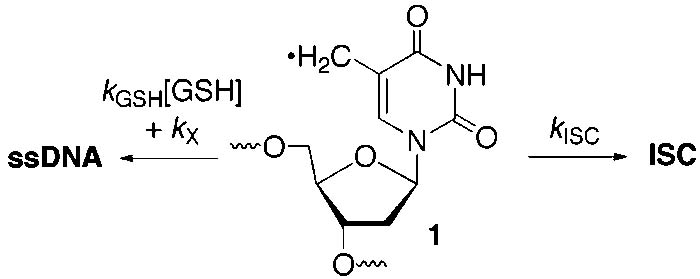
Competition between ISC Formation and GSH Trapping of 1
The disappearance of the cross-link product in the presence of azide can be explained via rate-limiting formation of an ion pair (IP, Scheme 5) or direct attack by the nucleophile on 3. In principle, these mechanisms are distinguishable from one another by measuring the observed rate constant for the disappearance of ISC as a function of azide concentration. Rate-limiting formation of an ion pair requires there to be a nonlinear dependence of kObs (eq 3) on azide concentration, provided that k−1 + k−2 are neither much greater nor much smaller than kAz[Az]. The rate constant for direct nucleophilic attack on the initially formed ISC product would vary linearly with azide concentration. We found that kObs varied linearly when [Az] varied from 50 to 500 mM (Figure 3). These data suggest that either azide reacts directly with 3 in the rate-determining step or that the relative magnitudes of k−1, k−2, and kAz[Az] prevent detecting the nonlinear dependence of kObs on azide ion concentration. Carrying out experiments in significantly higher concentrations of azide was deemed to be unfeasible.
Scheme 7.
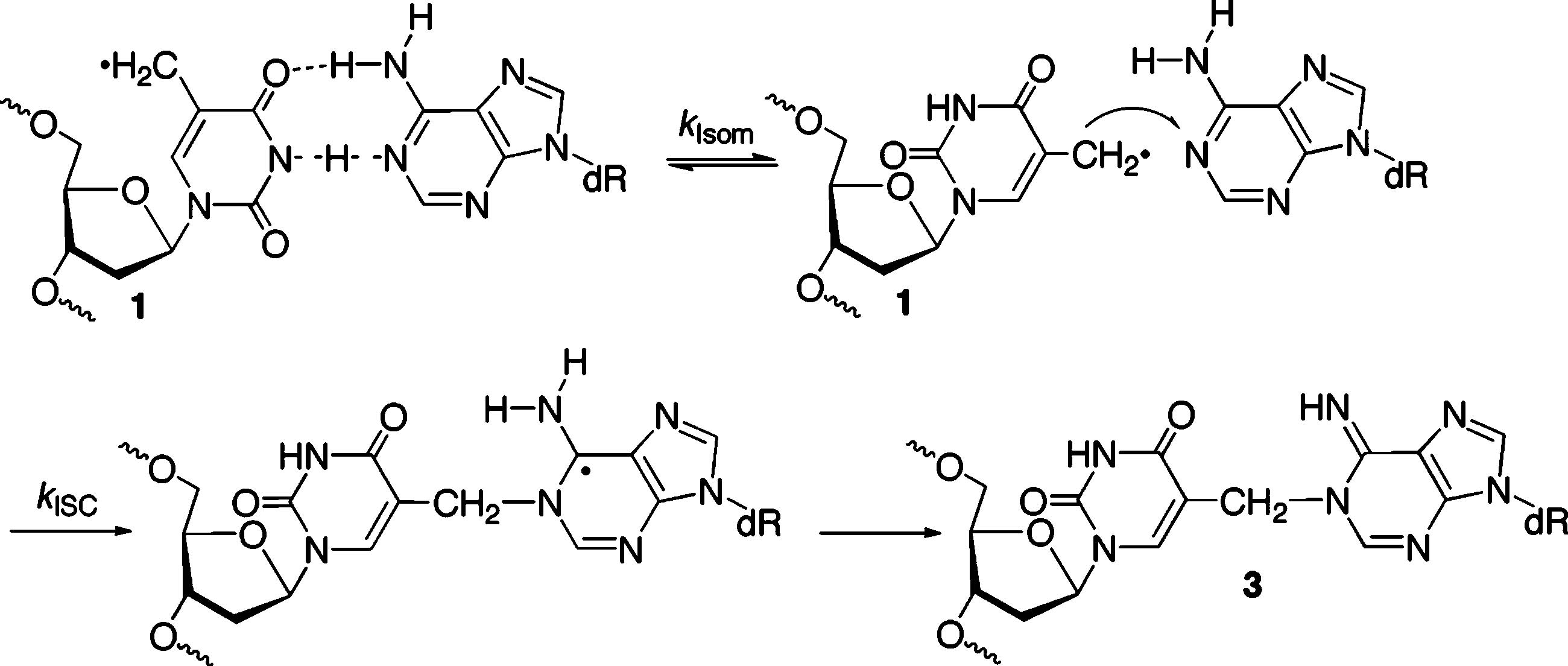
ISC Formation via Rotation about the Glycosidic Bond in the 5-(2′-Deoxyuridinyl)methyl Radical (1)
Figure 1.
