

Abstract
Here we report crystal structures of dimethylglycine oxidase (DMGO) from the bacterium Arthrobacter globiformis, a bifunctional enzyme that catalyzes the oxidation of N,N-dimethyl glycine and the formation of 5,10-methylene tetrahydrofolate. The N-terminal region binds FAD covalently and oxidizes dimethylglycine to a labile iminium intermediate. The C-terminal region binds tetrahydrofolate, comprises three domains arranged in a ring-like structure and is related to the T-protein of the glycine cleavage system. The complex with folinic acid indicates that this enzyme selectively activates the N10 amino group for initial attack on the substrate. Dead-end reactions with oxidized folate are avoided by the strict stereochemical constraints imposed by the folate-binding funnel. The active sites in DMGO are ∼40 Å apart, connected by a large irregular internal cavity. The tetrahydrofolate-binding funnel serves as a transient entry–exit port, and access to the internal cavity is controlled kinetically by tetrahydrofolate binding. The internal cavity enables sequestration of the reactive iminium intermediate prior to reaction with tetrahydrofolate and avoids formation of toxic formaldehyde. This mode of channelling in DMGO is distinct from other channelling mechanisms.
Keywords: ‘active’ formaldehyde/Arthrobacter globiformis/channelling/crystal structure/dimethylglycine oxidase
Introduction
Mammalian sarcosine dehydrogenase (SDH) and dimethylglycine dehydrogenase (DMGDH) are mitochondrial enzymes involved in choline and 1-carbon metabolism. SDH plays a key role in the glycine/sarcosine regulatory cycle and, when defective, leads to sarcosinaemia, a rare autosomal metabolic defect characterized by elevated levels of sarcosine in blood and urine (Eschenbrenner and Jorns, 1999). DMGDH deficiency similarly leads to elevated levels of N,N-dimethylglycine in the blood, leading to a fish-like body odour and chronic muscle fatigue (Binzak et al., 2001). These highly related enzymes bind FAD covalently and catalyse in a reductive half-reaction the oxidation of sarcosine and dimethylglycine to the corresponding products glycine and sarcosine, respectively (Cook et al., 1984; Porter et al., 1985). In the absence of tetrahydrofolate (THF), formaldehyde is the second product of catalysis, formed by hydrolysis of a labile iminium intermediate. In the cell, toxic formaldehyde is not produced and these enzymes catalyse the formation of 5,10-methylene-THF (i.e. ‘active’ formaldehyde) from THF and the labile iminium intermediate. Electron-transferring flavoprotein (ETF) links the activities of SDH and DMGDH to the respiratory redox chain in an oxidative half-reaction that completes the catalytic cycle (Steenkamp and Husain, 1982).
The primary structure of these enzymes suggests the presence of two active sites located in different regions of the protein. A nucleotide-binding motif is found towards the N-terminus of SDH and DMGDH in a region that suggests a distant relationship to bacterial monomeric sarcosine oxidase (MSOX) (Trickey et al., 1999). The C-terminal sequence is highly similar to that of the T-protein (an aminomethyltransferase) of the glycine cleavage system that catalyses a THF-dependent reaction analogous to that of SDH and DMGDH (Fujiwara et al., 1984). A number of mutations in the T-protein cause non-ketotic hyperglycinaemia, a metabolic disorder with autosomal recessive inheritance, and these effect severe, frequently lethal, neurological symptoms in the neonatal period (Toone et al., 2000).
Many microorganisms utilize the common metabolites sarcosine or betaine as a source of carbon and energy, and glycine betaine is also an osmoprotectant (Boch et al., 1994). Dimethylglycine oxidase (DMGO) from Arthrobacter globiformis is part of the betaine catabolism pathway and is ∼30% identical to mammalian SDH and DMGDH (Meskys et al., 2001) (Figure 1A). DMGO is also related to the complex bacterial enzyme heterotetrameric sarcosine oxidase. DMGO uses molecular oxygen rather than ETF in the oxidative half-reaction (Basran et al., 2002) but, as with SDH and DMGDH, substrate oxidation is linked to the formation of 5,10-methylene-THF or formaldehyde, depending on the availability of THF.



Fig. 1. Structure of DMGO. (A) Alignment of DMGO with human sarcosine dehydrogenase (SHD), human dimethylglycine dehydrogenase (DMGDH) and human T-protein or aminomethyltransferase (AMT). Residues identified as essential to the activity of active site 1 or 2 are shaded in grey. Conserved residues are depicted in bold. The positions of mutations that cause non-ketotic hyperglycinaemia are identified by underlining the corresponding residue in AMT. (B) Stereo view of a DMGO monomer. The FAD-binding domain is represented in blue strands and green α-helices, while the folate-binding domain is depicted in red strands and purple α-helices. FAD and folinic acid are rendered in CPK spheres coloured yellow and blue, respectively. The molecular surface is rendered transparent and coloured according to functional domain: the flavin oxidase domain in blue, and the N5,N10-methylene-THF synthase domain in pink. (C) Structure of the DMGO tetramer. The upper view displays the molecular surface of the tetramer, each monomer represented in a distinct colour; the bottom view displays the backbone in ribbons colour coded as (B). Both orientations are related by a 90° rotation along the horizontal. This and other figures have been generated using the programs Turbo-Frodo (Roussel and Cambillau 1996), Pymol (DeLano, 2002) and GRASP (Nicholls et al., 1991).
We have determined the crystal structure of DMGO from A.globiformis to elucidate the spatial relationships between both active sites and the mechanism of catalysis. Formaldehyde is toxic and highly reactive, and transfer of the C1 unit to THF prevents hydrolysis of the iminium intermediate. In principle, the efficient coupling of substrate oxidation to the formation of 5,10-methylene-THF might be achieved by close juxtaposition of the active sites or by channelling of the relatively unstable iminium intermediate between active sites. The structure of DMGO indicates a large separation (∼40 Å) between the oxidase and methylene transferase active sites, which requires the channelling of the iminium intermediate through a large irregular internal cavity. By analogy, we propose that similar internal cavities exist in mammalian SDH and DMGDH that protect the cell from the products of iminium hydrolysis.
