

Abstract
Although Kch of Escherichia coli is thought to be a K+ channel by sequence homology, there is little evidence that it actually conducts K+ ions in vitro or in vivo. We isolated gain-of-function (GOF) Kch mutations that render bacteria specifically sensitive to K+ ions. Millimolar added K+, but not Na+ or sorbitol, blocks the initiation or continuation of mutant growth in liquid media. The mutations are mapped at the RCK (or KTN) domain, which is considered to be the cytoplasmic sensor controlling the gate. Additional mutations directed to the K+-filter sequence rescue the GOF mutant. The apparent K+-specific conduction through the ‘loose-cannon’ mutant channel suggests that the wild-type Kch channel also conducts, albeit in a regulated manner. Changing the internal ATG does not erase the GOF toxicity, but removes kch’s short second product, suggesting that it is not required for channel function in vivo. The mutant phenotypes are better explained by a perturbation of membrane potential instead of internal K+ concentration. Possible implications on the normal function of Kch are discussed.
Keywords: bacteria/gain-of-function/Kch/potassium channels/RCK
Introduction
K+ is the major cation in all cytoplasms. Besides molecular ion pumps or exchangers that produce K+ gradients, cells are equipped with K+ channels that allow K+ to flow down the electrochemical gradient. This passive permeability of K+ governs the membrane potential and therefore excitability and behavior (Hille, 2001), making K+ channels a subject of intensive research in animal physiology. Plants apparently use certain K+ channels to take up K+ (Hirsch et al., 1998). Interestingly, recent genome sequencing has revealed that all bacteria and archea, except for a few parasitic forms, also contain K+-channel genes (Kumanovics et al., 2002). Although a few such genes produced electric conductances upon reconstitution into artificial lipid bilayers (Schrempf et al., 1995; Jiang et al., 2002a; Ruta et al., 2003) or heterologously expressed (Wolters et al., 1999; Sesti et al., 2003), none have been shown to produce actual K+ conduits in the original organisms in vivo. To date, no biological roles have been assigned to any single prokaryotic K+ channel.
In contrast to the paucity of our knowledge on their functions, the presumed prokaryotic channels have provided rich insights into channel structures, because they can be mass produced, purified and crystallized. We have learnt from KcsA that each K+ channel is a tetramer that encloses an aqueous pathway at the center. It narrows from the outside mouth into a selective filter of 2.85 Å diameter near the periplasmic surface (Doyle et al., 1998; Zhou et al., 2001). The filter is lined by horizontally arranged squares of carbonyl oxygens of the canonical amino acid sequence TXGYGD from each of the four subunits. These partially negative oxygens surround the K+ ion much like the eight water oxygens surround it in aqueous solution, accounting for both the high K+ selectivity and high throughput of the channel. The primary structure of many K+-channel subunits comprise six transmembrane α-helical segments (S1–S6) with the filter sequence between S5 and S6, trailing a short ‘pore’ helix (Figure 1A). In the 3D structure (Doyle et al., 1998), the central filter is cradled by the four pore helices in the form of an inverted teepee. Beneath the teepee is a water-filled cavity, which is sealed off at the bottom by the convergence of the four S6s when the channel closes. S6s therefore form the gate (Jiang et al., 2002b). The movement of S6 is linked to a ligand sensor, a cytoplasmic domain (Figure 1A) called RCK (Jiang et al., 2001) or KTN (Roosild et al., 2002) in many K+ channels, including MthK and Kch, below. Many but not all prokaryotic K+-channel genes, including kch, have an internal translational start site between the last transmembrane segment and the RCK domain, which can produce the latter alone. In a crystal, two MthK tetramers pack together through their RCK domains (Jiang et al., 2002a). Opinions differ as to how these domains govern channel gating (Jiang et al., 2002a; Roosild et al., 2002).

Fig. 1. The structure of Kch, a subunit of the putative K+ channel in E.coli. (A) Membrane topology of a Kch subunit is composed of a channel body (S1–S6, cylinder) with a pore region between S5 and S6, and a RCK domain (M240–N393) (Jiang et al., 2001) after S6. M240 also initiates the production of RCK protein (see Figure 6A). (B) Sequence alignment of the pore region from selected K+ channels with the conserved K+ signature sequence (GYG) highlighted in gray. Kch, E. coli (NCBI protein database code GI: 11354242); Shaker, Drosophila melanogaster (GI: 13432103); KcsA, Streptomyces lividans (GI: 8134524); MthK, Methanothermobacter thermautotrophicus (GI: 21542150); Kat1, Arabidopsis thaliana (GI: 421842); eag, Drosophila melanogaster (GI: 399253); Herg, Homo sapiens (GI: 7531135); Irk1, Mus musculus (GI: 6680530); mSlo1, Mus musculus (GI: 6754436).
Milkman (1994) recognized the first prokaryotic K+ channel, Kch of Escherichia coli, by its Shaker-like S1–S6 motif and TXGYG signature sequence (Figure 1B). The crystal structure of its RCK has recently been solved (Jiang et al., 2001). However, despite apparent attempts by several laboratories (Ungar et al., 2001; Munsey et al., 2002), there has been no report of electric conductance of Kch by functional reconstitution or by heterologous expression. Although overexpressing wild-type kch is toxic (Munsey et al., 2002), deleting kch presents no ‘loss-of-function’ phenotype discernible in the laboratory to date. In short, there is no clear evidence that Kch makes a functional K+ conduit in vitro or in vivo.
Our work takes advantage of E.coli genetics. By selecting, engineering and examining various mutants, we deduce that Kch most likely makes a functional K+-specific ion channel in live cells, and the structurally delineated ‘filter’, ‘gate’ and ‘sensor’ indeed function as such.