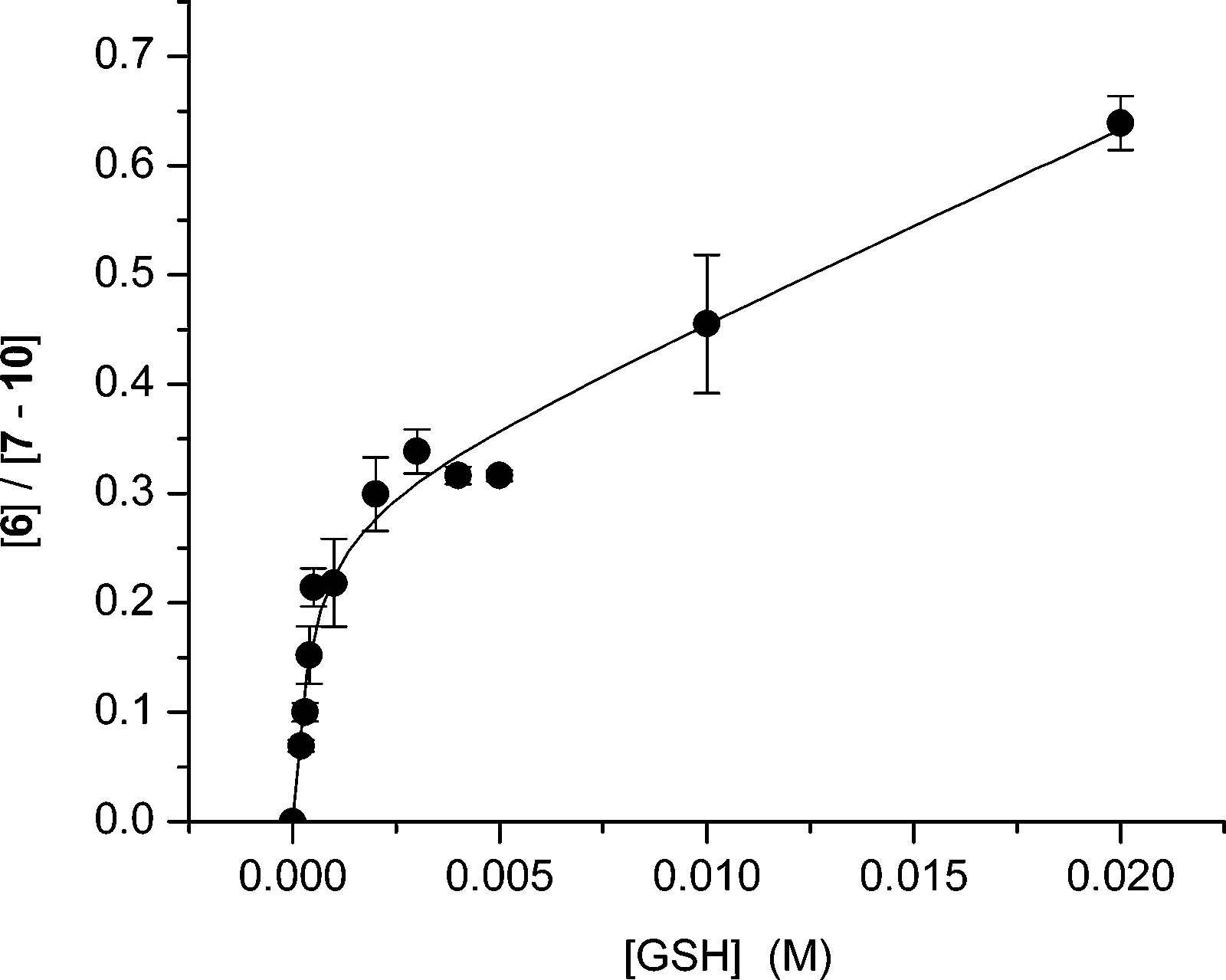
Product dependence of monomeric 5-(2′-deoxyuridinyl)methyl radical (1) derived from 12 under aerobic conditions as a function of GSH concentration.