Results and discussion
Overall structure of DMGO
The crystal structure of DMGO was determined to 1.60 Å using the MIRAS approach (Table I; Figure 1). The enzyme appears as a dimer of tightly packed dimers, with the four monomers related by 222 crystallographic point symmetry. Each monomer comprises two functionally distinct regions, as suggested from analysis of the protein sequence. The N-terminal region, which is responsible for the oxidation of dimethylglycine, comprises residues 1–420. The C-terminal region (residues 421–830) contains the second active site responsible for the formation of 5,10-methylene-THF (Figure 1B). A search for similar folds in the Protein Data Base (PDB) indicated strong structural similarity between the N-terminal region of DMGO and MSOX (Trickey et al., 1999), polyamine oxidase (PAO) (Binda et al., 1999) and p-hydroxybenzoate hydroxylase (PHBH) (Schreuder et al., 1994). Significant similarities were not observed with the C-terminal region of DMGO and other proteins in the PDB.
Table I. Data collection and refinement statistics.
| Data set | Acetate bound | Folinic bound | Folate bound | K2PtCl4 | Pb(CH3COO)2 |
|---|---|---|---|---|---|
| X-ray source | ESRF ID14.4 | DESY X11 | DESY X11 | ESRFID14.4 | ESRFID14.4 |
| Space group | C222 | ||||
| Cell parameters | a = 71.4 Å | ||||
| b = 226.7 Å | |||||
| c = 120.7 Å | |||||
| Solvent content | 54.3% | ||||
| Resolution (Å)a | 15–1.6 (1.65–1.6) | 20–2.1 (2.15–2.1) | 15–1.65 (1.7–1.65) | 20–2.0 (2.05–2.0) | 20–2.0 (2.05–2.0) |
| Unique reflections | 114 767 | 46 564 | 106 504 | 60 161 | 64 856 |
| Redundancy | 3.2 | 2.5 | 5.4 | 4.3 | 3.5 |
| Completeness (%)a | 97.2 | 88.3 | 99.3 | 99.8 | 98.6 |
| Rsym (%)a,b | 9.6 (52.1) | 7.3 (34.4) | 6.7 (34.6) | 10.6 (42.6) | 9.8 (35.8) |
| I/σIa | 7.4 (2.1) | 6.2 (2.2) | 11.9 (2.8) | 9.2 (3.8) | 9.1 (3.2) |
| Sites | 3 | 4 | |||
| Phasing power | 0.60/0.54 | 0.66/0.58 | |||
| (acentric/centric) | |||||
| Cullis (acentric/centric/anomal.) |
|
|
|
0.93/0.86/0.95 |
0.93/0.90/0.97 |
| Refinement | |||||
| PDB code | 1PJ5 | 1PJ7 | 1PJ6 | ||
| Resolution (Å) | 15–1.60 | 20–2.10 | 15–1.65 | ||
| R-factor (%)c | 16.0 (28.0) | 15.9 (20.0) | 15.8 (21.2) | ||
| Rfree (%)c | 19.8 (30.0) | 22.2 (27.4) | 19.3 (24.2) | ||
| R.m.s.ds | |||||
| Bond length (Å) | 0.017 | 0.021 | 0.016 | ||
| Bond angles (°) | 1.578 | 1.837 | 1.634 | ||
| Average B-factor | 22.9 | 33.3 | 25.3 | ||
| Atoms | 8097 | 7104 | 7985 | ||
| Ramachandran plot | |||||
| Core (%) | 92.1 | 91.3 | 91.9 | ||
| Allowed (%) | 7.6 | 8.6 | 8.0 | ||
| Generously (%) | 0.3 | 0.1 | 0.1 | ||
aValues in parentheses are for the highest resolution shell.
aRsym(I) = ΣhΣi|Ih,i – <Ih>|ΣhΣiIh,i, where I is the observed intensity and <I> is the average intensity of multiple observations of symmetry-related reflections.
bRcryst = Σ||Fo| –|Fc||/Σ|Fo|; Rfree is the same as Rcryst but was calculated using a separate validation set of 5% of the reflections that were excluded from the refinement process.
Quaternary structure
The 222 point symmetry of the crystal coincides with the internal symmetry of the tetramer. The entire tetramer resembles a flattened tetrahedron, with approximate dimensions 120 × 120 × 70 Å. The C-terminal domains reside at the centre of the tetrahedron, and the FAD domains occupy the four corners (Figure 1C). The interfaces between a monomer and two crystallographically related molecules are very different in both contact area and surface complementarity. The strongest interaction occurs with the -x,y,-z symmetry-related monomer, and is almost entirely made by the folate-binding domains. The interface is extensive, burying 3083 Å2 (11.2%) of the molecular surface area per monomer (calculated using GRASP; Nicholls et al. 1991). The shape complementarity (SC) value obtained using a 1.7 Å probe is 0.76, which is at the high end of values reported for oligomeric proteins (0.70–0.76) (Lawrence and Colman, 1993). The contact between monomer A and the -x,-y,z symmetry-related monomer is made by both functional regions and extends over 1667 Å2 (6.1%) of the molecular surface. In this case, the SC value is only 0.53 owing to the splitting of the contact surface into several independent and predominantly hydrophobic patches. Despite this low SC value, the separation of the observed tetramer along the weaker interaction into two dimers would require the breaking of 3334 Å2 (2 × 1667 Å2). Analytical gel filtration experiments have indicated the size of the protein to be slightly larger than what is predicted for a homodimer (Basran et al. 2002). It is possible that at low protein concentrations, the protein dissociates into dimers. There is no indication for any allosteric effects in DMGO, and tetramerization probably improves the stability of the enzyme. None of the interface contact residues in DMGO are conserved in the mammalian dehydrogenases despite the overall high sequence identity. This suggests that the quaternary organization is likely to be different in the mammalian enzymes.