Results
Isolation of the gain-of-function Kch mutants
In the context of channel biology, ‘gain-of-function’ (GOF) mutations are those that favor the open conformation of the channel protein, most probably by altering its gate, its element that receives the stimulus or the connection between the two (Loukin et al., 1997; Ou et al., 1998). We reasoned that, if wild-type Kch truly conducts K+ current in vivo, GOF mutations should make loose-cannon channels that misfire and their uncontrolled K+ flux across the cell membrane should block growth under certain circumstances. Figure 2A illustrates our approach. In short, plasmids were randomly mutagenized and their kch inserts excised and recloned into a virgin expression plasmid, pB11d (see Materials and methods), behind a LacUV5 promoter. In our first screen, the transformants were replicated onto permissive (LB) and various restrictive plates (LB + IPTG, K200 ± IPTG, Na200 ± IPTG and Sorbitol400 ± IPTG). Colonies whose replicas were missing in any of the restrictive plates were picked. Their plasmids were isolated and retransformed into fresh cells to reconfirm the non-growth phenotype (Figure 2B). Note that, in the presence of IPTG, even the LacUV5-driven wild-type kch is generally toxic (panel 1, row 2, arrowheads). Contrary to a recent report (Munsey et al., 2002), this toxicity is not relieved but is aggravated by the addition of K+ (panel 2, row 2). This aggravation is not K+-specific; Na+ also aggravates (panel 3, row 2). The effect seems to be related to ionic strength and not osmolarity, since equi-osmolar sorbitol did not seem to aggravate toxicity (panel 4, row 2). As shown below, this toxicity has to do with the overproduction of a membrane protein, Kch, and not with K+ permeation through Kch. To avoid the complication of overproduction, we only considered phenotypes in the absence of IPTG (Figure 2B, panels 5–8) derived from the leakage level of transcription by the uninduced LacUV5 promoter. Seven mutants were thus selected from ∼8000 colonies screened. Each of them had a single amino acid substitution: I247T, A307V, C312F, N325S, S331P, I361T and S364P. Interestingly, all of these mutations are located towards the C-terminal portion of the Kch protein, in the RCK domain (Jiang et al., 2001).

Fig. 2. Isolation of the GOF Kch mutants. (A) The experimental scheme. Kch coding region together with pBluescript vector were mutagenized in vivo using XL1-Red Cells. The mutated kchs were further cloned in pB11d behind IPTG-inducible LacUV5 promoter and used to transform kch-null FRAG1 E.coli. Transformed colonies whose replicas failed to grow on any of the restrictive plates were picked, DNA sequenced and phenotyped. (B) Growth phenotype on the permissive (panel 1) and various restrictive plates (panels 2–8) with (1–4) or without (5–8) IPTG. Each row shows the growth pattern of five 5-µl drops of inoculate from cultures of stationary cells (OD600 ∼ 3.5–4.5) diluted 102-, 103-, 104-, 105- and 106-fold.
K+-specific colony phenotype
In the absence of IPTG, the selected mutants grew like wild-type on regular LB plates (Figure 2B, panel 5), and did not grow on LB plates enriched with 200 mM KCl (K200), (Figure 2B, panel 6). This non-growth phenotype is K+-specific and is not due to the increased ionic strength or osmolarity, since cells grew normally on plates enriched with 200 mM NaCl (Na200) or 400 mM sorbitol (Sorbitol400) (Figure 2B, panels 7 and 8). These are phenotypes of the mutant kch gene expressed through the leakage of the LacUV5 promoter without induction. To avoid possible complications due to this artificial promoter, we constructed new plasmids with the promoter native to kch. The C312F, N325S and S331P mutants were chosen for further scrutiny. A 2 kb BamHI–PstI fragment containing the kch coding region and its native promoter was inserted into pGEM vector to create pGEM-WT (Figure 3A). A 396 bp SacII–BspEI subfragment of pGEM-WT was then replaced with that of pB11d-C312F, -N325S or -S331P to generate pGEM-C312F, -N325S or -S331P, respectively, that contains the original mutation. Again the bacteria bearing the C312F, N325S or S331P mutation in kch with the native promoter grew on LB plates enriched with NaCl (Na200) or sorbitol (Sorbitol400) but not on plates with KCl (K200) (Figure 3B). Thus, the natural promotion of GOF mutant kchs we selected confers the K+-specific sensitive phenotype.

Fig. 3. Expression of Kch from the native promoter. (A) A 2 kb E.coli genomic fragment containing the C-terminus of the neighboring gene yciI, kch’s native promoter and kch coding region was cloned in a multi-copy vector, pGEM, between BamHI and PstI. (B) Plate phenotype of the GOF Kch mutants expressed from native promoter on the LB-based plates. The bacteria with the naturally promoted C312F, N325S or S331P Kch mutants were sensitive to only K+ and not to Na+ or sorbitol. (C) On tryptone–agarose plates, the colony-forming units of these mutants decrease dramatically with the concentration of additional K+ between 1 and 10 mM.
By flame photometry, we determined that the regular LB agar used for the above plate assay contained ∼6.5 mM K+ ions, mostly from the added yeast extract (∼6 mM). To more precisely define the critical K+ concentration that restricts the mutants’ growth, we used agarose plates (0.75%) of a base medium containing only tryptone (total contamination: ∼0.5 mM K+). KCl, NaCl or sorbitol was then added to this base to generate various test media. Again, cells with pGEM-borne GOF mutants were sensitive to only K+ (Figure 3C) and not Na+ or sorbitol (data not shown). More interestingly, the K+ sensitivity is clearly dose-dependent, and the colony-forming units decreased >103-fold within 10 mM K+ increase (Figure 3C).
Millimolar K+ stops the initiation and continuation of growth in the GOF mutant
To examine closely the effect of added K+, we measured the growth curves of bacteria bearing pGEM-empty, pGEM-WT and pGEM-S331P plasmids in the tryptone-based liquid media (Figure 4). With stationary phase inocula, bacteria with the empty plasmid or wild-type plasmid (Figure 4A, left and middle) grew normally. However, those with the GOF-mutant plasmid (Figure 4A, right) were unable to initiate growth in the medium enriched with 5 mM KCl (open triangles), but grew normally with 5 mM NaCl (open circles) or 10 mM sorbitol (open diamonds). The K+-sensitive phenotype of these mutants depends on the pH of the medium. Although they could not initiate growth in 5 mM added KCl at pH 7.3 (Figure 4B, right, open triangles), this K+ sensitivity was suppressed at pH 5.8 (gray triangles), suggesting that the proton gradient counteracts the mutant defect (see Discussion).