Although cross-linking through N1 of dA (3) is the primary product, the ISC decomposition studies (Figure 2) do not eliminate the possibility that 3 is slowly converted to 4. Qualitative evidence for this was extracted by exploiting the anticipated difference in stability of the cross-link products. Although N6-alkylated deoxyadenosines are thermally stable, N1-alkylation is reversible.35-37 Therefore, we assumed that 3 would at least partially revert to single-stranded material upon heating. Indeed, heating a freshly cross-linked sample at 90 °C for 30 min converts approximately 25% of the product to single-stranded DNA (Figure 4). Similar treatment of aliquots of photochemically cross-linked material incubated at 37 °C taken over the course of 4 days revealed that the fraction of material converted to single-stranded DNA decreased over time (Figure 4), indicating that 3 does isomerize to 4 in solution. These data suggest that it will be important to elucidate the effects of 3 and 4 on DNA repair and replication.
Figure 2.
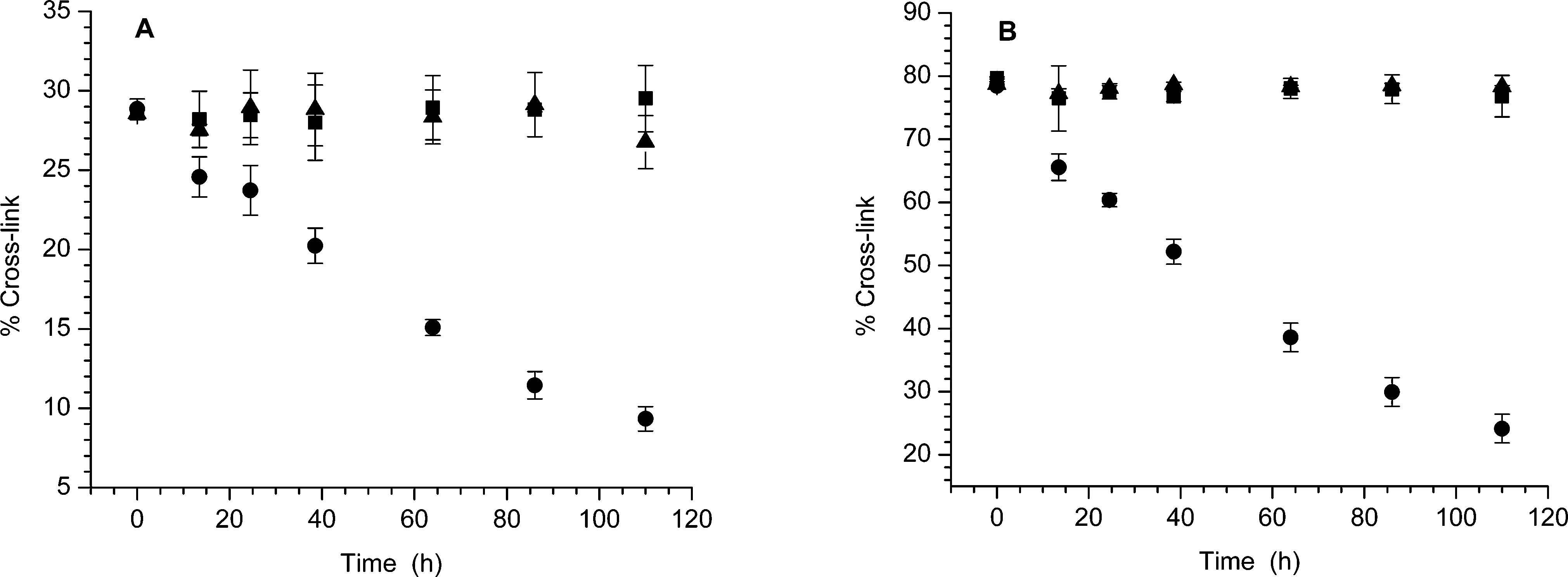
Stability of ISC from 14 in the absence and presence of NaN3 (0.3 M) or GSH (5 mM). (A) Photochemically generated ISC. (B) NaIO4 generated ISC. No additive (■), GSH (▲), NaN3 ([unk]).