FAD binding and active site structure
The FAD domain contains a typical FAD-binding motif common to the PHBH and glutathione reductase (GR) class of flavoproteins (Figure 2A). Excursions of the polypeptide chain into the catalytic region, a two-part, eight-stranded and predominantly antiparallel β-sheet, twice interrupt this motif. Comparison of the FAD domain with other available structures using the program DALI (Holm and Sander, 1999) identified MSOX (Trickey et al., 1999) (PDB code 1el5) as the most highly related protein (Z-score of 38.2). MSOX shares 17% sequence identity with the FAD domain of DMGO and superimposes with a root mean square deviation (r.m.s.d.) of 2.6 Å for 360 structurally equivalent Cα atoms. The FAD cofactor is covalently bound to DMGO by a bond between the C8α methyl group of the FAD and His48 NE2 atom, similar to that observed for SDH, DMGDH and the membrane-bound fumarate reductase/succinate dehydrogenase family (Cecchini et al., 2002). In addition, the flavin isoalloxazine ring is slightly bent. Butterfly bending and covalent attachment of the flavin probably modulate the reduction potential of the flavin to favour the oxidation of substrate. Electron density was observed during refinement and in the initial experimental electron density maps that resembled an acetate ion bound close to the FAD isoalloxazine ring (Figure 2B). This ion clearly mimics the carboxylate group of the substrate dimethylglycine, and its presence is consistent with perturbation of the electronic absorption spectrum of DMGO during titration with acetate (Basran et al., 2002). The acetate anion is bound firmly in the active site by a network of hydrogen bonds and a single ionic interaction. The latter is formed with Arg252 (2.7 Å), while the second oxygen atom of acetate is hydrogen bonded to Tyr272 (2.9 Å) and Tyr415 (2.5 Å). Additionally, a water molecule hydrogen-bonds with both acetate (3.0 Å) and the side chain of Thr363, in addition to the oxygen backbone atom of Leu52 and the nitrogen backbone atom of Phe54. These interactions position the acetate methyl group in van der Waals contact with the isoalloxazine ring and the side chain of Trp361. The electron density for Trp361 indicates a high degree of side chain flexibility. Bound acetate was removed from the crystals by soaking in mother liquor that was devoid of acetate. In the crystal structure of the acetate-free form of DMGO, Trp361 occupies a range of different conformations (Figure 2C). Several other residues including Phe335 and Phe337 in the active site also adopted multiple conformations, indicating that the binding of acetate (and by analogy dimethylglycine) restrains motion. The aromatic side chain of Trp361 seems to function as a lid, closing the active site cavity upon substrate binding and probably excluding water. While most residues involved in binding acetate are conserved in mammalian SDH and DMGDH (with the notable exception of Tyr415, Trp361 and Thr363) (Figure 1A), only three residues involved in the active site architecture are conserved throughout the family of SDH-like proteins (i.e. His225, Tyr259 and Gly270). These residues do not contact the bound acetate and are unlikely to be involved in the formation of the Michaelis complex with dimethylglycine. His225 is hydrogen bonded both to the backbone oxygen of Ser271 (2.8 Å) and to Tyr259 (2.8 Å). The latter is within weak hydrogen-bonding distance (3.3 Å) of the FAD N5 atom. The relative position of both His225 and Tyr259 only allows for a glycine residue at position 270, and this residue is again conserved throughout the family. The observed hydrogen-bonding pattern suggests that His225 is positively charged if Tyr259 acts as hydrogen bond donor to the flavin N5 and hydrogen bond acceptor to His225. However, His225 is uncharged if Tyr259 is a hydrogen bond donor to His225.




Fig. 2. Structure of the flavin oxidase domain of DMGO. (A) Comparison of the flavin oxidase domain of DMGO (left panel) and monomeric sarcosine oxidase (right panel). The FAD cofactor and sodium ion bound to DMGO are represented in CPK spheres, coloured yellow and grey, respectively. (B) Stereo view of the flavin oxidase active site. All residues lining the active site are depicted in ball-and-stick representation coloured according to atom type. The bound acetate ion is surrounded by the σA-weighted 2Fo – Fc electron density map contoured at 1σ, and key hydrogen bonds are depicted by black dotted lines. A model for the amine part of dimethylglycine is represented with grey-coloured carbons. (C) Stereo view of the active site residues that change conformation upon binding of actetate. The σA-weighted 2Fo – Fc electron density map is shown contoured at 1σ for the residues in the unbound conformation. Residues are depicted in grey for the actetate-bound conformation and in atom type colour for the unbound conformation. The FAD cofactor is represented in red. (D) Schematic representation of a possible mechanism for amine oxidation by DMGO. The initial deprotonation of the substrate amine by the His225–Tyr259 catalytic dyad is followed by the attack of the substrate on the FAD C4 position. A proton is abstracted from the covalent intermediate methyl group by the N5, with concomitant reduction of the isoalloxazine ring and release of the dimethylglycine iminium ion.
We modelled dimethylglycine in the active site based on the position of the acetate ion (Figure 2B). In this model, the substrate amine nitrogen atom is in close contact with the FAD C4a atom, similar to that observed for structures of MSOX in complex with inhibitors. The mechanism of amine oxidation by flavoproteins has been debated intensively over the years, and a number of mechanisms have been proposed (Jang et al., 1999). Our model suggests a mechanism for flavin reduction in which an adduct is formed by nucleophilic attack at the flavin C4a position (Figure 2D). Formation of this adduct leads to the development of a very strong base at the flavin N5 (pKa of ∼30) which is sufficiently basic to abstract a proton from a substrate methyl group. This mechanism of C–H bond cleavage has been advanced for monoamine oxidase (Miller and Edmondson, 1999) and trimethylamine dehydrogenase (Basran et al., 2001). However, this mechanism requires that the substrate amine nitrogen is deprotonated. At physiological pH, substrate is predominantly protonated in free solution (pKa ∼10) and the enzyme needs to either disfavour the binding of the protonated form or deprotonate the substrate when bound at the active site. In DMGO, the conserved Tyr259–His225 dyad located close to the flavin N5 might form a proton shuttle that is able to abstract a proton from the substrate amine. Tyr259 is positioned close to the flavin N5 and the amine nitrogen of the substrate (Figure 2B). In the unprotonated form, His225 might abstract a proton from Tyr259 that in its turn facilitates proton abstraction from the substrate amine prior to nucleophilic attack of the substrate at the flavin C4a atom. Consistent with the mechanisms proposed for monoamine oxidase and trimethylamine dehydrogenase, the flavin N5 would then abstract a proton from the substrate methyl group in a concerted reaction to yield dihydroFAD and the iminium product of active site 1. Mutating His225 to glutamine or Tyr259 to phenylalanine both have substantial effects on the catalytic behaviour of DMGO. Both mutant enzymes retain some activity (kcat 4.35/s for H225Q and 0.047/s for Y259F compared with 10.6/s for the WT), suggesting that the dyad is not involved directly in proton abstraction from the substrate methyl group. The Km values of both mutant enzymes are elevated (Km 59 mM for H225Q and 47 mM for Y259F compared with 2.4 mM for the wild-type), suggesting an important role in either binding or generating the deprotonated substrate, consistent with our proposal. Precise evaluation of the role of these residues, however, must await detailed stopped-flow kinetic studies of flavin reduction by substrate, the assignment of kinetically influential ionizations in wild-type and mutant enzymes, and also structural analyses of mutant enzymes.