Fig. 4. Growth in tryptone-based liquid media. Growth curves of cultures in various test media (see Materials and methods) inoculated with stationary-phase bacteria bearing pGEM-empty (left panels), pGEM-WT (middle) or pGEM-S331P (right). Mean ± SD given (n ≥ 3, SD smaller than symbol when not visible). (A) Growth of cells in the T medium (open squares, broken line), TK5 (open triangles), TNa5 (open circles) and TS10 (open diamonds). (B) Growth of cells in T medium (pH 7.3, open squares, broken line), TH5 (pH 5.8, gray squares, only pGEM-S331P was measured), TK5 (pH 7.3, open triangles) and TK5H5 (pH 5.8, gray triangles). The pH variations of the cultures remained within ±0.1 U in the first 4 h. (C) Growth of cells before and after the addition of 10 mM KCl (black triangles), NaCl (gray circles) or none (open squares, broken line), at the logarithmic-phase of growth (arrows).
We also tested the effect of K+ on cells in the exponential phase of growth. Addition of 10 mM KCl or NaCl had no effect on growing cultures of bacteria bearing pGEM-empty or pGEM-WT (Figure 4C, left and middle). However, the addition of KCl (Figure 4C, right, black triangles), but not NaCl (gray circles), immediately slowed and then stopped the doubling of those bearing the GOF-mutant plasmid.
Mutations in the canonical K+-filter sequence suppress the GOF phenotype
If the K+-sensitive phenotype of the GOF mutants depends upon K+ flux, their K+ sensitivity should be rescued by additional point mutations expected to block K+ flux. We therefore generated a large number of such mutations to rigorously test this notion. Our PCR-based mutagenesis began with oligonucleotides completely degenerate in one codon that corresponds to either Gly187, Tyr188 or Gly189 in the TXGYGD filter sequence. S331P mutant DNA was used as the PCR template. Bacteria transformed with the mutagenized plasmids were screened for growth on K200 plates, followed by western blot using antibody against the C-terminal peptide of Kch to confirm the expression. Of the 41 suppressor mutants isolated, each of 40 had a single missense mutation and one suffered a nonsense mutation. Among the 27 mutant kch genes randomly chosen for complete sequencing, 21 had no mutations elsewhere (Table I) [i.e. of the 41, 14 are sequenced only at the filter and found to be missense and six are entirely sequenced and found to have an additional mutation elsewhere, presumably due to PCR errors (data not shown)]. The plurality of missense mutations at the K+ filter, each alone being sufficient to suppress the GOF phenotype (Table I; Figure 5A, row 4), strongly argues that K+ passage is the cause of that phenotype. A caveat to this reasoning is that the growth on K200 plates could be due to the loss of Kch protein on the cell membrane, which necessitates loss of its K+ permeation. The presence of the double-mutated Kch proteins on the cell membrane was examined with a representative suppressed strain (S331P-G187Q). As shown in Figure 5B, the strain S331P-G187Q produced Kch proteins in the cell membrane as well as the parental S331P strain that did not survive the K200 plate.
Table I. Random mutations on three key residues of Kch’s filter (G187, Y188 and G189) and their effect on the S331P GOF mutant phenotype.
| Filter sequence |
||||||
|---|---|---|---|---|---|---|
| G187 |
Y188 |
G189 |
||||
| Codon |
A.A. |
Codon |
A.A. |
Codon |
A.A. |
|
| Wild type | GGC | G | TAT | Y | GGC | G |
| Mutations that suppress the GOF phenotype | TGC | C | ||||
| CAG | Q | |||||
| GGC | G | |||||
| ACT | T | |||||
| CGC | R | |||||
| ATA | I | |||||
| AAA | K | |||||
| CAA | Q | |||||
| TCC | S | |||||
| AGC | S | |||||
| GAC | D | |||||
| CTT | L | |||||
| TTC | F | |||||
| AGA | R | |||||
| AAT | N | |||||
| AGT | S | |||||
| TCA | S | |||||
| AAA | K | |||||
| TCC | S | |||||
| GCA | A | |||||
| GAG | E | |||||
| Do not suppress |
|
|
TTC |
F |
|
|
| TAT | Y | |||||
| TTC | F | |||||
| TAT | Y | |||||
| TTT | F | |||||

Fig. 5. Effect of representative filter mutations on the S331P GOF mutant. (A) Additional G187Q (S331P-G187Q) mutation on the K+ filter rescues the S331P mutant on K200 plates (bottom row). The Y188F (S331P-Y188F) mutation is the only K+ filter mutation we could isolate that did not rescue the S331P mutant on K200 plates (row 3). (B) Western blot analysis of the membrane fractions prepared from the cells carrying vectors: pGEM-empty (lane 1), pGEM-WT (lane 2), pGEM-S331P (lane 3) and pGEM-S331-G187Q (lane 4). All strains produce a comparable amount of Kch and RCK proteins in the bacterial membrane. RCK: RCK protein (see Figure 6A). M: MagicMark (Invitrogen, CA).
Mutations that preserve the K+-filter structure do not suppress the GOF phenotype
Thorough studies of the canonical filter sequence of three other K+ channels, Shaker (Heginbotham et al., 1994), Kat1 (Nakamura et al., 1997) and Kir2.1 (So et al., 2001), have shown that nearly all mutations disrupt the filter. One common exception is the GYG-to-GFG substitution that leaves the selectivity and permeation of the channels unaffected. To see whether Kch exhibits the same mutational tolerance, we tested the above bacteria with plasmids bearing a completely degenerated codon that encodes Tyr188 in the S331P kch gene. This time, however, we screened for colonies that remained unable to grow on K200 plates by replica plating. Among the 15 colonies we randomly picked and locally sequenced, three remained unchanged as having Y188 and 12 had the Y188F substitution. No other substitutions of Y188 were observed. Five out of six fully sequenced mutants had no mutation elsewhere (Table I). The S331P-Y188F mutant (Figure 5A, row 3) had the same K+ sensitivity as the S331 mutant (row 2).
