

Abstract
NFAT and SRF are important in the regulation of proliferation and cytokine production in lymphocytes. NFAT activation by the B cell receptor (BCR) occurs via the PLCγ–Ca2+–calcineurin pathway, however how the BCR activates SRF is unclear. We show here that like NFAT, BCR regulation of SRF occurs via an Src–Syk–Tec–PLCγ–Ca2+ (Lyn–Syk–Btk–PLCγ–Ca2+) pathway. However, SRF responds to lower Ca2+ and is less dependent on IP3R expression than NFAT. Ca2+-regulated calcineurin plays a partial role in SRF activation, in combination with diacylglycerol (DAG), while is fully required for NFAT activation. Signals from the DAG effectors protein kinase C, Ras and Rap1, and the downstream MEK–ERK pathway are required for both SRF and NFAT; however, NFAT but not SRF is dependent on JNK signals. Both SRF and NFAT were also dependent on Rac, Rho, CDC42 and actin. Finally, we show that Ca2+ is not required for ERK activation, but instead for its association with nuclear areas of the cell. These data suggest that combinatorial assembly of signaling pathways emanating from the BCR differentially regulate NFAT and SRF, to activate gene expression.
Keywords: calcineurin/DAG/ERK/IP3 receptor/Tec kinase
Introduction
Stimulation of lymphocyte antigen receptors (B cell receptor, BCR or T cell receptor, TCR) rapidly induces expression of a set of immediate early genes such as c-fos and egr-1 (Kaptein et al., 1995; Niiro and Clark, 2002), which are important for lymphocyte development and immune response. The serum response element (SRE) is a promoter element required for regulation of many immediate early genes (Johansen and Prywes, 1995; Chai and Tarnawski, 2002). The SRE contains a core CC(A/T)6GG motif (CArG box) which is specifically bound by the transcription factor serum response factor (SRF) (Treisman, 1986; Greenberg et al., 1987; Norman et al., 1988). SRF is ubiquitously expressed and has a DNA binding and dimerization domain at its N-terminal region and transactivation domain at its C-terminal region, both of which are required for transcriptional activity (Johansen and Prywes, 1995; Treisman, 1986; Norman et al., 1988; Chai and Tarnawski, 2002).
SRF has been extensively studied in various non-lymphocyte cell types using c-fos transcription as a model. During growth factor- and stress-induced c-fos expression, the ternary complex factor (TCF) of the Ets family of proteins including Elk-1, SAP-1 and SAP-2, is required to form a ternary complex with an SRF dimer. TCF alone cannot bind DNA and association of TCF to an Ets motif adjoining the CArG box at the c-fos SRE in response to growth factor stimulation requires DNA-bound SRF (Shaw et al., 1989; Schroter et al., 1990; Shaw, 1992). SRF can also regulate gene transcription independently of TCF. Serum or lysophosphatidic acid activates gene transcription by SRF independent of TCF in NIH 3T3 cells, mediated by RhoA and actin rearrangement (Hill et al., 1994; Sotiropoulos et al., 1999). Increases in intracellular Ca2+ by depolarization or Ca2+ ionophore ionomycin leads to SRF-dependent gene transcription mediated by CaMK II and IV in PC12 cells (Miranti et al., 1995).
The mechanism by which SRF regulates gene expression has been extensively investigated. It has been suggested that SRF directly regulates gene transcription rather than indirectly by tethering other transcription factors (Hill et al., 1993, 1994). Regulation of SRF activity through DNA binding seems unlikely since SRF has been found to associate with DNA constitutively and the DNA binding activity does not change upon stimulation in many cell types including Jurkat T cells (Prywes and Roeder, 1986; Treisman, 1986; Greenberg et al., 1987; Charvet et al., 2002). Signal-induced phosphorylation has been observed at multiple sites in SRF, however the significance of phosphorylation remains controversial (Johansen and Prywes, 1995).
BCR signals are essential for proliferation and maturation of B-lymphocytes. The BCR activates the Src, Syk and Tec families of non-receptor tyrosine kinases. Activated Src and Syk family kinases in concert lead to the activation of Tec family tyrosine kinase Btk (Afar et al., 1996; Rawlings et al., 1996; Baba et al., 2001). Downstream of these tyrosine kinases, a number of other important signaling molecules are activated including Ras GTPase, Rho small GTPases, protein kinase C (PKC) and intracellular Ca2+ increase (Hardy and Hayakawa, 2001). The Ca2+ response to BCR signals in B cells is regulated by phospholipase Cγ2 (PLCγ2), whose activation is dependent on Syk and Btk (Takata and Kurosaki, 1996; Rodriguez et al., 2001). PLCγ2 generates two messengers diacylglycerol (DAG) and inositol (1,4,5)-triphosphate (IP3). DAG can activate PKC as well as Ras and Rap1 (Ebinu et al., 2000; McLeod and Gold, 2001). IP3 induces intracellular Ca2+ release from intracellular Ca2+ stores through IP3 receptors (IP3Rs) (Berridge, 1993). Ca2+ signals regulate distinct downstream effectors depending on its amplitude, duration and frequency (Dolmetsch et al., 1997; De Koninck and Schulman, 1998).
It has been previously reported that the B cell superantigen Protein A activates the SRE in B cells (Das et al., 1999). In this report, we analyzed the regulation of SRF by BCR signals using DT40 cells, a chicken B cell line, as a model. We found that SRF is activated by BCR signaling via the Lyn–Syk–Btk–PLCγ pathway and that Ca2+ is essential for BCR-mediated SRF activation. However, while BCR mediates activation of both SRF and NFAT by the same Lyn–Syk–Btk–PLCγ pathway, these transcription factors are differentially sensitive to effectors of PLCγ such as Ca2+ levels and effectors of DAG. Thus NFAT activation is fully dependent on the presence of the IP3R, while SRF activation was only partially dependent in this protein and responds to lower levels of Ca2+ than NFAT. While the phosphatase calcineurin (CN), a downstream effector of Ca2+, is required for NFAT activation, it was only partially required for the activation of SRF. We also report that the DAG effector PKC, as well as Rap1 and Ras are involved in the activation of SRF and NFAT. However, whereas activation of both SRF and NFAT requires the ERK pathway, they differed in their requirement for the JNK pathway. Finally, we show that another potential role for Ca2+ in regulating SRF and NFAT may be in regulating BCR-induced nuclear association of ERK.
Results
Lyn and Syk are required for SRF activation by BCR ligation
To investigate the mechanism by which the BCR activates SRF, we chose DT40 cells, a chicken B cell line as a model, since the signaling pathway in these cells is well documented, and the availability of a number of mutants lacking signaling molecules in the BCR signaling pathway (Winding and Berchtold, 2001). Wild-type (WT) DT40 cells were transiently transfected with a SRF–luciferase reporter plasmid containing five tandem copies of a CArG sequence thus allowing it to be responsive to SRF independent of TCF (Schroter et al., 1990; Shaw, 1992). Cross-linking the BCR with anti-chicken IgM results in an increase of luciferase activity of ∼12-fold on average over non-stimulated controls (Figure 1A), showing that SRF is activated in response to BCR ligation. By contrast, these cells responded poorly to serum, while the same reporter was very responsive in Chinese hamster ovary cells (Supplementary figure 1A and B, available at The EMBO Journal Online), in agreement with other studies (Hill et al., 1994). We also confirmed the specificity of the reporter by demonstrating that a dominant negative mutant of SRF, SRFpm1 (Croissant et al., 1996), blocked BCR activation of the reporter in a dose-dependent manner (Figure 1A).