How Fast Is Interstrand Cross-Link Formation by the 5-(2′-Deoxyuridinyl)methyl Radical (1)? The rate constant for ISC formation (kISC) was estimated by measuring the dependence of its formation on the concentration of GSH under anaerobic conditions (Scheme 6, eq 4). This method assumes that the photochemical precursor (2) is completely consumed and that single-stranded products result from GSH trapping of 1 (kGSH) and other pathways that do not involve thiol (kX). The lack of an effect of extended irradiation (up to 90 min) on ISC yield noted above attests to the complete photochemical conversion of 2. The ratio of single-stranded product (ssDNA) to ISC obtained from photolysis of 14 varies linearly as a function of GSH concentration (Figure 5). The estimated rate constant for ISC formation from the 5-(2′- deoxyuridinyl)methyl radical (1) is calculated to be 3.8 × 104 s−1 or 2.6 × 104 s−1 (2 = 0.995), depending upon whether one uses the upper limit for alkyl radical trapping by a thiol (1 × 107 M−1 s−1) from the literature or that estimated from Figure 1 (6.9 × 106 M−1 s−1), respectively.28
Figure 4.
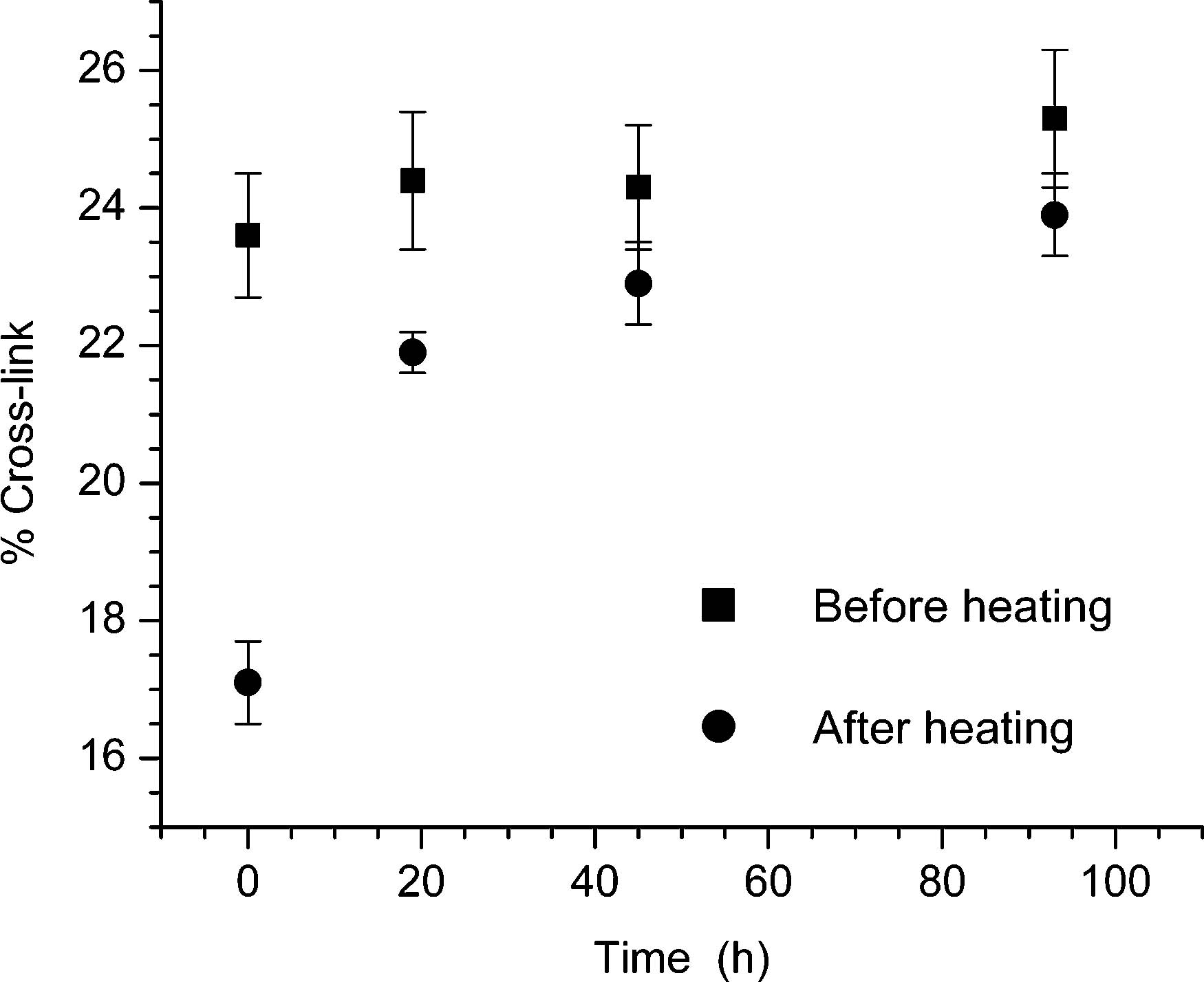
ISC stability to heating (90 °C, 30 min) upon incubation in aqueous buffer.
Figure 3.
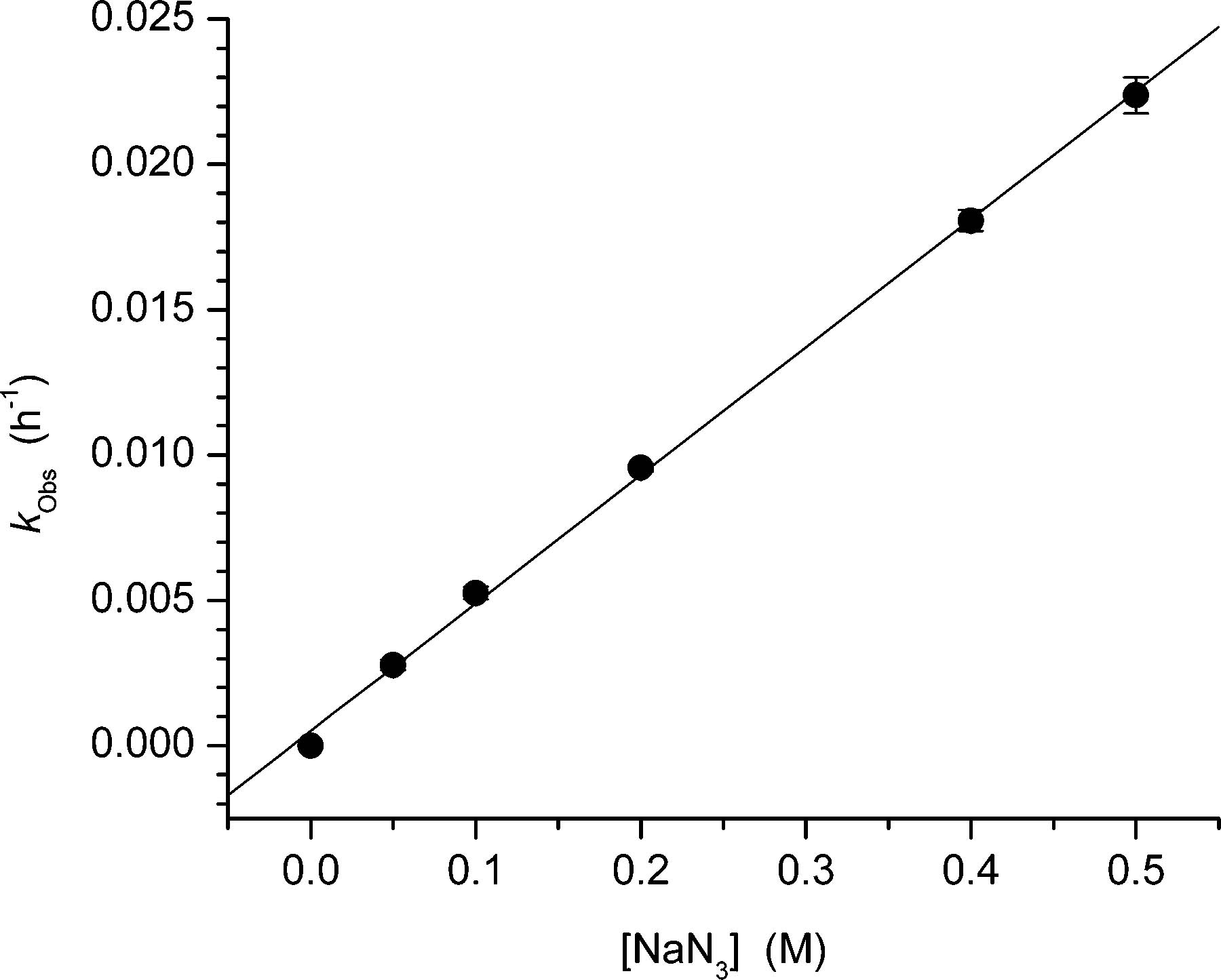
Dependence of the observed rate constant for the disappearance of ISC (kObs) on NaN3 concentration.