It is remarkable that MSOX catalyses an essentially identical reaction but uses a very different active site architecture (Trickey et al., 1999). In MSOX, the carboxylate groups of a number of inhibitors are bound by Arg52 and Lys348, and the central nitrogen group of these inhibitors is hydrogen bonded to the backbone oxygen of Gly344. In MSOX and PAO, a water molecule is held by hydrogen bonding between the FAD N5 and a conserved lysine residue, and is suggested to be involved in catalysis. In DMGO, a water molecule is not expected to bind close to either bound product or the substrate nitrogen atom. One might assume that by excluding an activated water molecule in the active site, DMGO and SDH-like enzymes might avoid the formation of formaldehyde by iminium hydrolysis in active site 1. However, the bacterial enzyme heterotetrameric sarcosine oxidase (Eschenbrenner et al., 2001) also generates 5,10-methylene-THF and, although structural information is lacking for this enzyme, sequence analysis suggests that it contains an MSOX-like active site. MSOX and DMGO clearly represent an unusual case of active site evolution within a similar protein scaffold, each finding highly similar, but geometrically distinct, solutions to catalysing the same chemistry. Of particular interest is the finding that the regulatory subunit of mitochondrial pyruvate dehydrogenase phosphatase that regulates the decarboxylase subunit of the pyruvate dehydrogenase multienzyme complex is related to members of the SDH family (Lawson et al., 1997). Sequence alignment of the bovine regulatory subunit of pyruvate dehydrogenase phosphatase with DMGO indicates that the former contains the essential residues His225, Tyr259 and Gly270, but lacks the carboxylate-binding residues. The regulation of the phosphatase is dependent on polyamines (e.g. spermidine), and these might be substrates of the regulatory subunit.
Folate-binding region
The C-terminal folate-binding region comprises three domains positioned in a cloverleaf-like arrangement around a central hole (Figure 3). No significant structural homology with known structures, including other folate-binding proteins, was identified. Domain 1 (residues 469–507 and 600–689) is a six-stranded antiparallel β-sheet containing a single Greek-key motif that is packed on one side by several α-helices. It is preceded by an irregular structure consisting of several small secondary elements packed in between both functional domains. Clear homology with the T-protein family is apparent from the start of domain 1 through to the C-terminus of the protein. Domain 1 is interrupted by a single excursion of the polypeptide chain to form domain 2 (residues 508–599), a five-stranded antiparallel β-sheet. Domains 1 and 2 have a similar topology and fold, with a Z-score of 6.3 and an r.m.s.d. of 1.8 Å for 76 Cα atoms, despite the lack of any obvious internal sequence homology (21%), suggesting early evolution by domain duplication followed by insertion of an additional β-strand in domain 1 (Figure 3B). The antiparallel β-sheets are loosely packed against one another, with the individual strands roughly aligned with the plane of the ring. The C-terminal domain 3 (residues 743–830) forms a distorted six-stranded jelly roll that packs perpendicular with the β-sheets of domains 1 and 2, closing the protein ring-like structure. The regions prior to domains 1 (residues 421–469) and 3 (residues 690–742) have irregular loop structures that pack against the three domains.


Fig. 3. Structure of the N5,N10-methylene-THF synthase domain of DMGO. (A) Stereo view of the N5,N10-methylene-THF synthase domain. The three domains are coloured blue (domain I), green (domain II) and red (domain III), with the bound folinic acid represented in atom type coloured spheres. (B) Overlay of domain II (in purple) on the structurally related domain I (in green). The positions of active site residues Glu658 and Asp552 are shown by a red and blue coloured asterisk, respectively. (C) Transparent molecular surface of the N5,N10-methylene-THF synthase domain on which mutations in the T-protein are mapped. The four mutations that cluster around the folate-binding funnel are represented in red. The green-coloured residue indicates the only residue mutated in the T-protein (E211K) within the homologous area that is not conserved in DMGO and is not involved in THF binding. The orientation of the N5,N10-methylene-THF synthase domain is similar to the views in (A) and (C).
Folate active site
The crystal structure of (S)-folinic acid (5-formyl-5,6,7,8-tetrahydrofolic acid) bound to DMGO clearly identifies active site 2 and the mode of tetrahydrofolate binding (Figure 4A and B). Folinic acid binds in the central cavity of the ring, effectively filling the cavity. The molecule is bound in a kinked conformation, with the pterin group facing the internal protein cavity. The binding of folinic acid is accompanied by several conformational changes in the enzyme, most of which result in improved hydrophobic packing with the folate aromatic ring system (Figure 4C). The hydrophobic side chains of Leu508, Tyr539 and Tyr651 adopt a different conformation on binding folate and become more ordered. Also, Arg694 adopts a new conformation and makes a new salt bridge with Asp705. The pterin group is strongly bound by a double hydrogen bond with the penultimate Glu658 (2.8 and 3.0 Å), in addition to an indirect hydrogen bond between the keto group and the acid through a water molecule. A similar interaction is observed between folinic acid and the formiminotransferase domain of formiminotransferase-cyclodeaminase (FCD; Kohls et al., 2000) (PDB 1QD1). Four additional hydrogen bonds are made between the pterin group and DMGO, in addition to hydrophobic stacking of the pterin ring against Met505 and Leu508. Of particular interest is the interaction between the oxygen backbone atom of Gly566 and the pterin N8 (2.8 Å), a contact that probably is responsible for selection of the reduced THF form. While folinic acid contains only one glutamyl group, both DMGO and related enzymes bind polyglutamylated THF in vivo. Several positively charged residues are located close to the entrance of the folate-binding funnel that could serve to bind the polyglutamate tail; none of these are conserved throughout the T-protein family. This indicates that in contrast to the conservation of those residues involved in binding the pterin, the flexible polyglutamate tail is differentially and perhaps only weakly bound.