Fig. 1. BCR-mediated SRF activation requires Lyn, Syk, Btk and PLCγ2. (A) WT DT40 B cells were transiently transfected with SRF– luciferase reporter plasmid along with empty vector, or increasing concentrations of plasmid carrying dominant negative SRF (SRFpm1, 10 and 20 µg DNA). The cells were then either stimulated with anti-IgM (black bar) or left unstimulated (open bar) then lysed for luciferase assay. *P < 0.05 versus empty vector control. (B) SRF transcriptional activity was determined in WT and mutant DT40 cells lacking Lyn, Syk, Btk and PLCγ2. The cells were either stimulated with anti-IgM (black bars), 10 ng/ml PMA/100 nM ionomycin (gray bars) or left unstimulated (open bars). *P < 0.05 versus WT cells. (C) Lipase activity of PLCγ is required for activation of SRF. SRF transcriptional activity was determined in PLCγ–/– DT40 cells transfected with empty vector, WT PLCγ, or lipase-deficient PLCγ, followed by stimulation with anti-IgM.
Src-family kinases Lyn, Blk and Fyn, and Syk-family kinase, Syk in B cells, initiate BCR signals. In DT40 cells Lyn and Syk are the only Src and Syk kinases expressed respectively. Both Lyn and Syk are required for initiation of BCR signaling in these cells (Takata et al., 1994). To confirm that activation of SRF by BCR cross-linking also required Lyn and Syk, we determined the transcriptional activation of SRF in Lyn–/– and Syk–/– DT40 cells and found that its activation was almost completely abrogated in Lyn–/– cells and completely abrogated in Syk–/– cells (Figure 1B). SRF was normally responsive to phorbol ester PMA and calcium ionophore ionomycin in these mutant cell lines (Figure 1B), suggesting that the downstream signaling machinery downstream of the BCR leading to activation of SRF is intact. These experiments demonstrate that BCR signals activate the SRF transcription factor and that Lyn and Syk kinases are required for this pathway.
Btk–PLCγ is the predominant downstream pathway leading to SRF in B cells
Btk, a Tec family kinase lies downstream of Lyn and Syk in the BCR signaling pathway and is essential for B cell development and activation (Kurosaki, 1999). Using Btk–/– DT40 cells, we found that BCR-mediated SRF activation is almost abolished in Btk–/– cells (Figure 1B). PLCγ2, a substrate of BTK, is the only PLCγ species expressed in these cells (Takata and Kurosaki, 1996), and we found that PLCγ2 null DT40 cells showed a severe defect in SRF activation, similar to that seen in the Btk–/– cells (Figure 1B). By contrast, DT40 cells lacking MEKK1 responded normally in activating SRF following BCR signaling (Supplementary figure 1C). The lipase activity of PLCγ is required for this activation as WT PLCγ, but not a lipase-deficient mutant, could rescue SRF activation in PLCγ null cells (Figure 1C). PMA and ionomycin could induce normal SRF activation in all the cells (Figure 1B). Taken together, these data show that the Btk–PLCγ2 pathway downstream of BCR–Lyn–Syk leads to activation of SRF.
DAG and its effectors are involved in BCR-induced activation of SRF
Our data suggest that the lipase activity of PLCγ is essential for BCR-mediated SRF activation. PLCγ2 regulates the production of DAG, which can activate downstream effectors such as PKC, Ras and Rap1. To test if DAG is a possible mediator for SRF activation, we pre-treated WT DT40 cells with calphostin C, which inhibits binding of DAG by its effectors, and found that calphostin C inhibited BCR-mediated SRF activation in a dose-dependent manner (IC50 ≈ 75 nM), and at a concentration of 300 nM, calphostin C fully blocked SRF activation (Figure 2A). These data show that DAG is required for BCR-mediated SRF activation in WT cells. To determine which downstream effector of DAG is involved in BCR-mediated SRF activation we used specific inhibitors and dominant negative constructs. A pan-specific PKC inhibitor (Gö6976, which inhibits DAG-dependent PKC species; Wilkinson et al., 1993) partially inhibited activation of both SRF and NFAT (Figure 2B), while dominant negative Ras and Rap1 both partially inhibited SRF and NFAT activation, suggesting that downstream of DAG, the activation of PKC as well as Ras and Rap1 are critical for the activation of these two transcription factors (Figure 2C). Similar results were obtained by overexpressing Rap1GAP (data not shown). Interestingly, Rap1 is a potent activator of B-Raf, and Brummer et al. have recently shown that in DT40 cells B-Raf is an important effector of the BCR signaling pathway (Brummer et al., 2002). PKC, Ras and Rap1 can activate the MEK–ERK pathway, and our analysis using a MEK-specific inhibitor (PD98059) indicates that this pathway is critical for the activation of SRF, NFAT, as well as the SRF–TCF complex binding to the full SRE (Figure 2D). By contrast, SRF was insensitive to JNK pathway inhibition (using the JNK1, 2 and 3 inhibitor SP600125), while NFAT was sensitive, suggesting that JNK is important for NFAT activation, but not for SRF activation (Figure 2E). [Note that the absence of MEKK1 does not affect BCR-induced activation of the SRF (Supplementary figure 1C) or the JNK pathway (Kwan et al., 2001).]

Fig. 2. DAG and downstream effectors involved in the regulation of SRF and NFAT downstream of the BCR. (A) DAG plays a role in the regulation of SRF via BCR signaling pathway. SRF transcriptional activity was determined in WT or IP3R–/– DT40 cells pre-treated with the indicated concentrations of calphostin C prior to no stimulation or stimulation with anti-IgM for 6 h. (B) Partial requirement for PKC. SRF and NFAT transcriptional activity was determined in WT DT40 cells pre-treated with the indicated concentrations of the pan-PKC inhibitor Gö6976 prior to BCR stimulation. (C) Partial requirement for Ras and Rap1. SRF and NFAT transcriptional activity was determined in WT DT40 cells transfected with 20 µg empty vector (control), dominant negative Ras (N17) or dominant negative Rap1 (N17). *P < 0.05 versus vector transfected control. (D) Full requirement for the MEK–ERK pathway. SRF, NFAT and SRE transitional activity was determined in WT DT40 cells pre-treated with the indicated concentrations of the MEK inhibitor PD98059 prior to BCR stimulation. (E) Differential requirement for JNK activity. SRF and NFAT transcriptional activity was determined in WT DT40 cells pre-treated with the indicated concentrations of the JNK inhibitor SP600125 prior to BCR stimulation.
