

Abstract
Sequence-specific protein recognition of single-stranded nucleic acids is critical for many fundamental cellular processes, such as DNA replication, DNA repair, transcription, translation, recombination, apoptosis and telomere maintenance. To explore the mechanisms of sequence-specific ssDNA recognition, we determined the crystal structures of 10 different non-cognate ssDNAs complexed with the Oxytricha nova telomere end-binding protein (OnTEBP) and evaluated their corresponding binding affinities (PDB ID codes 1PH1–1PH9 and 1PHJ). The thermodynamic and structural effects of these sequence perturbations could not have been predicted based solely upon the cognate structure. OnTEBP accommodates non-cognate nucleotides by both subtle adjustments and surprisingly large structural rearrangements in the ssDNA. In two complexes containing ssDNA intermediates that occur during telomere extension by telomerase, entire nucleotides are expelled from the complex. Concurrently, the sequence register of the ssDNA shifts to re-establish a more cognate-like pattern. This phenomenon, termed nucleotide shuffling, may be of general importance in protein recognition of single-stranded nucleic acids. This set of structural and thermodynamic data highlights a fundamental difference between protein recognition of ssDNA versus dsDNA.
Keywords: DNA recognition/protein–DNA interactions/single-stranded DNA/specificity/telomeres
Introduction
Specific recognition of single-stranded nucleic acids by proteins is crucial for maintaining, manipulating and utilizing the genetic material contained in cells. Single-stranded nucleic acid sequences participate in a large variety of fundamental dynamic biological functions, including telomere regulation, mRNA splicing, DNA damage repair, DNA replication, recombination, chromosomal condensation, transcription regulation, translation, apoptosis, stages in bacterial and viral life cycles, and retrotransposition (Hohjoh and Singer, 1997; Miller et al., 1997; Waga and Stillman, 1998; Handa et al., 1999; Deo et al., 1999; Gallia et al., 2000; Tateishi et al., 2000; Changela et al., 2001; Gasior et al., 2001; Gomis-Ruth et al., 2001; Negroni and Buc, 2001; Pennock et al., 2001). Consequently, protein recognition of single-stranded nucleic acids has been clearly implicated in myriad pathological processes in humans, such as cancer, aging, various infectious diseases, autoimmune illnesses and drug dependency (Vaziri et al., 1993; Osugi et al., 1996; Bodnar et al., 1998; Stevens and Glick, 1999).
In the past few years, several high-resolution structures of sequence-specific ssDNA and ssRNA complexes have been determined: Dm sex-lethal, HnUP1, BstTRAP, EcoRho transcription terminator, Oxytricha nova telomere end-binding protein (OnTEBP), HnSF1, Hn polyA-binding protein, FBP and the tumor suppressor BRCA2 (Antson et al., 1999; Bogden et al., 1999; Deo et al., 1999; Ding et al., 1999; Handa et al., 1999; Classen et al., 2001; Horvath and Schultz, 2001; Liu et al., 2001; Braddock et al., 2002; Peersen et al., 2002; Yang et al., 2002). The interactions observed in these structures hint at possible mechanisms for sequence-specific recognition, and several common themes have emerged (Theobald et al., 2003a). Single-stranded nucleic acids are bound on the surfaces of β-sheets. Phosphodiester groups are extensively solvent exposed, while DNA or RNA bases are in intimate contact with the protein. Bases are typically bound by stacking with aromatic side chains, and hydrogen bond donor and acceptor groups provide recognition of the edges of the bases.
In general, the molecular basis of sequence specificity has been inferred from the structure of the cognate complex. However, structural comparisons of both cognate and non-cognate complexes are required for elucidating the mechanisms of preferential binding, while parallel thermodynamic studies are necessary to evaluate the energetic importance of observed interactions (von Hippel, 1994). Accordingly, we have investigated the molecular basis of cognate ssDNA recognition via comparisons of 10 different non-cognate OnTEBP complexes, using both crystallographic studies and thermodynamic measurements (PDB ID codes 1PH1–1PH9 and 1PHJ). Two of these non-cognate ssDNAs correspond to DNA intermediates which occur transiently during extension of the telomere by the enzyme telomerase (Tel+T and Tel-3 in Table I) (Zahler and Prescott, 1988). This structural work, complemented by relevant energetic analyses, provides the most detailed information available revealing how any sequence-specific ssDNA–protein complex responds to non-cognate sequences.