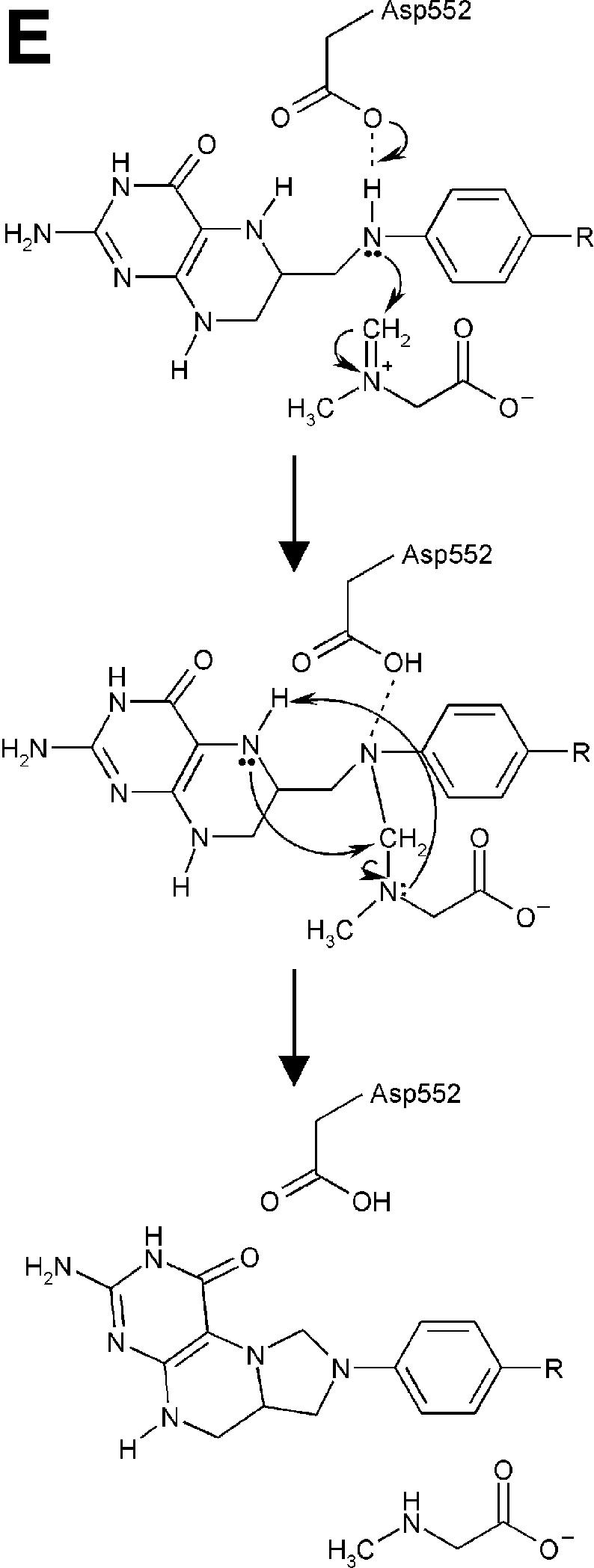
Fig. 4. Structure of the N5,N10-methylene-THF synthase active site. (A) Stereo view of the N5,N10-methylene-THF synthase active site with folinic acid bound. The Fo – Fc omit map is contoured at 3σ around the bound ligand. Residues lining the active site are in ball-and-stick representation coloured according to atom type. Key hydrogen bonds are depicted by black dotted lines. For comparison, the conformation of bound folic acid is represented in thin grey-coloured sticks. (B) Schematic representation of folinic acid bound to DMGO. Hydrogen bonds are indicated by dashed lines. (C) Stereo view of an overlay of the active site in unbound (all atoms grey coloured) and folinic acid-bound conformation (residues coloured according to residue type). (D) Stereo view of the N5,N10-methylene-THF synthase active site with folic acid bound. The Fo – Fc omit map is shown contoured at 3σ around the bound ligand; atoms are coloured according to atom type. Key hydrogen bonds are depicted by black dotted lines; for comparison, the conformation of bound folinic acid is represented in thin grey-coloured sticks. (E) Schematic representation of the N5,N10-methylene-THF synthase reaction. The bound THF attacks the iminium ion via the nucleophilic N10 group with concomitant proton abstraction by Asp552. The next step involves the attack by the N5 group on the covalent intermediate with concomitant deprotonation of the N5 by the nascent sarcosine. An alternative possibility is that the N5 position attacks first and donates a proton to the covalent intermediate, followed by the nucleophilic attack of the N10 group with release of sarcosine.
In close contact with the N5-formyl group is the side chain of Tyr651, the aromatic plane positioned perpendicular with respect to the pterin group. This residue probably stabilizes the initial binding of the iminium substrate through a cation–π interaction. In the folinic acid complex, the N10 nitrogen group is in direct hydrogen-bonding contact with Asp552 (3.0 Å), increasing the N10 nucleophilic character. This residue is conserved as either aspartate or glutamate in all T-protein-like enzymes. Further activation of the N10 functional group is achieved by a slight rotation of the amide group para to N10 with respect to the aromatic benzene ring, diminishing the electron-withdrawing properities. Conversion of the iminium intermediate to sarcosine and 5,10-methylene-THF is initiated by nucleophilic attack on the iminium by the N10 atom of the bound THF and concomitant protonation of Asp552 (Figure 4E). Following the initial nucleophilic attack, the covalent intermediate formed between THF and the iminium intermediate rearranges to form sarcosine and 5,10-methylene-THF. In contrast to catalytic mechanisms based on initial attack of the N5 functional group, this strategy presents the enzyme with a difficult problem. While the N5 group is only reduced in THF, there is no difference between THF and its oxidized precursor folic acid at the N10 amine function (Figure 4B). DMGO needs to discriminate between both molecules to avoid catalysing the initial N10 attack on folic acid, which would lead to a dead-end product owing to the lack of a second nucleophilic amine function (N5). The crystal structure of folic acid bound to DMGO indicates a major difference in the binding of folic acid and folinic acid (Figure 4D). While folic acid still binds in the folate-binding funnel, the molecule adopts a stretched conformation and is less deeply buried within the protein. None of the protein–pterin contacts observed in the complex with folinic acid are present, although the folic acid pterin group makes several (indirect) contacts with the same residues. Only one direct hydrogen bond is observed with the protein, between Glu658 and the amino pterin group (2.8 Å). This major difference in conformation is a likely consequence of the narrow folate-binding funnel and the strict requirement for a hydrogen bond donor at the Gly566 position, which safeguards the enzyme from catalysing the non-productive reaction with oxidized folate precursors.