Extracellular Ca2+ is essential but not sufficient for SRF activation in WT DT40 cells
PLCγ2 also regulates the production of IP3, which induces calcium release from intracellular calcium stores via the IP3R. Stimulation of the BCR results in a Ca2+ response consisting of two coupled components: an initial rapid Ca2+ increase caused by IP3-mediated intracellular Ca2+ release followed by a slow but sustained extracellular Ca2+ influx through store operated channels, the nature of which are still unclear (Putney and McKay, 1999; Putney et al., 2001). To test whether extracellular Ca2+ also plays a role in regulation of SRF, we used EGTA to chelate extracellular calcium and thus block sustained Ca2+ increase in WT cells and found that BCR-mediated SRF activation is almost fully abolished (Figure 3A), which was rescued by adding equimolar Ca2+ into the medium (Figure 3A). EGTA did not affect cell viability during the time of our assay (data not shown), nor did it non-specifically block general transcription and/or translation as it did not block PKA-induced CRE–luciferase activity (Supplementary figure 2). Thus, activation of SRF in WT DT40 cells requires extracellular Ca2+ influx.

Fig. 3. Extracellular Ca2+ influx is essential but not sufficient for BCR-mediated SRF activation in WT DT40 cells. (A) BCR-mediated SRF activation is abrogated by chelating extracellular Ca2+ with EGTA. SRF transcriptional activity was determined in WT DT40 cells treated with or without anti-IgM in normal Ca2+ concentration, lacking extracellular Ca2+ (by adding EGTA) or in the presence of EGTA and reconstituted with Ca2+ (equimolar EGTA and Ca2+ concentrations). *P < 0.05 versus control without EGTA. (B) NiCl2 blocks BCR-induced intracellular Ca2+ elevation. Intracellular Ca2+ response induced by anti-IgM stimulation with (bold line) or without (thin line) 1 mM NiCl2 added at the indicated time in WT DT40 cells. The results are representative of three independent experiments. (C) NiCl2 inhibits BCR-induced SRF activation. SRF transcriptional activity was determined in WT DT40 cells stimulated with anti-IgM in the absence or presence of the indicated concentrations of NiCl2. The SRF–luciferase activity is shown as % activation (0 and 100% activation refer to no stimulation or stimulation with anti-IgM in normal medium, respectively, and are the mean ± SD of two independent experiments each performed in triplicate). (D) Intracellular Ca2+ elevation is not sufficient for SRF activation. SRF transcriptional activity was determined in WT DT40 cells treated with the indicated concentrations of ionomycin before harvesting for luciferase assay.
To further confirm this, we used a calcium channel blocker, Ni2+, which blocks calcium channels without affecting extracellular free Ca2+ concentration. Unlike other divalent cations, Ni2+ does not penetrate the plasma membrane. Furthermore millimolar levels of Ni2+ can block calcium influx in a dose-dependent manner (Winegar et al., 1991). Intracellular calcium measurements using Fura-2 showed that NiCl2 abolished BCR-mediated intracellular calcium increase in a dose-dependent manner (Figure 3B and data not shown). Note that the difference in the Ca2+ peaks are within the normal variation observed between experiments and is not significant. However, the decrease in Ca2+ seen after the addition of NiCl2 was only observed in the presence of NiCl2. SRF activation in WT DT40 cells was greatly inhibited by 0.5 mM Ni2+ and is almost completely blocked at 1 mM (IC50 ≈ 0.5 mM, Figure 3C), while NFAT activation was more sensitive to Ni2+ (IC50 ≈ 0.25 mM, Figure 3C). These data show that extracellular calcium influx is essential for BCR-mediated SRF activation in WT DT40 cells. However, Ca2+ increase alone was not sufficient to activate SRF (Figure 3D).
IP3-induced intracellular Ca2+ release is not essential for SRF but required for NFAT activation
Extracellular Ca2+ influx results from depletion of intracellular Ca2+ through IP3Rs by IP3 upon BCR stimulation and a mutant DT40 cell line lacking all three subtypes of IP3R (IP3R–/–) does not exhibit Ca2+ increase upon BCR cross-linking (Sugawara et al., 1997 and Figure 4A). To confirm our observation on the requirement of extracellular Ca2+ influx for SRF activation, we performed SRF reporter assays in this mutant cell line after BCR stimulation. Surprisingly, we found that SRF is still activated, although less efficiently than WT cells (∼30–50% of WT, Figure 4B). We verified that these cells are fully defective in intracellular Ca2+ increase (Figure 4A) and NFAT activation upon BCR stimulation (Figure 4B), agreeing with other studies (Sugawara et al., 1997). These results show that unlike NFAT, IP3-induced intracellular calcium release is not essential for SRF activation via the BCR. The DAG pathway was responsible for the remaining ability of these cells to activate SRF as calphostin C also inhibited BCR-mediated SRF activation in a dose-dependent manner (IC50 ≈ 75 nM) in these cells (Figure 2A). Thus the two downstream effectors of PLCγ, Ca2+ and DAG, are essential for BCR-mediated SRF activation and full activation of SRF requires the cooperative action of Ca2+ and DAG.

Fig. 4. Differential regulation of SRF and NFAT in IP3R null DT40 cells. (A) Absence of intracellular Ca2+ response to BCR stimulation in IP3R–/– DT40 cell. IP3R–/– DT40 cells were stimulated with anti-IgM. One mM EGTA and 1 mM Ca2+ treatment was added at the indicated times. Representative trace of three independent experiments. (B) SRF but not NFAT is partially activated in IP3R null mutant DT40 cells. SRF and NFAT transcriptional activity were determined in WT and IP3R–/– DT40 cells. (C) SRF transcriptional activity was determined in IP3R–/– DT40 cells stimulated with or without anti-IgM in the presence of the indicated concentration of EGTA or equimolar concentration of EGTA and Ca2+. *P < 0.05 versus control without EGTA. (D) SRF transcriptional activity was determined in IP3R–/– DT40 cells stimulated with anti-IgM in the absence or presence of indicated concentrations of NiCl2.
SRF activation in IP3R–/– cells also requires calcium
The seeming discrepancy between IP3R–/– and WT DT40 cells in their requirement of calcium for SRF activation led us to explore whether IP3R–/– cells use a Ca2+-independent mechanism to regulate SRF. Intriguingly, similar to WT DT40 cells, EGTA and Ni2+ could also fully abrogate SRF activation in the IP3R null cells to levels similar to that seen in the PLCγ2–/– mutant cells, suggesting that activation of SRF in IP3R–/– cells is also dependent on extracellular Ca2+ influx (Figure 4C and D).
The requirement of extracellular Ca2+ influx for SRF activation seems contradictory to the partial activation of SRF in IP3R–/– cells, which shows no intracellular Ca2+ increase upon BCR ligation (Figure 4A; Sugawara et al., 1997). One possible explanation for this paradox is that the basal level of intracellular Ca2+ is sufficient to partially activate SRF and extracellular calcium entry is required for maintaining the basal level of Ca2+. This is probably the case, since both EGTA and Ni2+ decreased intracellular Ca2+ to below the resting levels in both IP3R–/– and WT cells (Figures 3B and 4A, and data not shown). This suggests that extracellular Ca2+ is critical for the maintenance of intracellular Ca2+ levels at resting state. Taken together these data suggest that in DT40 B cells, SRF is regulated not only by increased levels but also basal levels of intracellular Ca2+, both of which are dependent on extracellular Ca2+. By contrast, NFAT activation is dependent on increased levels of intracellular calcium.