Interstrand cross-link formation requires that 1 adopt the syn-conformation and cannot be faster than rotation about the glycosidic bond (Scheme 7). The estimated rate constants for ISC formation from 1 are faster than what one might expect based upon NMR determination of A·T base pair dynamics. Although A·T base pair lifetimes less than 1 ms have been detected, typical values are in the millisecond (kIsom 103 s−1) range.38,39 Some factors that might help explain this difference are that the cross-linking experiments are carried out at higher temperature and lower pH. In addition, we cannot discount the possibility that the phenyl selenide precursor produces the 5-(2′-deoxyuridinyl)methyl radical (1) in a conformation that facilitates adoption of the syn-conformation.
Figure 5.
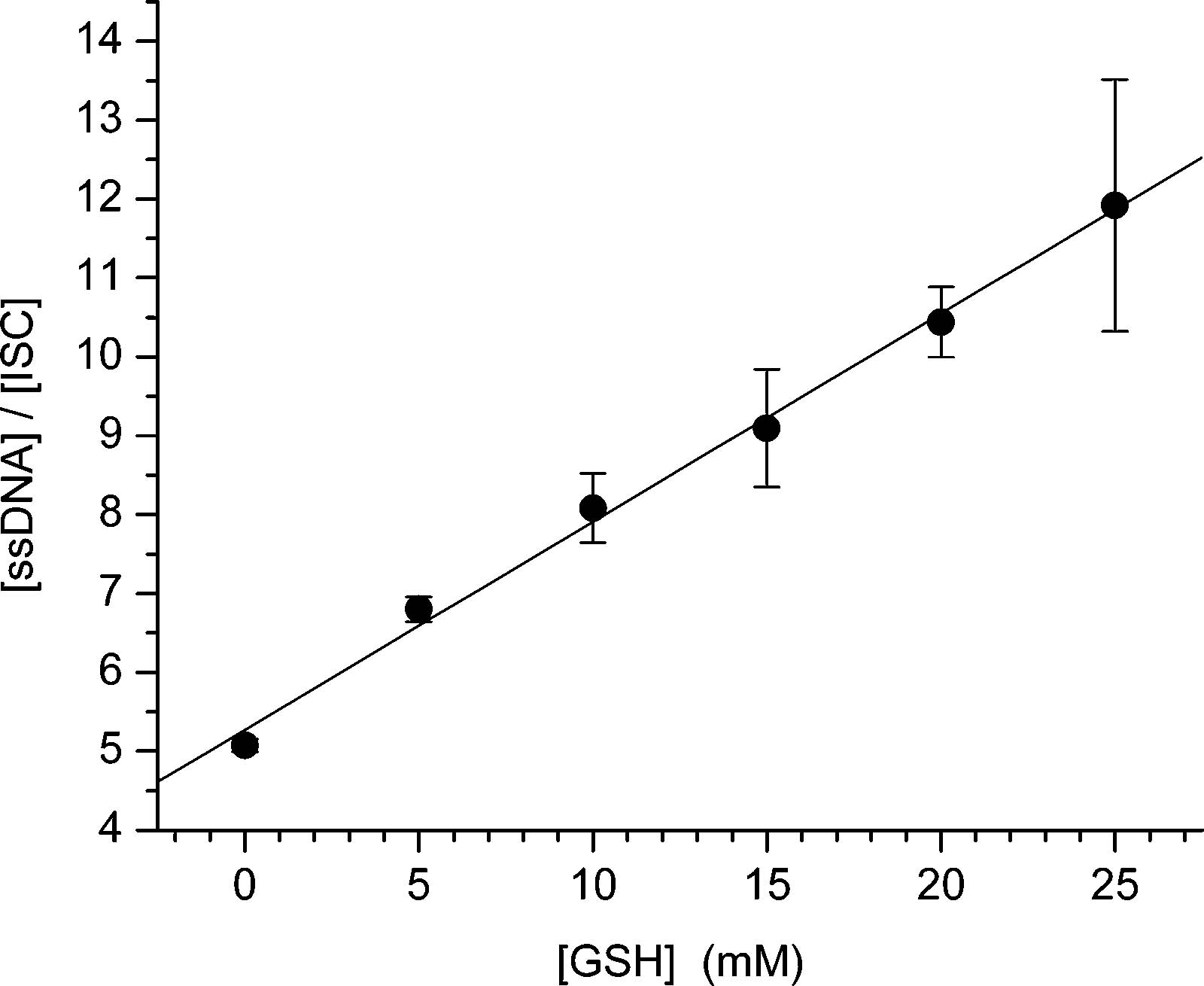
Ratio of ssDNA:ISC produced from photolysis of 14 as a function of GSH concentration.
Conclusions
A 5-(2′-deoxyuridinyl)methyl radical (1) was generated from three different photochemical precursors in a variety of sequence contexts. In all instances the radical produces interstrand cross-links (ISCs). ISC formation is independent of O2 because peroxyl radical formation (13) is reversible. Analysis of the stability of the ISC product indicates that the product resulting from radical addition to N1 of dA is the primary product but that this molecule isomerizes in solution to the thermodynamically more stable N6-adduct.35-37
| (4) |
The 5-(2′-Deoxyuridinyl)methyl radical (1) is the first example of a DNA radical that produces interstrand cross-links. ISC formation in the absence of O2 is potentially important given the hypoxic nature of tumor cells. The important biological effects of other cross-links provide the impetus to determine the consequences of 3, as well as its yield in DNA exposed to various forms of oxidative stress. In addition, the accessibility and low BDE of the respective carbon–hydrogen bond in thymidine make 1 an attractive target for synthetic DNA damaging agents.
Supplementary Material
Acknowledgment
We are grateful for support of this research by the National Institute of General Medical Sciences (GM-054996).
Footnotes