Non-ketotic hyperglycinaemia is caused by a mutation in the genes encoding the components of the glycine cleavage multienzyme system (Toone et al., 2000). More than 80% of patients have defects in the gene encoding the P-protein, whereas the rest have defects in the gene encoding the T-protein. Several clinical mutations of the human T-protein can be mapped onto the DMGO C-terminal domain (Figure 3D). This reveals most mutations to cluster either around the folate-binding cavity or at the N-terminal region of the folate-binding domains. The latter region is situated on the pterin ring structure and is presumably involved in docking with the substrate H-protein. Mutations around the folate-binding site are likely to result in loss of either catalytic activity or the ability to bind folate. Four of the seven residues that on mutation lead to the onset of non-ketotic hyperglycinaemia (http://uwcmml1s.uwcm.ac.uk/uwcm/mg/search/132138.html) are conserved in DMGO (Figure 1A). These residues cluster around the folate-binding site: N568I (DMGO numbering), G698D, D705H and R747H. Asn568 is only conserved within the T-protein family, and is in close proximity to the bound pterin group. It is within hydrogen-bonding distance of the backbone oxygen of Asp552 and the adjacent side chain of Ser551. At the C-terminal end of domain 2, the polypeptide chain makes an abrupt turn at position 698, leading to a strict requirement for a glycine or alanine residue at this position. The conserved Asp705 makes an ionic interaction with Arg694 on binding folate. The fourth mutation, R747H, found near the folate-binding site, is probably involved in binding the polyglutamated region of folate.
A large channel connects both active sites
In the absence of THF, DMGO and the mammalian SDH and DMGDH enzymes generate formaldehyde during catalysis with their respective substrates. Hydrolysis of the iminium intermediate by water is probably non-enzymatic. The rates of hyrolysis of iminium cations similar to that formed by DMGO are in the region of per minute (Cordes and Jencks, 1963; for substituted benzylidene-1,1-dimethylethylamines). In the cell, efficient coupling of the reactions at active site 1 (iminium formation) and active site 2 (iminium decay on forming 5,10-methylene-THF) is needed if the accumulation of formaldehyde is to be avoided through iminium hydrolysis. Given that the rate of hydrolysis is slow, the enzyme only needs to retain the iminium ion prior to the more rapid reaction at active site 2. In principle, channelling of the iminium intermediate from active site 1 to active site 2, or close juxtaposition of the two active sites can achieve this. A distance of ∼40 Å separates both active sites, indicating the need to channel the iminium intermediate from active site 1 to active site 2. The active sites are internally connected by a large, irregular cavity (Figure 5A), which is made up in equal parts by both functional domains, with the ring structure of the C-terminal domain effectively docking onto a ring-shaped ridge of the flavin domain (Figure 5B and C). This architecture extends the folate-binding funnel to the FAD active site. The internal cavity is water filled with a volume of >5000 Å3 (calculated using GRASP; Nicholls et al., 1991), which is much larger than that needed to transport a single iminium intermediate. Although minor conformational changes occur at both active sites upon binding of substrate, the overall shape of the internal cavity remains the same, with no evidence of allosteric communication between active sites. Notably, there are no clear access channels for substrate entry or product release except in the region of the THF-binding funnel, which connects the internal cavity with bulk solvent. Since the binding of THF blocks this entry site, dimethylglycine is competing with THF for entrance into the central cavity. Kinetic studies indicate that steady-state production of hydrogen peroxide (a product of the reaction at active site 1) is compromised as the THF concentration is increased (Figure 6). This indeed suggests that the binding of THF in the folate-binding funnel hinders transfer of dimethylglycine to active site 1, consistent also with the lag phase observed in the kinetic progress curves. We attribute the lag phases to the establishment of a steady-state local concentration of dimethylglycine in the internal cavity. Without THF, formaldehyde is generated owing to hydrolysis of the iminium intermediate but, in the presence of THF, 5,10-methylene-THF is formed at the expense of formaldehyde, indicating strong coupling between the active sites. Rapid formation of 5,10-methylene-THF at active site 2 is essential if iminium hydrolysis is to be avoided in the cavity, and the relatively slow turnover kinetics of the FAD domain (∼10/s; Basran et al., 2002) should avoid excessive accumulation of the iminium intermediate in the internal cavity. Similarly, release of the iminium from the internal cavity to the external solution must be prevented if the cell is to be protected from the potentially harmful effects of formaldehyde. Again, iminium ions are competing with both THF and dimethylglycine for passage through the folate-binding funnel. DMGO probably avoids unwanted leakage of iminium ions by balancing the kinetic activity of both active sites, with the folate-binding funnel acting as a ‘kinetic filter’.


Fig. 5. Structure of the internal cavity of DMGO. (A) Stereo representation of a DMGO monomer with the internal cavity depicted as a transparent blue surface. The bound FAD cofactor and folate ligand are depicted in CPK spheres coloured yellow and green, respectively. (B) Footprint of the folate-binding region on the FAD domain. The molecular surface of the latter is depicted and coloured according to distance to the folate-binding region, white being within van der Waals distance and blue outside. The backbone of the folate-binding region is shown in red. (C) Footprint of the FAD-binding-domain on the folate-binding region. The molecular surface of the latter is depicted and coloured according to distance to the FAD domain, white being within van der Waals distance and blue outside. The backbone of the FAD domain is shown in green. (D) Schematic representation of channelling of the iminium ion in DMGO (scheme I) compared with channelling of intermediates in CPS or TS (scheme II) and the electrostatic highway in FCD (scheme III). Numbered spheres represent active sites, and black lines indicate the boundaries of the connecting tunnel/cavity.

Fig. 6. Main panel: effect of THF on hydrogen peroxide formation during steady-state turnover with DMGO. Reaction conditions: 70 nM DMGO, 40 IU of HRP, 0.05% o-dianisidine, 40 mM dimethylglycine and the required concentration of THF in 10 mM pyrophosphate buffer pH 8.5. Absorbance changes at 430 nm were monitored in the absence of THF (black trace); 10 µM THF (blue trace); 25 µM THF (red trace); 50 µM THF (green trace); 75 µM THF (magenta trace); and 100 µM THF (yellow trace). Inset: formation of 5,10-methylene-THF during dimethylglycine oxidation in the presence of THF. Reaction conditions: 7.5 µM DMGO, 0.04 U of 5,10-methylene-THF dehydrogenase, 2 mM NADP+, 40 mM dimethylglycine in 50 mM phosphate buffer pH 7.0. Reaction at A340 monitored in the presence of 100 µM THF (curve 1) and in the absence of THF (curve 2). All reactions were performed at 25°C.