SRF and NFAT are differentially sensitive to Ca2+ signals
Our data demonstrate that BCR-mediated SRF activation, like NFAT, is Ca2+ dependent. However we were intrigued as to the differential requirement for the IP3R in activating these two transcription factors. One possible explanation is that SRF is sensitive to lower intracellular Ca2+ levels than NFAT, such that resting intracellular levels of Ca2+ is sufficient for partial activation of SRF (note that SRF induction in IP3R–/– cells is only 30–50% of that in WT cells) but not sufficient for NFAT induction. To confirm the differential sensitivities of SRF and NFAT to Ca2+ levels, we induced various levels of intracellular Ca2+ increase by using different concentrations of ionomycin and analyzed the degree of SRF and NFAT activation (in the presence of a fixed dose of PMA, required for activation of NFAT and SRF). Intracellular Ca2+ increase positively correlates with the concentration of ionomycin used (Dolmetsch et al., 1997, and data not shown). As expected, SRF induction peaked between 100–200 nM ionomycin (EC50 ∼50 nM ionomycin), then dropped down to the basal level at 1000 nM of ionomycin (Figure 5A). By contrast, NFAT activation peaked at 500 nM ionomycin (EC50 ∼100 nM ionomycin, Figure 5A). These data illustrate that SRF and NFAT are differentially sensitive to Ca2+ levels, and that SRF is more responsive to lower levels of intracellular Ca2+ than NFAT.

Fig. 5. Differential regulation of SRF and NFAT by Ca2+. (A) SRF (solid diamonds) or NFAT (open circles) transcriptional activity was determined in WT DT40 cells stimulated with the indicated concentrations of ionomycin along with 5 ng/ml PMA. (B) CN is a downstream effector of Ca2+ in the regulation of SRF by BCR signaling. SRF or NFAT transcriptional activity was determined in WT DT40 cells pre-treated with the indicated concentrations of the CN inhibitor CsA prior to stimulation with anti-IgM. The activity of the non-stimulated and anti-IgM stimulated cells were set as 0 and 100% activation respectively. (C) SRF transcriptional activity was determined in IP3R–/– DT40 cells pre-treated with 400 nM CsA prior to stimulation with anti-IgM. (D) SRF transcriptional activity was determined in IP3R–/– DT40 cells transfected with 15 µg empty vector or 10 or 15 µg CN-A and stimulated with 5 ng/ml PMA.
Calcineurin is a downstream effector of Ca2+ in the activation of SRF by the BCR
The above data show that Ca2+ plays an essential role in regulation of SRF via BCR signals. We next asked what was the downstream effector of Ca2+ in this function. In lymphocytes, intracellular Ca2+ increase can activate the phosphatase CN (Baksh and Burakoff, 2000). We investigated whether CN is a possible downstream effector of Ca2+ in regulating SRF in DT40 cells by we pre-treating WT DT40 cells with the CN inhibitor, cyclosporin A (CsA) followed by anti-IgM stimulation. CsA partially inhibited anti-IgM-induced SRF activation in a dose-dependent manner (Figure 5B). However, unlike NFAT, CsA did not fully block SRF activation (∼50–70% inhibition) (Figure 5B). This is probably due to residual activation of SRF at basal levels of intracellular Ca2+. This is confirmed by the insensitivity of BCR-mediated SRF activation to CsA in IP3R–/– (Figure 5C). Note that the level of CsA-insensitive SRF activation in WT cells is similar to that seen when we stimulated IP3R–/– cells with the BCR, 30–50% of WT. Indeed, a constitutively active CN (Clipstone et al., 1994), could enhance PMA-induced SRF activation in IP3R null cells (Figure 5D).
Ca2+ is not required for BCR-induced ERK activation but for its nuclear association
Chelation of Ca2+ by EGTA leads to complete inhibition of both SRF and NFAT activation, and we were curious to determine whether this treatment affected signal transduction by the BCR. Stimulating cells in the presence of EGTA did not affect early tyrosine phosphorylation in these cells (Figure 6A). Given the importance of the ERK pathway in SRF activation, we next analyzed the effect of EGTA on ERK activation. To our surprise, EGTA did not affect either the magnitude or the duration of ERK activation (Figure 6B). However, EGTA blocked the translocation of ERK to nuclear areas of the cells following BCR stimulation, suggesting that EGTA may block access to critical ERK substrates in regulating SRF (Figure 6C).

Fig. 6. Calcium regulates ERK nuclear association, but not its activation. (A) EGTA does not affect tyrosine phosphorylation during BCR signaling. WT DT40 cells were either not stimulated (lane 1) or stimulated with anti-IgM for 5 min in the absence (lane 2) or presence (lane 3) of 1 mM EGTA, and protein tyrosine phosphorylation by anti-phosphotyrosine western blotting. (B) ERK activation is not affected by EGTA. WT DT40 cells were either not stimulated (lane 1) or stimulated for the indicated times with anti-IgM in the absence (lanes 2–7) or presence (lanes 9–14) of 1 mM EGTA, and lysates probed for activated ERK (anti-phospho-ERK, top panel), or total ERK (bottom panel) by western blotting. Lane 8 contains lysates from cells treated with EGTA but not stimulated. (C) ERK nuclear association is affected by EGTA. WT DT40 cells were either not stimulated (top left panel) or stimulated for 10 min with anti-IgM in the absence (bottom left panel) or presence (bottom right panel) of 1 mM EGTA and stained for ERK as described in Materials and methods, then analyzed by confocal microscopy. The top right panel contains cells treated with EGTA but left unstimulated. In each panel, the left side is a confocal fluorescent image and the right is a phase contrast image of the same cells.
Role of Rho family GTPases and actin in BCR-mediated SRF activation
Rho family GTPases and actin have been shown to regulate SRF activation in fibroblasts (Hill et al., 1994; Sotiropoulos et al., 1999). We found that dominant negative mutants of the Rho family GTPases, Rac, RhoA and CDC42 all inhibited BCR activation of SRF and NFAT to varying degrees (Figure 7A). For instance, SRF was more dependent on CDC42 than NFAT for full activation (Figure 7A). These latter results suggest that perhaps the actin superstructure may be involved in the activation of these two transcription factors, and indeed, overexpression of actin lead to inhibition of both SRF and NFAT (Figure 7B). However, this was not via a Rho–ROK pathway as a ROK inhibitor had no effect on SRF or NFAT activation (data not shown). Neither of these dominant negative GTPases or actin inhibited BCR activation of Elk-1 (data not shown).

Fig. 7. Rho family GTPases and actin in BCR-mediated SRF activation. (A) Differential requirement for Rho, Rac and CDC42. SRF and NFAT transcriptional activity was determined in WT DT40 cells transfected with empty vector (control), dominant negative RhoA, Rac or CDC42. These dominant negative GTPases did not affect BCR-induced Elk-1 activation (data not shown). (B) A role for actin. SRF and NFAT transcriptional activity was determined in WT DT40 cells transfected with GFP (control) or actin–GFP (actin).