Table I. ssDNA sequences.

All oligonucleotides are numbered beginning from 1 at the 5′ end; –, abasic; N, random mixture of all four nucleotides.
OnTEBP recognizes a 16-nucleotide 3′-terminal single-stranded T4G4T4G4 telomeric DNA with high affinity (Raghuraman and Cech, 1990; Fang et al., 1993) and strong sequence specificity (Price and Cech, 1987; Gray, 1992; Classen et al., 2003) (Figure 1). OnTEBP contains two protein subunits, a 56 kDa α-subunit and a 41 kDa β-subunit, which together form a remarkably stable α:β:ssDNA ternary complex with 1:1:1 stoichiometry. In the structure of the cognate complex the ssDNA is bound in a cleft, formed from the surfaces of β-sheets presented by multiple OB-folds, between the N-terminal domain of α and the globular portion of β (Horvath and Schultz, 2001). The DNA is folded into a contorted configuration in which generally the bases are enclosed within the complex and the phosphodiester groups are solvent exposed. The 3′-terminal deoxyribose is recognized via burial within the ternary complex, a useful feature for these studies as it positions the DNA in a specific register.

Fig. 1. Cognate OnTEBP complex. The crystal structure of OnTEBP ternary complex, PDB ID 1OTC (Horvath and Schultz, 2001). (A) A 12-mer ssDNA (G4T4G4) is bound in a cleft between the N-terminal domain of α (mauve) and the globular portion of β (blue). (B) OnTEBP recognizes a single-stranded 16-nucleotide 3′-overhang, shown boxed in magenta. (C) Positive FO–FC electron density map of Tel+T complex, contoured at 2σ, in a stereoview similar to Figure 2B. The ssDNA was omitted during the map calculation. All images were prepared with Molscript (Kraulis, 1991), Raster3D (Merritt and Bacon, 1997) and Bobscript (electron density) (Esnouf, 1999).
Sequence specificity and binding affinity of non-cognate sequences
The high degree of specificity exhibited by OnTEBP binding to its cognate sequence was confirmed using a competition assay (Sidorova and Rau, 1996). The assay directly measures Kspec, which is the ratio of the affinity constant of the protein for cognate DNA to the affinity constant of the protein for non-specific DNA. Previous work has shown that various non-specific ssDNAs give an ∼Kspec ≥ 105 for the OnTEBP ternary complex (Gray, 1992), and we determined that Kspec = 8.5 × 104 for a cognate sequence relative to a pool of random ssDNA of the same length. Thus the OnTEBP complex is highly sequence specific, with a Kspec comparable with that of classical double-stranded DNA binding proteins such as EcoRI (Lesser et al., 1990), BamHI (Engler, 1998), lac repressor (Mossing and Record, 1985) and Cro protein (Takeda et al., 1992).
All 10 of the non-cognate ssDNAs bind with significantly weaker affinity than the cognate sequence, with the single exception of OxyG10I (Table II). Relative to the cognate sequence, the single base substitutions and modifications result in increases in KD of up to a factor of 100. A Krel of 6700 [Krel = KD(noncognate)/KD(cognate)] is observed for a ssDNA in which the three 3′-most cognate nucleotides are missing (OxyTel-3), equivalent to an average increase in KD of 19-fold per missing nucleotide. To elucidate the molecular basis of the energetic perturbations seen with these non-cognate ssDNAs, we solved the crystal structures of the 10 corresponding non-cognate complexes.
Table II. Summary of binding and structural data for OnTEBP non-cognate complexes corresponding to oligos in the crystal structures.