The mechanism of action of DMGO can be likened to the gastrovascular cavity of simple Parazoan organisms (Figure 5D). The folate-binding funnel, ‘the mouth’, serves as a transient entry–exit port, and access to the internal cavity is controlled kinetically by THF binding. THF binds in the folate funnel and effectively seals the inner cavity, ensuring reaction of the iminium ion with THF. This stratagem is strikingly different from the mechanism of channelling in tryptophan synthase (TS) and carbamoyl phosphate synthase (CPS) (Huang et al., 2001). Channelling in TS and CPS can be likened to the more advanced alimentary canals of higher organisms, rather than the more primitive Parazoan gut (Figure 5D). TS and CPS have an entry point at active site 1 from which product leaves and is delivered to active site 2. A narrow internal tunnel connects both active sites, and the channelled intermediates are likely to be uncharged. In CPS, channelling avoids hydrolysis of a very unstable intermediate. Transfer of a stable and hydrophobic indole intermediate through the narrow tunnel in TS prevents loss through the cellular membrane. The ionic nature of the intermediate formed by DMGO is probably incompatible with transfer along a long and narrow tunnel. The iminium ion would have to be stripped from associated water molecules and counter-ions prior to entering the tunnel, and such a tunnel would require appropriate counter-charges placed along the path without creating unwanted high affinity binding sites. However, the creation of a large internal cavity through the fusion of two catalytic regions enables ‘unguided’ transfer by Brownian motion of the iminium intermediate between active sites and maintains ownership of the reactive iminium prior to its conversion to sarcosine and 5,10-methylene-THF at active site 2. This type of channelling is also distinct from the ‘electrostatic highway’ in FCD in which the negatively charged polyglutamylated folate intermediate is retained at the surface of the protein and moves along a positively charged surface patch to active site 2 (Kohls et al., 2000).
Conclusions
Amine oxidation is widespread in biology, and a number of different enzymes have evolved to catalyse these reactions. The crystal structure of DMGO from the bacterium A.globiformis represents the first crystal structure of a novel class of amine-oxidizing enzymes that generate 5,10-methylene-THF. The FAD-binding domain is related to MSOX, another amine-oxidizing enzyme. Despite catalysing almost identical reactions and being related in fold, the active sites of DMGO and MSOX are clearly different. The folate-binding region is the first structure for a member of the T-protein family from which the effects of clinical mutations leading to non-ketotic hyperglycinaemia can be rationalized. The complex with folinic acid indicates that these enzymes selectively activate the N10 amino group for initial attack on the substrate. Dead-end reactions with the oxidized precursor folic acid are avoided by the strict stereochemical constraints imposed by the folate-binding funnel. The crystal structures reveal a large internal cavity connecting both active sites, which is open to the outside solution only via the folate-binding funnel. The internal cavity with a single access point at active site 2 enables sequestration of the reactive iminium intermediate prior to reaction with folate and release into bulk solution. The mode of channelling in DMGO is distinct from the classical mechanisms seen in TS and CPS, and the ‘electrostatic highway’ in FCD.
Materials and methods
Crystallization
The protein was expressed in Escherichia coli and purified as described previously (Meskys et al., 2001). Long rod-like crystals were grown over several days using the sitting-drop vapour diffusion method by mixing 4 µl of a 15 mg/ml protein solution with 2 µl of reservoir solution. The reservoir solution contained 15% PEG 5000 monomethylether (MME) and 0.2 M MgCl2, and was buffered at pH 7.5 with 0.1 M HEPES. Acetate-bound crystals were obtained by exchange of the MgCl2 salt by Mg(CH3COO–)2. Crystals of DMGO complexed with either folic acid or folinic acid were obtained by soaking crystals in a ligand-saturated solution containing 15% PEG 5000 MME and 0.3 M NaCl for 10 min.
Data collection, structure solution and refinement
Data were collected at several X-ray sources (Table I) and processed and scaled using the HKL2000 package (Otwinowski and Minor, 1997). The structure was solved using MIRAS with two independent derivatives. Both derivatives were obtained by short soaking for 10 min in 10 mM heavy metal-supplemented reservoir solutions. Initial heavy metal sites were found using the program RSPS and verified using the program MLPHARE (CCP4, 1994). Additional sites were found through difference Fourier methods, and final phases were calculated using MLPHARE. The phases were extended to full resolution by density modification with DM and then submitted to automatic model building using WARP (Perrakis et al., 1999). The resultant model was refined further through consecutive rounds of manual rebuilding and refined using REFMAC5 (Murshudov et al., 1997). The structures of the ligand–DMGO complexes were solved by difference Fourier analysis using the coordinates for the refined unliganded form as a starting model. The refinement parameters are presented in Table I.
Kinetics
DMGO-catalysed production of hydrogen peroxide for both wild-type and mutant forms was monitored using a horseradish peroxidase (HRP)-coupled assay described previously (Basran et al., 2002) using dimethylglycine as reducing substrate. The production of 5,10-methylene-THF was monitored using 5,10-methylenetetrahydrofolate dehydrogenase as described (Kvalnes-Krick and Jorns, 1987)] with the following modifications: reactions were performed in an anaerobic environment [using a Jasco V-530 uv/vis spectrophotometer with a 1 cm light path housed in a glove box (Belle Technology)] and dimethylglycine (40 mM) was used as the reducing substrate.
Coordinates
Atomic coordinates and structure factors have been deposited with the Protein Data Bank (accession codes 1PJ5, 1PJ6 and 1PJ7 for the acetate-bound, folic acid-bound and folinic acid-bound form of DMGO, respectively).
Acknowledgments
Acknowledgements
We gratefully acknowledge the use of beamlines at the DESY EMBL outstation, Hamburg and at ESRF, Grenoble. We are grateful to Dr Rolandas Meskys (Institute of Biochemistry, Vilnius, Lithuania) for supplying the expression clone encoding DMGO. The work was funded by the Biotechnology and Biological Sciences Research Council and the Lister Institute of Preventive Medicine. N.S.S. is a Lister Institute Research Professor.