Removal of the internal translational initiation site does not suppress the GOF phenotype
The kch gene encodes two proteins (Figure 6A and B). It contains an internal translational start site in a position similar to the one in mthK whose product has been crystallized (Jiang et al., 2002a). To test the requirement of the smaller protein for Kch function in vivo, we replaced the internal methionine with another small non-polar amino acid, leucine, and prevented the production of the RCK protein (Figure 6B, lanes 3, 5, 7 and 9). This additional M240L mutation had no effect on the wild-type Kch phenotype (Figure 6C, row 2). More importantly, M240L also did not suppress the K+ sensitivity of the N325S, C312F and S331P mutants (Figure 6C, rows 4, 6 and 8), suggesting that the RCK protein is not necessary for Kch function in vivo.

Fig. 6. Removing the internal start codon eliminates the RCK protein but does not suppress the toxicity of the GOF mutations. (A) Diagram of the relative position of the internal start and the two translation products, Kch and RCK protein. The protein secondary structures are indicated as gray rectangles (α-helix) and arrows (β-sheet). (B) Western blot analysis of the Kch proteins. The samples were prepared by directly boiling the stationary cells in SDS–PAGE sample buffer. As expected, both the larger (Kch) and the smaller (RCK) proteins are found in the wild-type (lane 2) and the GOF mutants (lanes 4, 6 and 8). Genetically engineered removal of the internal translational initiation removes the smaller peptide in both the wild-type (lane 3) and the GOF mutants (lanes 5, 7 and 9). M: MagicMark (Invitrogen, CA). (C) Growth analysis. Removal of the RCK proteins does not alter the growth phenotype of the wild-type (rows 1 and 2). It also does not affect the toxic phenotype of the GOF mutants (rows 3–8).
The toxicity of overexpressing wild-type Kch is not due to K+ leakage through its filter
As shown in Figure 2B (panels 1–4, second row), when wild-type kch is overexpressed from the LacUV5 promoter, it blocks growth. We also used mutations to examine whether this effect is due to overwhelming K+ flux through the wild-type Kch channel proteins. Recall that G189Q (Table I) is one of the filter mutations in the kch of pGEM-S331P that suppresses the S331P mutant’s K+ sensitivity in K200, most likely due to blockage of K+ flux through the filter. We subcloned this mutation into pB11d-WT (without the S331P defect). As shown in Figure 7 (second row) this mutation was unable to suppress the sensitivity to high ionic strength due to the overexpression of kch upon IPTG induction. Even in the absence of high ionic-strength, overexpressing wild-type Kch interferes with growth (Figure 2B, panel 1, arrowheads). The degree of interference from overexpression varied from mutant to mutant (Figure 2B, panel 1; Figure 7, second panel).

Fig. 7. Mutation on the K+ filter does not rescue bacteria from the toxicity of Kch overproduction. When wild-type Kch is overproduced it is toxic to bacteria. A mutation directed at its K+ filter (G189Q) to block the K+ flux is unable to erase this toxicity.
Discussion
Much attention has been paid to the putative K+ channel in the popular model organism, E.coli (Milkman, 1994; Johansson and von Heijne, 1996; Ungar et al., 2001). Despite sequence homology and crystallographic structural analysis (Jiang et al., 2001), Kch has never been shown to pass ionic current electrophysiologically. Its biological role is also in doubt because kch-knockout bacteria present no discernible laboratory phenotype. Here we provide genetic evidence that Kch forms a K+-specific conduit in the E.coli membrane in vivo and speculate on its possible function.
The model
Kch has a K+-specific filter (GYGD sequence) and a gating structure (S6 and sensor), illustrated diagrammatically as functional parts in Figure 8A. The sensor (the cytoplasmic domain called RCK or KTN) binds an as yet undefined ligand and changes conformation, thereby pulling the S6 gate open (Figure 8A, top). We found that certain mutations in the sensor cause K+-specific sensitivity, and surmise that this phenotype reflects K+-specific leakage due to inappropriate opening of the gate (Figure 8A, middle). This idea is supported by the observation that blocking the K+ flux by mutating the filter suppresses the K+ sensitivity (Figure 8A, bottom). In general, ion channels are tightly controlled and are usually closed, since their ion passage down the gradient wastes energy. The open conformation is favored only when the sensor receives its stimulus (‘the gating principle’ such as ligand, voltage or stretch force). Here it is likely that the mutated sensor biases the gate towards opening even without the usual ligand.