| ssDNA name | Summary of structural differences | Krela | ΔΔGrel (kJ/mol) | ΔCSAb (Å2) |
|---|---|---|---|---|
| Tel+T | Backbone conformation; extruded nucleotide; – 1 H-bond; +1 water-mediated interaction; + 2 intra-DNA H-bonds | 5.3 | 4.2 | +28 |
| Tel-3 | Major differences (see text) | 6700 | 22 | –352 |
| G3X | Backbone conformation; extruded nucleotide | 56 | 10 | 0 |
| G10I | – 1 H-bond | 0.5 | –2.0 | –12 |
| G10X | – 2 H-bonds; – 1 base stacking | 100 | 11 | –111 |
| G10A | – 2 H-bonds | 31 | 8.8 | –23 |
| G10C | – 1 H-bond; smaller base stacking | 37 | 9.0 | 0 |
| G10T | – 1 H-bond; + 1 H-bond; smaller base stacking | 12 | 6.2 | +9 |
| G11C | – 1 H-bond; + 1 water-mediated interaction; smaller base stacking | 81 | 11 | –31 |
| G11T | – 1 H-bond; + 1 water-mediated interaction; smaller base stacking | 44 | 9.4 | –27 |
X, abasic; CSA, contact surface area.
aKrel = KD(non-cognate)/KD(cognate).
bΔCSA = non-cognate CSA – cognate CSA.
Crystal structures of 10 non-cognate On TEBP complexes
Our analysis of 10 independent crystal structures of OnTEBP complexed with non-cognate ssDNAs revealed a surprising malleability of the complex in accommodating non-cognate nucleic acids. In all complexes, the overall protein conformation remains nearly identical to that of the cognate ternary complex. The ssDNA, on the other hand, exhibits dramatic differences in several of the non-cognate complexes. In the complexes with Tel+T, Tel-3 and G3X, the ssDNA undergoes an unprecedented conformational rearrangement involving a shift in the register of the ssDNA of up to nine nucleotides, which re-establishes a more cognate-like ssDNA recognition pattern. This register shift is accompanied by a nucleotide ‘flip’, wherein an entire nucleotide swings out of the complex. The other seven non-cognate complexes display interesting subtle structural changes illustrating how a protein can accommodate various non-cognate nucleotide substitutions.
Register shifting and nucleotide shuffling in non-cognate complexes
Addition of a thymine nucleotide at the 3′ end of the cognate ssDNA (Tel+T complex) (Krel = 5.3). The 3′-OH of the cognate ssDNA is buried deep within the complex and hydrogen bonds to the backbone amide of αK66 (Figure 2A). There is no apparent room for an additional nucleotide. Therefore it was surprising to discover that Tel+T, a ssDNA with a single thymine nucleotide added to the 3′ end of the cognate sequence, binds with nearly the same affinity as the cognate ssDNA. Notably, Tel+T ssDNA occurs as an intermediate during polymerization of the telomeric DNA by telomerase (Zahler and Prescott, 1988).

Fig. 2. Tel+T complex. The OnTEBP α subunit is shown in blue and the β-subunit is shown in mauve. Carbon atoms are steel blue in the cognate ssDNA and white in the non-cognate ssDNA. Protein side chains and nucleotides G1–T6 are omitted for clarity. (A) The 3′-end region of the cognate complex. Note the phosphodiester backbone in the region of T7 and T8, and the adjacent T8:G12 stack. (B) The corresponding 3′-end region of the Tel+T complex. Relative to the cognate structure, G9–T13 is shifted in register one nucleotide towards the 5′ end (counterclockwise in this figure). This register shift is arrested at T7; T8 is ejected from the complex. A tilted G9:T13 stack replaces the T8:G12 stack in the cognate complex. (C) Stereoview of the Tel+T complex superimposed with the cognate complex. The cognate ssDNA is shown in dark blue, and the Tel+T ssDNA in yellow. Note the close superposition at base positions at G11, G10 and G9, though T7 has reoriented slightly. Phosphorus atoms 5′ to the cognate G9 position and 3′ to the cognate T7 position are in very similar positions despite the shuffled nucleotide.
The structure of the Tel+T complex reveals that OnTEBP binds the 3′-OH of Tel+T in precisely the same protein site as does the cognate complex (Figure 2B). The DNA register shifts one base towards the 5′ end compared with the cognate sequence in order to maintain this 3′-OH interaction. However, this register shift is not propagated throughout the entire length of the DNA. Rather, T8 flips entirely out of contact with the protein and into solution. This nucleotide expulsion arrests the register shift initiated by the ‘extra’ thymidine (T13) and re-establishes the proper cognate register for nucleotides G1–T7.