Induction of SRF-regulated genes is dependent on PLCγ, the IP3R and calcium
To determine whether endogenous SRF expression was regulated by the BCR, the absence of PLCγ or the IP3R, or the presence of EGTA, we probed lysates from WT, PLCγ or IP3R null cells for SRF following BCR stimulation. We found that endogenous SRF expression was not affected over the course of our analysis (note that we detected the two forms of the chicken SRF that has been previously reported; Croissant et al., 1996), nor by the lack of PLCγ or the IP3R (Figure 8A and B, nor the presence of EGTA, data not shown). Similar analysis of SRF DNA binding indicated that SRF constitutively bound DNA, and the BCR signal, the presence of EGTA, or the absence of PLCγ or the IP3R did not affect this activity. Specific oligonucleotide competitors and antibody supershifts confirm that the complex we observe is SRF (Figure 8C and D).
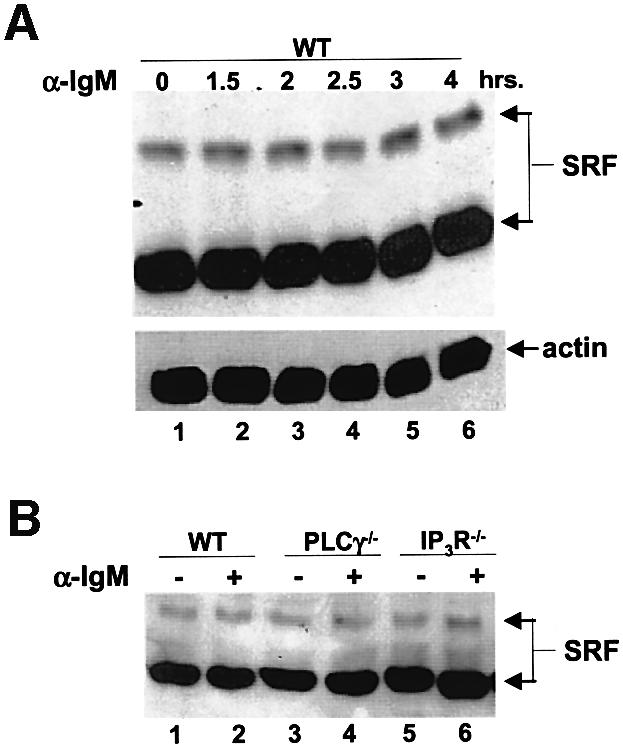

Fig. 8. SRF expression and DNA binding activity does not change during BCR signaling. (A) WT DT40 cells were either not stimulated (lane 1) or stimulated for the indicated times with anti-IgM, and lysates probed for SRF (top panel) or actin as a loading control (bottom panel) by western blotting. Note that we detect two forms of SRF in these chicken cells similar to previously published reports (Croissant et al., 1996). (B) SRF expression is not affected by the lack of PLCγ or IP3R. WT (lanes 1 and 2), PLCγ–/– (lanes 3 and 4) or IP3R–/– (lanes 5 and 6) were either left unstimulated (lanes 1, 3 and 5) or stimulated for 4 h with anti-IgM (lanes 2, 4 and 6), and lysates probed for SRF. Probing for actin as a loading control showed that there was equal amount of proteins in each lane (data not shown). (C) SRF DNA binding is not altered by BCR stimulation. WT DT40 cells were either not stimulated (odd numbered lanes) or stimulated for 1 h with anti-IgM (even numbered lanes), nuclear extracts prepared and EMSA performed. Extracts in lanes 3 and 4 were incubated in the presence of 50-fold excess of cold SRF oligo. Extracts in lanes 5 and 6 were incubated with antibodies to SRF. Extracts in lanes 7–10 were incubated with unlabeled oligos containing binding sites for NFAT (lanes 7 and 8) or NF-κB (lanes 9 and 10) as non-specific competitors. (D) The presence of EGTA or absence of PLCγ or IP3R does not affect DNA binding ability of SRF. WT (lanes 1–4, 9–14), PLCγ–/– (lanes 5 and 6) or IP3R–/– DT40 cells (lanes 7 and 8) were either not stimulated (odd numbered lanes) or stimulated for 10 min with anti-IgM (even numbered lanes), nuclear extracts prepared and EMSA performed. Note that in lanes 3 and 4, WT DT40 cells were incubated in the presence of 1 mM EGTA. Extracts in lanes 9 and 10 were incubated in the presence of 50-fold excess of cold SRF binding site oligo. Extracts in lanes 11 and 12 were incubated with 50-fold excess of cold NFAT binding site oligo. Extracts in lanes 13 and 14 were incubated with antibodies to SRF. The arrow indicates the SRF-specific DNA complex.
We then determined whether endogenous SRF-regulated genes were affected by the absence of PLCγ, IP3R and EGTA. Both c-fos and egr-1 are induced following BCR stimulation within 1.5 h, and the expression of c-fos was significantly affected by the presence of EGTA during the stimulus (Figure 9A, C and D). The absence of PLCγ1 also significantly affected the expression of these genes. However, although SRF was activated ∼4-to 5-fold in the IP3R null cells, this was insufficient for the activation of expression of these SRF-regulated genes in these cells (Figure 9B and D). We also show that these genes are indeed regulated by SRF since a dominant negative mutant of SRF significantly reduced the BCR-induced expression of both genes (Figure 9B and D). Finally, we show that downstream effects of c-fos expression, namely AP-1 complex activation, is also blocked by the dominant negative SRF, while the transcriptional activity of NFAT (or Elk-1, data not shown) was not affected (Figure 9E).


Fig. 9. EGTA, absence of PLCγ or IP3R or dominant negative SRF affect the induction of SRF-regulated genes. (A) Time course of egr-1 induction by the BCR. WT DT40 cells were left unstimulated (lane 1) or stimulated with anti-IgM antibodies for the indicated time periods (lanes 2–6), and lysates probed for the presence of egr-1 (top panel) or actin (bottom panel). (B) WT, PLCγ2–/– or IP3R–/– cells were left unstimulated (odd numbered lanes) or were stimulated with anti-IgM antibodies (even numbered lanes) for 2 h, and lysates probed for egr-1 (top panel) or actin (bottom panel). Lanes 9 and 10 contain lysates from WT DT40 cells transfected with dominant negative SRF (SRFpm1). (C) Time course of c-fos induction by the BCR. WT DT40 cells were left unstimulated (lane 1) or stimulated with anti-IgM antibodies for the indicated time periods (lanes 2–6), and lysates probed for the presence of c-fos. NS: non-specific band below the c-fos band. (D) WT, PLCγ2–/– or IP3R–/– cells were left unstimulated (odd numbered lanes), or were stimulated with anti-IgM antibodies (even numbered lanes) for 2 h, and lysates probed for c-fos. Lanes 3 and 4 contains lysates from WT cells incubated with 1 mM EGTA. Lanes 9 and 10 contain lysates from WT DT40 cells transfected with dominant negative SRF (SRFpm1). NS: non-specific band below the c-fos band. (E) Dominant negative SRF inhibits AP-1 activation. AP-1 or NFAT transcriptional activity was determined in WT DT40 cells transfected with empty vector (control) or with SRFpm1. *P < 0.05 versus control.