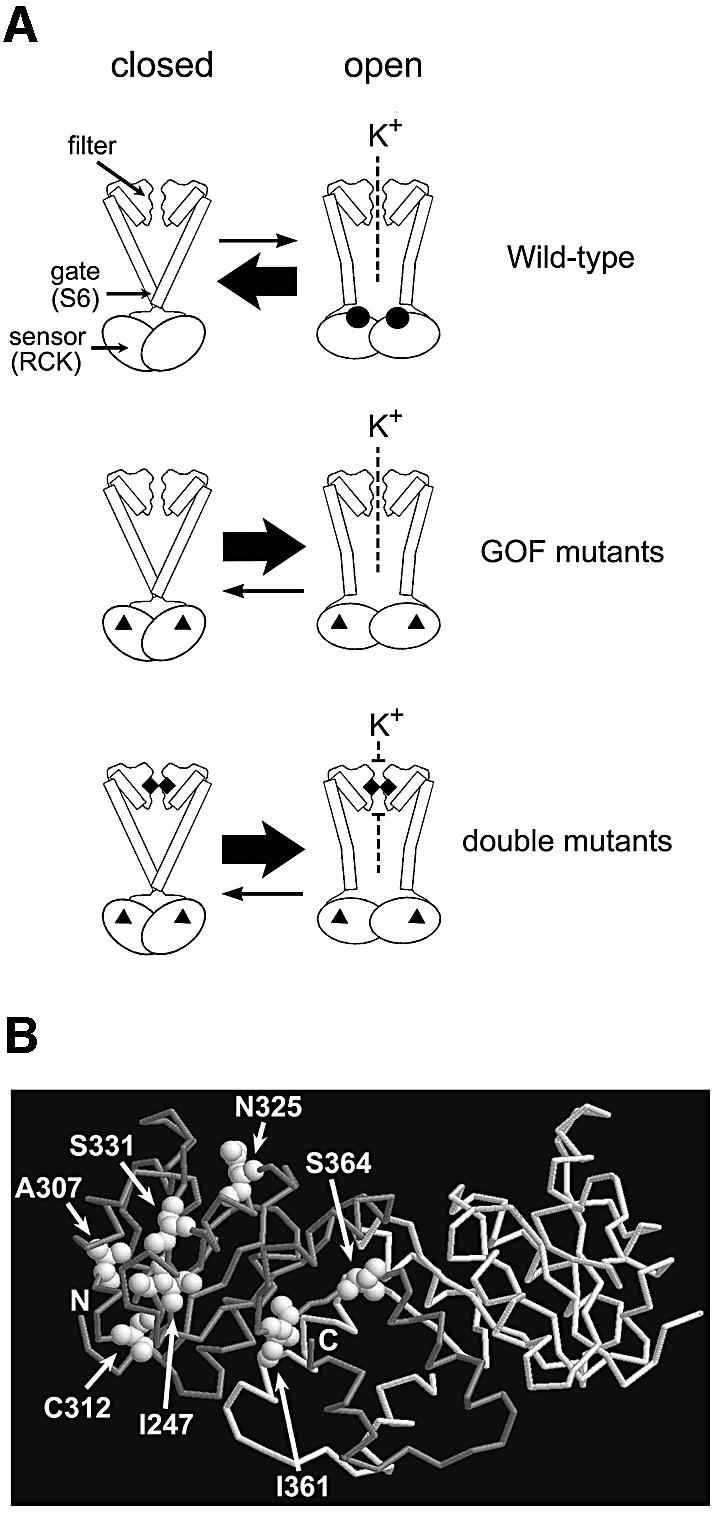
Fig. 8. A model of the functional parts of Kch and the locations of the GOF mutations in RCK. (A) Diagram of the wild-type, the GOF mutant and the double mutant Kch channels. Top: the wild-type Kch only opens briefly when ligands (black circles) bind to its cytoplasmic sensor domain (RCK). Middle: the GOF mutations (black triangles) in the RCK domain bias the sensor towards its open configuration even without the ligand. Bottom: a mutation (black diamonds) in the K+ filter disrupts the filter and blocks the K+ permeation. (B) A map of the GOF mutations in the RCK dimer of Kch (Jiang et al., 2001; PDB code 1ID1). I247, A307 and C312 are toward the core of the Rossmann fold. N325 and S331 are on the hydrophobic interface. I361 and S364 are at the hinge/flexible interface.
The positions of the GOF mutations indicate that the RCK (or KTN) domain is the sensor that controls the gate
Note that our mutagenesis (Figure 2) is not hypothesis-driven. We did not site-direct the mutations to the domain suggested by crystallography to be the sensor (Jiang et al., 2002a; Roosild et al., 2002). Instead, the entire kch-bearing plasmid was randomly mutagenized before screening for K+-sensitive mutants among the transformants. There are therefore no preconceived biases as to where such mutations should be located. Although the mutagenic experiment has not been carried out to saturation, all seven mutations we recovered that caused the K+-specific sensitivity were found to be in the RCK domain. This coincidence strongly supports the view that this domain is indeed the functional sensor governing gating.
In the reported 3D structures (Jiang et al., 2001, 2002a; Roosild et al., 2002) two RCK/KTN domains are dimerized through a β–α motif that follows a Rossmann fold in a handshake (Figure 8B). Upon ligand binding, two domains move relative to each other through this motif, called hinge (Roosild et al., 2002) or flexible interface (Jiang et al., 2002a). In the Roosild et al. model, two pairs of the KTN dimers further tetramerize through a hydrophobic interface to form a functional KTN tetramer. In the Jiang et al. model, a functional ‘gating ring’ is formed by four pairs of this RCK dimer (eight RCKs are required) also through the hydrophobic interface. In both models, the relative domain movements reposition the N-termini of RCK/KTN and pull open the bottom gate formed by S6. The positions of our GOF mutations (Figure 8B): I247T, A307V and C312F towards the core of the Rossmann fold; N325S and S331P at the hydrophobic interface; and I361T and S364P around the hinge/flexible interface, suggest that the stability of the RCK/KTN dimers and the flexibility of the relative domain movement are indeed important for the sensor to function properly. Interestingly, the positions of our GOF mutations are comparable with those of the spontaneous K+-leaking mutants reported from the KefC K+ efflux system (Miller et al., 1997), suggesting a conservation of this gating mechanism among RCK/KTN-containing K+ transport systems.
The K+ filter of Kch is like those of eukaryotic K+ channels
The extreme conservation of the TXGYGD sequence in a variety of animal K+ channels was recognized early (Schwarz et al., 1988) and shown later by genetic and crystallographic experiments to be the sequence that forms the K+-specific filter when tetramerized (Doyle et al., 1998). This extreme conservation reflects the precision needed to construct the filter that presents surrogate oxygens to exactly coordinate the dehydrated K+ ion in transit. This exquisite structure can easily be disrupted by mutations. As expected, extensive genetic and electrophysiological work on the Drosophila Shaker K+ channel (Heginbotham et al., 1994), the Arabidopsis Kat1 (Nakamura et al., 1997) and the murine Kir2.1 (So et al., 2001) showed that, with rare exceptions, filter-mutations are not tolerated. Although we have no direct electrophysiological evidence, our filter mutations (Table I) with the exception of Y188F all suppress the K+ sensitivity of the GOF mutants (Figure 5). This finding is consistent with the view that they block the K+ permeation by disrupting the filter. Doyle et al. (1998) observed that a ring of aromatic side chains surrounds the KcsA filter, and the tyrosine in the TXGYGD sequence interacts with them, forming a cuff that is thought to stabilize the filter. One exceptional filter mutation that still preserves its structure and function is the GYG-to-GFG substitution in Shaker, Kat1 and Kir2.1 K+ channels (Heginbotham et al., 1994; Nakamura et al., 1997; So et al., 2001), presumably because of the conservation of the aromatic ring between Y and F. As shown in Table I this mutation is the only one we isolated among the 15 colonies we randomly picked that does not suppress the K+ sensitivity of the Kch GOF mutant on K200 plates. Once again, these are not site-directed mutations. If there were other changes that would have maintained the filter integrity, they should have appeared among the isolates. This result implies that the filter and its immediate surrounding in the prokaryotic Kch are very similar to those of the eukaryotic channels, Shaker, Kat1 and Kir2.1, in 3D structure and in function, and that K+ leakage through the filter of the Kch GOF mutants is the cause of the K+-specific sensitivity.