Supporting Information Available: Complete ref 4. Experimental procedures for the synthesis of the phosphoramidite precursors to 11 and 12 and the synthesis of 21 and 22. Histogram of Fe·EDTA cleavage of cross-linked 18. ESI-MS of modified oligonucleotides. HPLC and ESI-MS describing the formation of 22 from 20. Kinetic plots describing first-order decay of ISC in the presence of sodium azide. NMR spectra of previously unreported compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.DePinho RA. Nature. 2000;408:248–254. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- 2.Schar P. Cell. 2001;104:329–332. doi: 10.1016/s0092-8674(01)00220-3. [DOI] [PubMed] [Google Scholar]
- 3.Loeb LA, Loeb KR, Anderson JP. Proc. Natl. Acad. Sci. U.S.A. 2003;100:776–781. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kujoth GC, et al. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 5.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. Cell. 2005;120:435–567. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 6.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 7.von Sonntag C. The Chemical Basis of Radiation Biology; 1987; London: Taylor & Francis; [Google Scholar]
- 8.Pogozelski WK, Tullius TD. Chem. Rev. 1998;98:1089–1107. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 9.Burrows CJ, Muller JG. Chem. Rev. 1998;98:1109–1151. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 10.Hong IS, Greenberg MM. J. Am. Chem. Soc. 2005;127:3692–3693. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]
- 11.Schärer OD. Chem. Biol. Chem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- 12.Dronkert ML, Kanaar R. Mutat. Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 13.Brown JM, Wilson WR. Nat. Rev. Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 14.Douki T, Cadet J. Int. J. Radiat. Biol. 1999;75:571–581. doi: 10.1080/095530099140212. [DOI] [PubMed] [Google Scholar]
- 15.Wagner JR, van Lier JE, Johnston LJ. Photochem. Photobiol. 1990;52:333–343. doi: 10.1111/j.1751-1097.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- 16.Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free Radical Biol. Med. 2002;32:1102–1115. doi: 10.1016/s0891-5849(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 17.Anderson AS, Hwang JT, Greenberg MM. J. Org. Chem. 2000;65:4648–4654. doi: 10.1021/jo000271s. [DOI] [PubMed] [Google Scholar]
- 18.Romieu A, Bellon S, Gasparutto D, Cadet J. Org. Lett. 2000;2:1085–1088. doi: 10.1021/ol005643y. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Wang Y. J. Am. Chem. Soc. 2003;125:12795–12802. doi: 10.1021/ja034866r. [DOI] [PubMed] [Google Scholar]