Discussion
The signaling pathways leading to activation of SRF-mediated gene transcription has been extensively studied in non-lymphoid cells. However, how SRF is activated in lymphocytes is unclear. In this study, we show that SRF is constitutively able to bind DNA in the presence or absence of BCR signals, and that these signals activate SRF predominantly through a Lyn–Syk–Btk–PLCγ2 pathway (Figure 10). This pathway bifurcates and effectors of the two second messengers, DAG and Ca2+ downstream of PLCγ2, are involved in SRF activation. We found that PKC, Ras and Rap1, and CN respectively are involved in SRF activation. Downstream of PKC, Ras and Rap1, the MEK–ERK pathway is required for activation of SRF. The small GTPases Rac, Rho and CDC42 are also involved, as well as actin, although we have ruled out a role for the Rho–ROK–LIMK pathway as a ROK inhibitor does not affect BCR-induced SRF activation (data not shown). Our data also show that while NFAT and SRF are both activated by BCR signals via the same PLCγ pathway, they differ in their dependence on Ca2+, the IP3R and elements of the MAPK pathway and are differentially regulated by the Rho family of small G proteins. Furthermore, while we could detect a 4- to 5-fold increase in SRF activation in IP3R null cells, this was not sufficient for activation of endogenous gene expression in these cells. Charvet et al. (2002) recently showed that Rac1 is involved in TCR-mediated SRF activation in Jurkat T cells. Our data also suggest that Rac1 is involved in regulation of SRF by the BCR, however, there is an absolute dependence on PLCγ2 for activation of SRF as measured by transcriptional assays as well as analysis of endogenous gene expression. The relationship between PLCγ activation and Rho family GTPases is unclear.

Fig. 10. BCR signaling leading to SRF activation. Model of BCR signals leading to SRF based on the data presented (see text).
Ca2+ increase is regulated by PLCγ, and can regulate a large number of downstream molecules including transcription factors such as NF-κB and NFAT in lymphocytes. Both NF-κB and NFAT are also activated by the same signaling pathway, Syk–BTK–PLCγ, as SRF in B-lymphocytes. A long-standing question has been how the same signals from a single BCR can lead to the activation of different transcription factors that ultimately lead to different cell fates, and different transcription factors have been found to be responsive to different parameters of Ca2+ signals (Dolmetsch et al., 1997). We have shown that basal levels of intracellular Ca2+ can partially function to activate SRF via BCR signals, while these levels of Ca2+ are not sufficient for NFAT activation. Our data also suggests that Ca2+ may regulate nuclear association of activated ERK in these cells, and since this pathway is critical in BCR-induced SRF activation, this may be crucial for its ability to regulate SRF transcription.
SRF has the potential to regulate cell growth as well as survival or apoptosis by regulating the expression of specific genes involved in these processes (Bertolotto et al., 2000). Indeed, SRF can regulate the IL-2 receptor α chain (CD25) (Kuang et al., 1993; Algarte et al., 1995; Pierce et al., 1995), which is expressed on activated B and T cells, and can provide survival signals to these cells. In addition, SRF can regulate the expression of a member of the Bcl family of anti-apoptotic genes MCL-1 (Townsend et al., 1999), which is highly expressed in resting B-lymphocytes and may regulate survival of peripheral B-lymphocytes (Lomo et al., 1996). SRF has been reported to undergo cleavage in cells undergoing apoptosis, and this cleavage has been suggested to alter the ability of these cells to survive (Drewett et al., 2001). Our analysis of SRF expression suggest that cleavage of SRF does not occur in response to BCR signals, and that other mechanisms of regulation play a role in these cells. The regulation of the balance between cell death and survival by Ca2+ signals may involve differential activation of SRF, which along with other transcription factors such as NF-κB and NFAT, may differentially regulate the expression of crucial genes that control these processes. Our data suggest that common early signals from the BCR diverge and can act in a combinatorial fashion to differentially activate different transcription factors, leading to gene expression. This may be one mechanism by which single BCR signals regulate diverse cell functions.
Materials and methods
Cells, reporter plasmids and reagents
WT and mutant DT40 cells were cultured in growth medium: RPMI 1640 supplemented with 10% fetal bovine serum, 1% chicken serum (Sigma, St Louis, MO), penicillin, streptomycin, glutamine and 50 µM β-mercaptoethanol. Lyn, Syk, Btk, PLCγ2 and IP3R null DT40 cells were generated as described (Kurosaki, 1999) and the MEKK1–/– DT40 cells were a kind gift from Genhong Cheng (Kwan et al., 2001; UCLA, Los Angeles, CA). The SRF–, CRE–, SRE– and AP-1–luciferase reporters were from Stratagene (Stratagene, La Jolla, CA), and contained a luciferase gene driven by tandem copies of the SRF-binding element CArG box (GTCCATATTAGGAC)5, CREB-binding element (AGCC TGACGTCAGAG)4, SRE-binding element (AGGATGTCCATATT AGGACATCT)5 or AP-1-binding element (TGACTAA)7. The NFAT–luciferase reporter was a kind gift of Gary Koretzky (University of Pennsylvania, Philadelphia, PA). The following plasmids were kind gifts: constitutively active CN (Neil Clipstone, Northwestern University, Chicago, IL), dominant negative mutants Ras(N17), Rac(N17), RhoA(N19) and CDC42(N14) (Ray Birge, UMDNJ, Newark, NJ), dominant negative Rap1(N17) and Rap1GAP (Lawrence Quilliam, Indiana University, Indianapolis, IN), dominant negative SRF mutant (SRFpm1, Ron Prywes, Columbia University, New York, NY) and GFP–actin (Andrew Henderson, Penn State University, University Park, PA). WT and PLCγ lipase-deficient mutant have been previously described (Hashimoto et al., 1998). Antibodies to activated ERK (anti-phospho-ERK) and ERK was from Cell Signaling (Beverly, MA). Anti-actin, -c-fos and -egr-1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Calphostin C, pan-specific PKC inhibitor Gö6976 and JNK inhibitor SP600125 were from Calbiochem (Irvine, CA). The MEK inhibitor PD98059 was from Research Biochemicals International (Natick, MA) and CsA was from Sigma.
Western blot
Western blots were performed as previously described (Hao and August, 2002). Blots were probed with anti-phosphotyrosine, -phospho-ERK, -ERK, -c-fos, -egr-1 or -actin primary antibodies and appropriate secondary antibody conjugated with HRP and detected using the ECL system (Amersham).
Ca2+ measurements
In brief, 107 cells were suspended in 1 ml PBS containing 20 mM HEPES pH 7.2, 5 mM glucose, 0.025% BSA and 1 mM CaCl2, and loaded with 5 µg Fura-2AM at 37°C for 30 min. Cells were then washed twice and adjusted to 106 cells/ml. Continuous fluorescence was detected at 22°C with a Hitachi F-2000 fluorospectrophotometer (Hitachi, San Jose, CA) at excitation wavelengths of 340 and 380 nm and an emission wavelength of 510 nm. Relative intracellular Ca2+ is shown as the ratio of fluorescence intensity at 340 to 380 nm excitation.