The mutants’ K+ sensitivity could be due to losing membrane potential (Vm)
Although the above evidence supports the model that it is K+ permeation through the loose-cannon Kch channels that stops growth, we are not entirely certain how this happens. All other K+ uptake (Kdp, Trk, Kup) and K+ efflux systems (KefB, KefC) are intact in these mutant strains. Within experimental error, these GOF mutants also do not have any less internal K+ than the wild-type bacteria in LB media, as measured by flame photometry (data not shown). It is therefore unlikely that the GOF mutations generate permanently open pores that leak K+ constantly to perturb the cytoplasmic concentration of K+ ([K+]in). Such a mutation would presumably be unconditionally lethal and has not been reported in ion-channel literature, to our knowledge. It seems more likely that the GOF mutations bias the Kch channel towards its open conformation, leaking enough K+ to affect the membrane potential (Vm) but not the bulk [K+]in. It is possible that the opening of GOF-mutant channels increases the K+ permeability so as to keep the Vm near the K+ equilibrium potential (EK). The shallow Vm (interior less negative) could reduce the proton motive force below that needed for growth. Alternatively, the counteracting feedback increase in H+ extrusion may become energetically untenable for growth. These may explain why toxicity parallels the external K+ concentration ([K+]out) (Figure 3C), since an increase in [K+]out makes EK shallower. In liquid media, the exquisite K+-specific sensitivity of the GOF mutants is especially striking. Ten mM added K+ stops exponential growth (Figure 4C) and 5 mM K+ blocks growth initiation (Figure 4A). Raising the [K+]out from ∼0.5 mM (the estimated contaminant in the tryptone medium) to ∼5.5 mM would change the EK from –150 mV to –90 mV, assuming 200 mM [K+]in. This would have no effect on a bacterium with no passive K+ permeability (with wild-type Kch). If the GOF Kch channels indeed open uncontrollably, their permeability will tend to clamp the Vm at EK. This degree of depolarization will require the change equivalent to one pH unit to counteract. The energy wasted by the H+ pump in a futile effort to establish a large enough proton motive force may be growth prohibitive. Consistent with this notion is the observation that external acidity suppresses the K+-sensitive phenotype (Figure 4B) since external H+ provides the proton motive force. Animal cells have Vm resting near the EK due to their resting K+-channel permeability (Hille, 2001). Escherichia coli, however, is clearly under a selective pressure against such a device. This is evident from the appearance of stable revertants that would eventually populate K+-enriched liquid medium originally inoculated with the GOF mutants (data not shown). Some K+ channels apparently function in the uptake of bulk K+ in plants (Hirsch et al., 1998). To our knowledge, there has not been any proposal on the natural physiological role of Kch in the literature. If the above speculation on the GOF mutants is correct, the wild-type Kch may also function to adjust the proton motive force by adjusting the Vm and not the bulk [K+]in under certain circumstances. It would be interesting to discover the conditions in which this adjustment becomes necessary.
Overexpression of Kch in physiological study
Munsey et al. (2002) observed that overexpressing wild-type kch is toxic and additional K+ suppresses this toxicity. We also encountered this toxicity when Kch was promoted by LacUV5 in the presence of IPTG, but we found that the toxicity is aggravated by additional K+ (Figure 2B, panel 2, row 2). This toxicity correlates with ionic strength and persists even after the K+ filter is destroyed by a mutation (Figure 7) which argues that K+ conduction through Kch is not the cause of the toxicity here. Whether this general toxicity is due to large misfolded Kch aggregates, congestion of proteins in the membrane, or other reasons is currently unknown. As it is not germane to the thesis of this paper, our strategy is to study the wild-type and mutant kch genes controlled by kch’s native promoter (Figures 3–6). Bacteria vastly over-producing channel protein have proven useful in crystallography. In the context of cell physiology in vivo, however, results from such bacteria should be interpreted with caution.
Materials and methods
Strain and plasmids
The wild-type FRAG1 and its kch-null E.coli strains were gifts of Dr Ian R.Booth (University of Aberdeen). The empty pB11d plasmid (ampicillin resistance) was created by substituting the multiple cloning site-containing PstI–XbaI fragment of pB10b (Yoshimura et al., 1999) with that of pET21d (Novagen, USA).
To create pGEM-WT (Figure 3A), a BamHI-tagged 5′ primer (5′-CGGGATCCGATTTACTGGCTCAACCGTTATTGC-3′) and a PstI-tagged 3′ primer (5′-AACTGCAGTCCTTTTGAAAGCGCATTGTTAT GAG-3′) were used for PCR to clone the kch coding and flanking region from wild-type FRAG1 genomic DNA, and the resulting BamHI–PstI fragment was inserted in pGEM-3Zf(+) vector (Promega, USA). The correct insertion was confirmed by sequencing. For cloning kch open reading frame in pBluescript II KS– vector, a BspHI-containing 5′ primer (5′-GGATCCACATCATGAGTCACTGGGCTAC-3′) and an XhoI-tagged 3′ primer (5′-CCGCTCGAGCTATTTTTGCGCCGATTCTTT AC-3′) were used for PCR with pGEM-WT as the template. The resulting PCR fragment was inserted in pBluescript II KS– vector (Stratagene, USA) between EcoRV and XhoI. For the GOF screen, the BspHI–XhoI fragments containing mutated kchs were inserted in pB11d between NcoI and XhoI.