References
- Basran J., Sutcliffe,M.J. and Scrutton,N.S. (2001) Optimising the Michaelis complex of trimethylamine dehydrogenase: identification of interactions that perturb the ionization of substrate and facilitate catalysis with trimethylamine base. J. Biol. Chem., 276, 42887–42892. [DOI] [PubMed] [Google Scholar]
- Basran J., Bhanji,N., Basran,A., Nietlispach,D., Mistry,S., Meskys,R. and Scrutton,N.S. (2002) Mechanistic aspects of the covalent flavoprotein dimethylglycine oxidase of Arthrobacter globiformis studied by stopped-flow spectrophotometry. Biochemistry, 41, 4733–4743. [DOI] [PubMed] [Google Scholar]
- Binda C., Coda,A., Angelini,R., Federico,R., Ascenzi,P. and Mattevi,A. (1999) A 30-angstrom-long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure Fold. Des., 7, 265–276. [DOI] [PubMed] [Google Scholar]
- Binzak B.A. et al. (2001) Cloning of dimethylglycine dehydrogenase and a new human inborn error of metabolism, dimethylglycine dehydrogenase deficiency. Am. J. Hum. Genet., 68, 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch J., Kempf,B. and Bremer,E. (1994) Osmoregulation in Bacillus subtilis: synthesis of the osmoprotectant glycine betaine from exogenously provided choline. J. Bacteriol., 176, 5364–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchini G., Schroder,I., Gunsalus,R.P. and Maklashina,E. (2002) Succinate dehydrogenase and fumarate reductase from Escherichia coli.Biochim. Biophys. Acta, 1553, 140–157. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cook R.J., Misono,K.S. and Wagner,C. (1984) Identification of the covalently bound flavin of dimethylglycine dehydrogenase and sarcosine dehydrogenase from rat liver mitochondria. J. Biol. Chem., 259, 12475–12480. [PubMed] [Google Scholar]
- Cordes E.H. and Jencks,W.P. (1963) The mechanism of hydrolysis of Schiff bases derived from aliphatic amines. J. Am. Chem. Soc., 85, 2843–2848. [Google Scholar]
- DeLano W.L. (2002) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA. http://www.pymol.org [Google Scholar]
- Eschenbrenner M. and Jorns,M.S. (1999) Cloning and mapping of the cDNA for human sarcosine dehydrogenase, a flavoenzyme defective in patients with sarcosinemia. Genomics, 59, 300–308. [DOI] [PubMed] [Google Scholar]
- Eschenbrenner M., Chlumsky,L.J., Khanna,P., Strasser,F. and Jorns,M.S. (2001) Organization of the multiple coenzymes and subunits and role of the covalent flavin link in the complex heterotetrameric sarcosine oxidase. Biochemistry, 40, 5352–5367. [DOI] [PubMed] [Google Scholar]
- Fujiwara K., Okamura-Ikeda,K. and Motokawa,Y. (1984) Mechanism of the glycine cleavage reaction. Further characterization of the intermediate attached to H-protein and of the reaction catalyzed by T-protein. J. Biol. Chem., 259, 10664–10668. [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1999) Protein folds and families: sequence and structure alignments. Nucleic Acids Res., 27, 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Holden,H.M. and Raushel,F.M. (2001) Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem., 70, 149–180. [DOI] [PubMed] [Google Scholar]
- Jang M.H., Basran,J., Scrutton,N.S. and Hille,R. (1999) The reaction of trimethylamine dehydrogenase with trimethylamine. J. Biol. Chem., 274, 13147–13154. [DOI] [PubMed] [Google Scholar]
- Kohls D., Sulea,T., Purisima,E.O., MacKenzie,R.E. and Vrielink,A. (2000) The crystal structure of the formiminotransferase domain of formiminotransferase-cyclodeaminase: implications for substrate channeling in a bifunctional enzyme. Structure Fold. Des., 8, 35–46. [DOI] [PubMed] [Google Scholar]
- Kvalnes-Krick K. and Jorns,M.S. (1987) Interaction of tetrahydrofolate and other folate derivatives with bacterial sarcosine oxidase. Biochemistry, 26, 7391–7395. [DOI] [PubMed] [Google Scholar]
- Lawrence M.C. and Colman,P.M. (1993) Shape complementarity at protein–protein interfaces. J. Mol. Biol., 234, 946–950. [DOI] [PubMed] [Google Scholar]
- Lawson J.E., Park,S.H., Mattison,A.R., Yan,J. and Reed,L.J. (1997) Cloning, expression and properties of the regulatory subunit of bovine pyruvate dehydrogenase phosphatase. J. Biol. Chem., 272, 31625–31629. [DOI] [PubMed] [Google Scholar]
- Meskys R., Harris,R.J., Casaite,V., Basran,J. and Scrutton,N.S. (2001) Organization of the genes involved in dimethylglycine and sarcosine degradation in Arthrobacter spp.: implications for glycine betaine catabolism. Eur. J. Biochem., 268, 3390–3398. [DOI] [PubMed] [Google Scholar]
- Miller J.R. and Edmondson,D.E. (1999) Structure–activity relationships in the oxidation of para-substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry, 38, 13670–13683. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin,A.A. and Dodson,E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris,R. and Lamzin,V.S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol., 6, 458–463. [DOI] [PubMed] [Google Scholar]
- Porter D.H., Cook,R.J. and Wagner,C. (1985) Enzymatic properties of dimethylglycine dehydrogenase and sarcosine dehydrogenase from rat liver. Arch. Biochem. Biophys., 243, 396–407. [DOI] [PubMed] [Google Scholar]
- Roussel A. and Cambillau,C. (1996) TURBO-FRODO Manual. AFMB-CNRS, Marseille. [Google Scholar]
- Schreuder H.A., Mattevi,A., Obmolova,G., Kalk,K.H., Hol,W.G., van der Bolt,F.J. and van Berkel,W.J. (1994) Crystal structures of wild-type p-hydroxybenzoate hydroxylase complexed with 4-amino benzoate,2,4-dihydroxybenzoate and 2-hydroxy-4-aminobenzoate and of the Tyr222Ala mutant complexed with 2-hydroxy-4-aminobenzoate. Evidence for a proton channel and a new binding mode of the flavin ring. Biochemistry, 33, 10161–10170. [DOI] [PubMed] [Google Scholar]
- Steenkamp D.J. and Husain,M. (1982) The effect of tetrahydrofolate on the reduction of electron transfer flavoprotein by sarcosine and dimethylglycine dehydrogenases. Biochem. J., 203, 707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toone J.R., Applegarth,D.A., Coulter-Mackie,M.B. and James,E.R. (2000) Biochemical and molecular investigations of patients with nonketotic hyperglycinemia. Mol. Genet. Metab., 70, 116–121. [DOI] [PubMed] [Google Scholar]
- Trickey P., Wagner,M.A., Jorns,M.S. and Mathews,F.S. (1999) Monomeric sarcosine oxidase: structure of a covalently flavinylated amine oxidizing enzyme. Structure Fold. Des., 7, 331–345. [DOI] [PubMed] [Google Scholar]