Cell transfection and luciferase activity assay
Cells (20 × 106) were mixed with 4 µg SRF–luciferase or 6 µg NFAT–luciferase plasmid in 400 µl RPMI 1640 supplemented with 10 mM HEPES and transferred to a 4 mm-gap electroporation cuvette and electroporated (240 V, 60 ms, 1 pulse, BTX electro-square-porator T820). After recovering in growth medium for 8 h, cells were starved in serum-free RPMI 1640 supplemented with 10 mM HEPES pH 7.2 for 12 h and then equally split into different groups which were either stimulated by adding 4 µg/ml anti-chicken IgM, other stimulants as indicated, or no stimulation as control for 8 h unless stated otherwise. Cells treated with inhibitors were pre-treated for 30 min prior to stimulation. Cells were then harvested, counted and lysed for luciferase assay using Promega luciferase assay kit, and for checking the expression of dominant negative constructs by western blot. The results are normalized as fold over non-stimulated control and are the mean ± SD of 3–6 independent experiments performed in triplicate unless otherwise indicated. Statistical significance was determined using student’s t-test.
Electrophoretic mobility shift assays
In brief, nuclear extracts were obtained from cells stimulated or not with anti-chicken IgM under the conditions listed in the figure legends. Extracts were incubated with a double-stranded oligonucleotide (5′-tcgaGTCCATATTAGGAC, annealed and labeled by filling in the extra four base overhangs with [32P]dCTP) in the presence or absence of 50-fold cold specific competitor (the SRF-binding site), or non-specific competitors, either an NF-κB or an NFAT-binding site that was not labeled, or with antibodies to SRF as indicated. Following a 20 min incubation, reactions were separated on a 6% native polyacrylamide gel and exposed to film.
Immunofluorescence analysis
WT DT40 cells were either not stimulated or stimulated for 10 min with 4 µg/ml anti- chicken IgM in the absence or presence of 1 mM EGTA, fixed with 3% paraformaldehyde, permeabilized with 0.1% Triton X-100, then ERK detected with anti-ERK polyclonal antibodies and FITC-conjugated goat anti-rabbit antibodies. Cells were then analyzed by confocal microscopy.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank members of the Immunology Research Labs at Penn State for their critical feedback. We also thank Julie Cook for help with the EMSA assays, and the investigators mentioned in the materials section for the kind gifts of crucial reagents. This work was supported by start up funds from the Department of Veterinary Science at Penn State University and a Johnson & Johnson Focused Giving Program Award (to A.A.). S.H. is a graduate fellow of the Huck Institute for Life Sciences at Penn State University.
References
- Afar D., Park,H., Howell,B., Rawlings,D., Cooper,J. and Witte,O. (1996) Regulation of Btk by Src family tyrosine kinases. Mol. Cell. Biol., 16, 3465–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algarte M., Lecine,P., Costello,R., Plet,A., Olive,D. and Imbert,J. (1995) In vivo regulation of interleukin-2 receptor alpha gene transcription by the coordinated binding of constitutive and inducible factors in human primary T cells. EMBO J., 14, 5060–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba Y., Hashimoto,S., Matsushita,M., Watanabe,D., Kishimoto,T., Kurosaki,T. and Tsukada,S. (2001) BLNK mediates Syk-dependent Btk activation. Proc. Natl Acad. Sci. USA, 98, 2582–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baksh S. and Burakoff,S. (2000) The role of calcineurin in lymphocyte activation. Semin. Immunol., 12, 405–415. [DOI] [PubMed] [Google Scholar]
- Berridge M. (1993) Inositol trisphosphate and calcium signalling. Nature, 261, 315–325. [DOI] [PubMed] [Google Scholar]
- Bertolotto C., Ricci,J., Luciano,F., Mari,B., Chambard,J. and Auberger,P. (2000) Cleavage of the serum response factor during death receptor-induced apoptosis results in an inhibition of the c-FOS promoter transcriptional activity. J. Biol. Chem., 275, 12941–12947. [DOI] [PubMed] [Google Scholar]
- Brummer T., Shaw,P., Reth,M. and Misawa,Y. (2002) Inducible gene deletion reveals different roles for B-Raf and Raf-1 in B-cell antigen receptor signalling. EMBO J., 21, 5611–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai J. and Tarnawski,A.S. (2002) Serum response factor: discovery, biochemistry, biological roles and implications for tissue injury healing. J. Physiol. Pharmacol., 53, 147–157. [PubMed] [Google Scholar]
- Charvet C., Auberger,P., Tartare-Deckert,S., Bernard,A. and Deckert,M. (2002) Vav1 couples T cell receptor to serum response factor-dependent transcription via a MEK-dependent pathway. J. Biol. Chem., 277, 15376–15384. [DOI] [PubMed] [Google Scholar]
- Clipstone N.A., Fiorentino,D.F. and Crabtree,G.R. (1994) Molecular analysis of the interaction of calcineurin with drug–immunophilin complexes. J. Biol. Chem., 269, 26431–26437. [PubMed] [Google Scholar]
- Croissant J., Kim,J., Eichele,G., Goering,L., Lough,J., Prywes,R. and Schwartz,R. (1996) Avian serum response factor expression restricted primarily to muscle cell lineages is required for alpha-actin gene transcription. Dev. Biol., 177, 250–264. [DOI] [PubMed] [Google Scholar]
- Das T., Sa,G. and Ray,P. (1999) Mechanisms of protein A superantigen-induced signal transduction for proliferation of mouse B cell. Immunol. Lett., 70, 43–51. [DOI] [PubMed] [Google Scholar]
- DeKoninck P. and Schulman,H. (1998) Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science, 279, 227–230. [DOI] [PubMed] [Google Scholar]
- Dolmetsch R.E., Lewis,R.S., Goodnow,C.C. and Healy,J.I. (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature, 386, 855–858. [DOI] [PubMed] [Google Scholar]
- Drewett V., Devitt,A., Saxton,J., Portman,N., Greaney,P., Cheong,N., Alnemri,T., Alnemri,E. and Shaw,P. (2001) Serum response factor cleavage by caspases 3 and 7 linked to apoptosis in human BJAB cells. J. Biol. Chem., 276, 33444–33451. [DOI] [PubMed] [Google Scholar]
- Ebinu J. et al. (2000) RasGRP links T-cell receptor signaling to Ras. Blood, 95, 3199–3203. [PubMed] [Google Scholar]
- Greenberg M.E., Siegfried,Z. and Ziff,E.B. (1987) Mutation of the c-fos gene dyad symmetry element inhibits serum inducibility of transcription in vivo and the nuclear regulatory factor binding in vitro. Mol. Cell. Biol., 7, 1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S. and August,A. (2002) The proline rich region of the Tec homology domain of ITK regulates its activity. FEBS Lett., 525, 53–58. [DOI] [PubMed] [Google Scholar]
- Hardy R. and Hayakawa,K. (2001) B cell development pathways. Annu. Rev. Immunol., 19, 595–621. [DOI] [PubMed] [Google Scholar]