Culture media
All media used in this study were supplemented with 100 µg/ml ampicillin. For plates with IPTG, 1 µg/ml was added. The LB-based plates (called ‘LB’, ‘K200’, ‘Na200’ and ‘Sorbitol400’ here) contained basic ingredients: 10 g/l tryptone (Bacto Tryptone), 5 g/l yeast extract (Bacto Yeast Extract), 10 mM NaCl and 1.5% agar (Bacto Agar; all from Becton, Dickinson and Co., USA). In addition, 75.6 mM NaCl (5 g/l final), 200 mM KCl, 200 mM NaCl and 400 mM sorbitol were added to make ‘LB’, ‘K200’, ‘Na200’ and ‘Sorbitol400’ plates, respectively. The tryptone–agarose plates contained: 10 g/l tryptone (Bacto Tryptone; Becton, Dickinson and Co.); 0.75% agarose (BP160-500; Fisher Scientific, USA). The liquid media (‘TK5’, ‘TNa5’, ‘TS10’, ‘TH5’ and ‘TK5H5’ in Figure 4) all contained a basic ingredient of 10 g/l tryptone (‘T’ medium). In addition, 5 mM KCl, 5 mM NaCl, 10 mM sorbitol, 5 mM HCl or 5 mM KCl and HCl were added to make ‘TK5’, ‘TNa5’, ‘TS10’, ‘TH5’ and ‘TK5H5’ media, respectively. At 37°C, the pH of T, TK5, TNa5 and TS10 media is 7.3, and that of TH5 and TK5H5 media is 5.8.
Gain-of-function screen
The entire kch was first cloned into pBluescript II KS– vector and randomly mutagenized by in vivo XL1-Red Cell System (Stratagene, USA). The mutated kch was then cloned into a fresh expression vector, pB11d (between NcoI and XhoI), behind the IPTG-inducible LacUV5 promoter, and used to transform kch-null FRAG1 E.coli to eliminate mutations other than those in kch. The transformants were plated on LB plates and replicated to the restriction plates. Colonies missing on any of the restriction plates were picked, their plasmids were retransformed in fresh bacteria, and the phenotype was re-examined.
Phenotype assay
For all plate assays, a freshly streaked single colony was inoculated in 2 ml of LB medium, and grown aerobically at 37°C up to stationary phase (OD600 ∼ 3.5–4.5). The stationary cells were then diluted 102-, 103-, 104-, 105- and 106-fold in the background media of the assay plates. Drops of cells (5 µl) from each dilution tube were inoculated on assay plates and the plates were incubated in 37°C for 16 h (LB-based plates) or 24 h (tryptone-based plates) before photographs were taken.
For the growth curves in liquid media from stationary inocula (Figure 4), single colonies of fresh transformants bearing various plasmids were individually inoculated into 2 ml of LB medium and grew aerobically to stationary. Cells were spun down and resuspended in fresh tryptone medium (T) before being diluted to OD600 0.02 into 8 ml (Figure 4A and B) or 12 ml (Figure 4C) of test media in 25 × 150 mm test tubes, shaking at 275 r.p.m., 15° angle and 37°C. Growths were measured at OD600 every 1.5 h. If the cell density exceeded an OD600 of 0.4, samples were diluted with the same growth media before measuring. To test the effect of K+ on exponential growth (Figure 4C), a culture in such growth in the tryptone medium (T) was divided into three subcultures at 3 h (OD600 ∼ 0.15). Their growths were monitored continually with no addition, or with the addition of 10 mM KCl or NaCl.
Mutageneses
To randomly mutagenize the G187 codon, the pGEM-S331P plasmid was used as the template in the first-round PCR. A degenerated 5′ primer (5′-CATGTCAACCGTCNNNTATGGCGATATTG-3′) and the PstI-tagged 3′ primer were used to generate a PstI-tagged 3′ megaprimer, and the BamHI-tagged 5′ primer and a degenerated 3′ primer (5′-CAATATCG CCATANNNGACGGTTGACATG-3′) were used to generate a BamHI-tagged 5′ megaprimer. In the second-round PCR, the BamHI-tagged 5′ primer and the PstI-tagged 3′ primer together with the two megaprimers were mixed for PCR with the megaprimers themselves as the template. The final PCR product, a BamHI–PstI fragment, was inserted in pGEM vector. Random mutagenesis at the Y188 and the G189 codon was carried out using the same strategy with appropriate degenerated primers in first PCRs. The M240L mutation was generated similarly using oligos (5′-AACAATCATACACTGCATCGTAAAG-3′ and 5′-TTTACGATGCA GTGTATGATTGTTTC-3′) in the first-round PCRs to generate the megaprimers.
Membrane fractionation
The bacteria carrying the pGEM constructs were grown aerobically at 37°C in LB medium to stationary phase (OD600 ∼ 3.5), washed once in 4°C Pi buffer containing 50 mM Na-phosphate buffer pH 7.2, 10 mM KCl and 5 mM MgCl2, then French-pressed twice at 18 000 p.s.i. in the same buffer supplemented with 1 mM DTT, 0.5 mg/ml lysozyme, 5 µg/ml DNase, 2 µg/ml aprotinin, 2 µg/ml leupetin and 1 mM phenylmethylsulfonyl fluoride (PMSF). Unbroken cells and cell debris were pelleted at 19 000 g at 4°C for 5 min twice, and the membranes were harvested by ultracentrifugation at 4°C in the SW50.1 rotor of a Beckman centrifuge (Fullerton, CA) for 30 min at 45 000 r.p.m. The membrane pellets were then dissolved in Pi buffer supplemented with 1% SDS and 1 mM PMSF for immunoblot analysis.
Immunoblot
The membrane fraction or whole-cell lysate samples were run on a 10% SDS–PAGE gel for western blot analysis using anti-TKADSKESAQK antibody, a gift of Dr Ian R.Booth (University of Aberdeen) and ECL reagents (Amersham Biosciences, UK) for detection.
Acknowledgments
Acknowledgements
This work was supported by NIH47856. We thank Dr Ann Batiza for criticisms and Dr Ian R.Booth for the wild-type FRAG1, its kch– E.coli strains and the Kch monoclonal antibody.