- 20.Tallman KA, Greenberg MM. J. Am. Chem. Soc. 2001;123:5181–5187. doi: 10.1021/ja010180s. [DOI] [PubMed] [Google Scholar]
- 21.Carter KN, Greenberg MM. J. Am. Chem. Soc. 2003;125:13376–13378. doi: 10.1021/ja036629u. [DOI] [PubMed] [Google Scholar]
- 22.Hong IS, Carter KN, Greenberg MM. J. Org. Chem. 2004;69:6974–6978. doi: 10.1021/jo0492158. [DOI] [PubMed] [Google Scholar]
- 23.Hong IS, Greenberg MM. Org. Lett. 2004;6:5011–5013. doi: 10.1021/ol047754t. [DOI] [PubMed] [Google Scholar]
- 24.Hong IS, Greenberg MM. J. Am. Chem Soc. 2005;127:10510–10511. doi: 10.1021/ja053493m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hildenbrand K, Schulte-Frohlinde D. Int. J. Radiat. Biol. 1997;71:377–385. doi: 10.1080/095530097143996. [DOI] [PubMed] [Google Scholar]
- 26.Dussy A, Meggers E, Giese B. J. Am. Chem. Soc. 1998;120:7399–7403. [Google Scholar]
- 27.von Sonntag C, Schuchmann H-P. Angew. Chem., Int. Ed. Engl. 1991;30:1229–1253. [Google Scholar]
- 28.Newcomb M. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 29.Adam W, Arnold MA, Nau WM, Pischel U, Saha-Moeller CR. J. Am. Chem. Soc. 2002;124:3893–3904. doi: 10.1021/ja017600y. [DOI] [PubMed] [Google Scholar]
- 30. See Supporting Information.
- 31.Millard JT, Weidner MF, Kirchner JJ, Ribeiro S, Hopkins PB. Nucleic Acids Res. 1991;19:1885–1892. doi: 10.1093/nar/19.8.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reich HJ, Yelm KE, Wollowitz S. J. Am. Chem. Soc. 1983;105:2503–2504. [Google Scholar]
- 33.David SS, Williams SD. Chem. Rev. 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 34.Stivers JT, Jiang YL. Chem. Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 35.Veldhuyzen WF, Shallop AJ, Jones RA, Rokita SE. J. Am. Chem. Soc. 2001;123:11126–11132. doi: 10.1021/ja011686d. [DOI] [PubMed] [Google Scholar]
- 36.Veldhuyzen WF, Parde P, Rokita SE. J. Am. Chem. Soc. 2003;125:14005–14013. doi: 10.1021/ja036943o. [DOI] [PubMed] [Google Scholar]
- 37.Zhou Q, Rokita SE. Proc. Natl. Acad. Sci. U.S.A. 2003;100:15452–15457. doi: 10.1073/pnas.2533112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leroy J-L, Charretier E, Kochoyan M, Guéron M. Biochemistry. 1988;27:8894–8898. doi: 10.1021/bi00425a004. [DOI] [PubMed] [Google Scholar]
- 39.Guéron M, Leroy J. Methods Enzymol. 1995;261:383. doi: 10.1016/s0076-6879(95)61018-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.