- Hashimoto A., Okada,H., Jiang,A., Kurosaki,M., Greenberg,S., Clark,E. and Kurosaki,T. (1998) Involvement of guanosine triphosphatases and phospholipase C-gamma2 in extracellular signal-regulated kinase, c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J. Exp. Med., 188, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill C.S., Marais,R., John,S., Wynne,J., Dalton,S. and Treisman,R. (1993) Functional analysis of a growth factor-responsive transcription factor complex. Cell, 73, 395–406. [DOI] [PubMed] [Google Scholar]
- Hill C.S., Wynne,J. and Treisman,R. (1994) Serum-regulated transcription by serum response factor (SRF): a novel role for the DNA binding domain. EMBO J., 13, 5421–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen F.E. and Prywes,R. (1995) Serum response factor: transcriptional regulation of genes induced by growth factors and differentiation. Biochim. Biophys. Acta, 1242, 1–10. [DOI] [PubMed] [Google Scholar]
- Kaptein J., Yang,C., Lin,C., Nguyen,T., Chen,F. and Lad,P. (1995) Synergy between signal transduction pathways is obligatory for expression of c-fos in B and T cell lines: implication for c-fos control via surface immunoglobulin and T cell antigen receptors. Immunobiology, 193, 465–485. [DOI] [PubMed] [Google Scholar]
- Kuang A., Novak,K., Kang,S., Bruhn,K. and Lenardo,M. (1993) Interaction between NF-kappa B- and serum response factor-binding elements activates an interleukin-2 receptor alpha-chain enhancer specifically in T lymphocytes. Mol. Cell. Biol., 13, 2536–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T. (1999) Genetic analysis of B cell antigen receptor signaling. Annu. Rev. Immunol., 17, 555–592. [DOI] [PubMed] [Google Scholar]
- Kwan R., Burnside,J., Kurosaki,T. and Cheng,G. (2001) MEKK1 is essential for DT40 cell apoptosis in response to microtubule disruption. Mol. Cell. Biol., 21, 7183–7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomo J., Smeland,E.B., Krajewski,S., Reed,J.C. and Blomhoff,H.K. (1996) Expression of the Bcl-2 homologue Mcl-1 correlates with survival of peripheral blood B lymphocytes. Cancer Res., 56, 40–43. [PubMed] [Google Scholar]
- McLeod S.J. and Gold,M.R. (2001) Activation and function of the Rap1 GTPase in B lymphocytes. Int. Rev. Immunol., 20, 763–789. [DOI] [PubMed] [Google Scholar]
- Miranti C.K., Ginty,D.D., Huang,G., Chatila,T. and Greenberg,M.E. (1995) Calcium activates serum response factor-dependent transcription by a Ras- and Elk-1-independent mechanism that involves a Ca2+/calmodulin-dependent kinase. Mol. Cell. Biol., 15, 3672–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niiro H. and Clark,E.A. (2002) Decision making in the immune system: regulation of B-cell fate by antigen-receptor signals. Nat. Rev. Immunol., 2, 945–956. [DOI] [PubMed] [Google Scholar]
- Norman C., Runswick,M., Pollock,R. and Treisman,R. (1988) Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell, 55, 989–1003. [DOI] [PubMed] [Google Scholar]
- Pierce J.W., Jamieson,C.A., Ross,J.L. and Sen,R. (1995) Activation of IL-2 receptor alpha-chain gene by individual members of the rel oncogene family in association with serum response factor. J. Immunol., 155, 1972–1980. [PubMed] [Google Scholar]
- Prywes R. and Roeder,R.G. (1986) Inducible binding of a factor to the c-fos enhancer. Cell, 47, 777–784. [DOI] [PubMed] [Google Scholar]
- Putney J.W. Jr and McKay,R.R. (1999) Capacitative calcium entry channels. BioEssays, 21, 38–46. [DOI] [PubMed] [Google Scholar]
- Putney J.W. Jr, Broad,L.M., Braun,F.J., Lievremont,J.P. and Bird,G.S. (2001) Mechanisms of capacitative calcium entry. J. Cell Sci., 114, 2223–2229. [DOI] [PubMed] [Google Scholar]
- Rawlings D., Scharenberg,A., Park,H., Wahl,M., Lin,S., Kato,R., Fluckiger,A., Witte,O. and Kinet,J. (1996) Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science, 271, 822–825. [DOI] [PubMed] [Google Scholar]
- Rodriguez R. et al. (2001) Tyrosine residues in phospholipase cγ 2 essential for the enzyme function in B-cell signaling. J. Biol. Chem., 276, 47982–47992. [DOI] [PubMed] [Google Scholar]
- Schroter H., Mueller,C.G., Meese,K. and Nordheim,A. (1990) Synergism in ternary complex formation between the dimeric glycoprotein p67SRF, polypeptide p62TCF and the c-fos serum response element. EMBO J., 9, 1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P.E. (1992) Ternary complex formation over the c-fos serum response element: p62TCF exhibits dual component specificity with contacts to DNA and an extended structure in the DNA-binding domain of p67SRF. EMBO J., 11, 3011–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P.E., Schroter,H. and Nordheim,A. (1989) The ability of a ternary complex to form over the serum response element correlates with serum inducibility of the human c-fos promoter. Cell, 56, 563–572. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos A., Gineitis,D., Copeland,J. and Treisman,R. (1999) Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell, 98, 159–169. [DOI] [PubMed] [Google Scholar]
- Sugawara H., Kurosaki,M., Takata,M. and Kurosaki,T. (1997) Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J., 16, 3078–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M. and Kurosaki,T. (1996) A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J. Exp. Med., 184, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M., Sabe,H., Hata,A., Inazu,T., Homma,Y., Nukada,T., Yamamura,H. and Kurosaki,T. (1994) Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J., 13, 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend K.J., Zhou,P., Qian,L., Bieszczad,C.K., Lowrey,C.H., Yen,A. and Craig,R.W. (1999) Regulation of MCL1 through a serum response factor/Elk-1-mediated mechanism links expression of a viability-promoting member of the BCL2 family to the induction of hematopoietic cell differentiation. J. Biol. Chem., 274, 1801–1813. [DOI] [PubMed] [Google Scholar]
- Treisman R. (1986) Identification of a protein-binding site that mediates transcriptional response of the c-fos gene to serum factors. Cell, 46, 567–574. [DOI] [PubMed] [Google Scholar]
- Wilkinson S.E., Parker,P.J. and Nixon,J.S. (1993) Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem. J., 294, 335–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winding P. and Berchtold,M. (2001) The chicken B cell line DT40: a novel tool for gene disruption experiments. J. Immunol. Methods, 249, 1–16. [DOI] [PubMed] [Google Scholar]
- Winegar B.D., Kelly,R. and Lansman,J.B. (1991) Block of current through single calcium channels by Fe, Co and Ni. Location of the transition metal binding site in the pore. J. Gen. Physiol., 97, 351–367. [DOI] [PMC free article] [PubMed] [Google Scholar]