References
- Doyle D.A., Cabral,J.M., Pfuetzner,R.A., Kuo,A.L., Gulbis,J.M., Cohen,S.L., Chait,B.T. and MacKinnon,R. (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science, 280, 69–77. [DOI] [PubMed] [Google Scholar]
- Heginbotham L., Lu,Z., Abramson,T. and Mackinnon,R. (1994) Mutations in the K+ channel signature sequence. Biophys. J., 66, 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. (2001) Ion Channels of Excitable Membranes. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Hirsch R.E., Lewis,B.D., Spalding,E.P. and Sussman,M.R. (1998) A role for the AKT1 potassium channel in plant nutrition. Science, 280, 918–921. [DOI] [PubMed] [Google Scholar]
- Jiang Y.X., Pico,A., Cadene,M., Chait,B.T. and MacKinnon,R. (2001) Structure of the RCK domain from the E.coli K+ channel and demonstration of its presence in the human BK channel. Neuron, 29, 593–601. [DOI] [PubMed] [Google Scholar]
- Jiang Y.X., Lee,A., Chen,J.Y., Cadene,M., Chait,B.T. and MacKinnon,R. (2002a) Crystal structure and mechanism of a calcium-gated potassium channel. Nature, 417, 515–522. [DOI] [PubMed] [Google Scholar]
- Jiang Y.X., Lee,A., Chen,J.Y., Cadene,M., Chait,B.T. and MacKinnon,R. (2002b) The open pore conformation of potassium channels. Nature, 417, 523–526. [DOI] [PubMed] [Google Scholar]
- Johansson M. and von Heijne,G. (1996) Membrane topology of Kch, a putative K+ channel from Escherichia coli. J. Biol. Chem., 271, 25912–25915. [DOI] [PubMed] [Google Scholar]
- Kumanovics A., Levin,G. and Blount,P. (2002) Family ties of gated pores: evolution of the sensor module. FASEB J., 16, 1623–1629. [DOI] [PubMed] [Google Scholar]
- Loukin S.H., Vaillant,B., Zhou,X.L., Spalding,E.P., Kung,C. and Saimi,Y. (1997) Random mutagenesis reveals a region important for gating of the yeast K+ channel Ykc1. EMBO J., 16, 4817–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkman R. (1994) An Escherichia coli homolog of eukaryotic potassium channel proteins. Proc. Natl Acad. Sci. USA, 91, 3510–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S., Douglas,R.M., Carter,P. and Booth,I.R. (1997) Mutations in the glutathione-gated KefC K+ efflux system of Escherichia coli that cause constitutive activation. J. Biol. Chem., 272, 24942–24947. [DOI] [PubMed] [Google Scholar]
- Munsey T.S., Mohindra,A., Yusaf,S.P., Grainge,A., Wang,M.H., Wray,D. and Sivaprasadarao,A. (2002) Functional properties of Kch, a prokaryotic homologue of eukaryotic potassium channels. Biochem. Biophys. Res. Commun., 297, 10–16. [DOI] [PubMed] [Google Scholar]
- Nakamura R.L., Anderson,J.A. and Gaber,R.F. (1997) Determination of key structural requirements of a K+ channel pore. J. Biol. Chem., 272, 1011–1018. [DOI] [PubMed] [Google Scholar]
- Ou X.R., Blount,P., Hoffman,R.J. and Kung,C. (1998) One face of a transmembrane helix is crucial in mechanosensitive channel gating. Proc. Natl Acad. Sci. USA, 95, 11471–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosild T.P., Miller,S., Booth,I.R. and Choe,S. (2002) A mechanism of regulating transmembrane potassium flux through a ligand-mediated conformational switch. Cell, 109, 781–791. [DOI] [PubMed] [Google Scholar]
- Ruta V., Jiang,Y.X., Lee,A., Chen,J.Y. and MacKinnon,R. (2003) Functional analysis of an archaebacterial voltage-dependent K+ channel. Nature, 422, 180–185. [DOI] [PubMed] [Google Scholar]
- Schrempf H., Schmidt,O., Kummerlen,R., Hinnah,S., Muller,D., Betzler,M., Steinkamp,T. and Wagmer,R. (1995) A prokaryotic potassium-ion channel with two predicted transmembrane segments from Streptomyces lividans. EMBO J., 14, 5170–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz T.L., Tempel,B.L., Papazian,D.M., Jan,Y.N. and Jan,L.Y. (1988) Multiple potassium-channel components are produced by alternative splicing at the Shaker locus in Drosophila. Nature, 331, 137–142. [DOI] [PubMed] [Google Scholar]
- Sesti F., Rajan,S., Gonzalez-Colaso,R., Nikolaeva,N. and Goldstein,S.A.N. (2003) Hyperpolarization moves S4 sensors inward to open MVP, a methanococcal voltage-gated potassium channel. Nat. Neurosci., 6, 353–361. [DOI] [PubMed] [Google Scholar]
- So I., Ashmole,I., Davies,N.W., Sutcliffe,M.J. and Stanfield,P.R. (2001) The K+ channel signature sequence of murine Kir2.l: mutations that affect microscopic gating but not ionic selectivity. J. Physiol., 531, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar D., Barth,A., Haase,W., Kaunzinger,A., Lewitzki,E., Ruiz,T., Reilander,H. and Michel,H. (2001) Analysis of a putative voltage-gated prokaryotic potassium channel. Eur. J. Biochem., 268, 5386–5396. [DOI] [PubMed] [Google Scholar]
- Wolters M., Madeja,M., Farrell,A. and Pongs,O. (1999) Bacillus stearothermophilus lctB gene gives rise to functional K+ channels in Escherichia coli and in Xenopus oocytes. Recept. Channels, 6, 477–491. [PubMed] [Google Scholar]
- Yoshimura K., Batiza,A., Schroeder,M., Blount,P. and Kung,C. (1999) Hydrophilicity of a single residue within MscL correlates with increased channel mechanosensitivity. Biophys. J., 77, 1960–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.F., Morais-Cabral,J.H., Kaufman,A. and MacKinnon,R. (2001) Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 angstrom resolution. Nature, 414, 43–48. [DOI] [PubMed] [Google Scholar]