The shift in register of the 3′-loop region effectively results in two base substitutions. G12 and T8 of the cognate sequence are replaced with T13 and G9, respectively, of Tel+T. Therefore the T8:G12 stack that closes the 3′ loop in the cognate complex is exchanged for a G9:T13 stack in the Tel+T complex. Otherwise, the molecular conformations of the cognate and non-cognate complexes are nearly identical, including the portions of the DNA backbone that flank the flipped-out nucleotide Tel+T T8 (Figure 2C). All aromatic stacking interactions are maintained.
The Tel+T non-cognate ssDNA results in a strikingly small reduction in binding energy relative to the cognate ssDNA (ΔΔGrel = 4.2 kJ/mol). Though the Tel+T ssDNA has undergone a register shift involving six nucleotides, two effective nucleotide substitutions and a dramatic repositioning of the ssDNA backbone, the complex maintains nearly all cognate ssDNA–protein interactions. If the crystal structure of the non-cognate Tel+T complex had been unavailable, binding results for the Tel+T DNA might have been misinterpreted as a lack of 3′-end binding rather than shuffling of nucleotides.
Structure of an OnTEBP complex with three 3′-nucleotides removed (Tel-3) (Krel = 6700). We were similarly surprised to obtain crystals of a complex containing a DNA in which three nucleotides at the 3′ end were entirely removed (Tel-3). Similar to Tel+T, the Tel-3 ssDNA corresponds to a telomeric intermediate that occurs during extension of the chromosomal terminus by telomerase. As shown in Figure 3B, OnTEBP binds to the Tel-3 ssDNA by shifting the DNA register one nucleotide towards the 3′ end relative to the cognate sequence, leaving the two 3′-most nucleotide binding sites entirely unoccupied. This register shift, unlike that seen in Tel+T and G3X, is propagated for the entire length of the DNA sequence (involving a total of nine nucleotides). This results in base substitutions at two positions: G4 in the Tel-3 complex replaces cognate T5, and T8 in the Tel-3 complex replaces cognate G9.

Fig. 3. Tel-3 complex. (A) Cognate ssDNA bound in the complex (stereoview). (B) Corresponding view of the Tel-3 complex, which lacks the final three guanosine nucleotides at the 3′ end. The entire ssDNA has shifted towards the 3′ end by one nucleotide. G9 occupies the cognate G10 protein binding pocket, G4 replaces T5 and T8 replaces G9. T7 no longer has a stacking partner and is swung away from the complex into solution (towards the viewer). Though omitted for clarity, there are only minor differences involving protein residues.
Three nucleotide binding sites, corresponding to T8, G11 and G12 of the cognate complex, are completely unoccupied in the Tel-3 structure, yet the protein side-chain conformations are very similar to those in the cognate complex. If the Tel-3 ssDNA had not shifted towards the 3′ end by one nucleotide, the cognate G10 binding pocket would also have remained vacant. Nevertheless, the register shift observed in the Tel-3 complex allows G9 to occupy the cognate G10 binding site.
As one might predict, Tel-3 results in the largest energetic destabilization of the non-cognate ssDNAs (ΔΔGrel = 22 kJ/mol, or ∼7 kJ/mol per removed nucleotide). The DNA–protein contacts found in the G10 binding site appear strong enough to outweigh the introduction of non-cognate interactions at the T5 and G9 binding sites arising from the shift of the ssDNA. Relative to the cognate complex, the Tel-3 complex has lost 352 Å2 of contact surface area between the protein and ssDNA, amounting to nearly a quarter of the contact surface area in the cognate complex. Presumably, the majority of the ΔΔGrel for the Tel-3 complex arises from the evacuation of the predominantly non-polar T8, G11 and G12 binding sites.
Abasic G3X nucleotide substitution at position G3 (Krel = 56). Substitution of G3 with an abasic nucleotide also results in a large unanticipated change in the ssDNA conformation (Figures 4B and C). The abasic nucleotide is entirely expelled from the complex, shifting the register of the ssDNA in the 3′ direction. G1 and G2 occupy the protein binding sites that G2 and G3 occupy in the cognate crystal structure. The abasic G3X ribose is flipped out of contact with the α-protein subunit and into solution with virtually no repositioning of the adjoining bases. All the interactions observed between the protein and the ssDNA observed in the cognate complex are maintained in the non-cognate G3X complex.
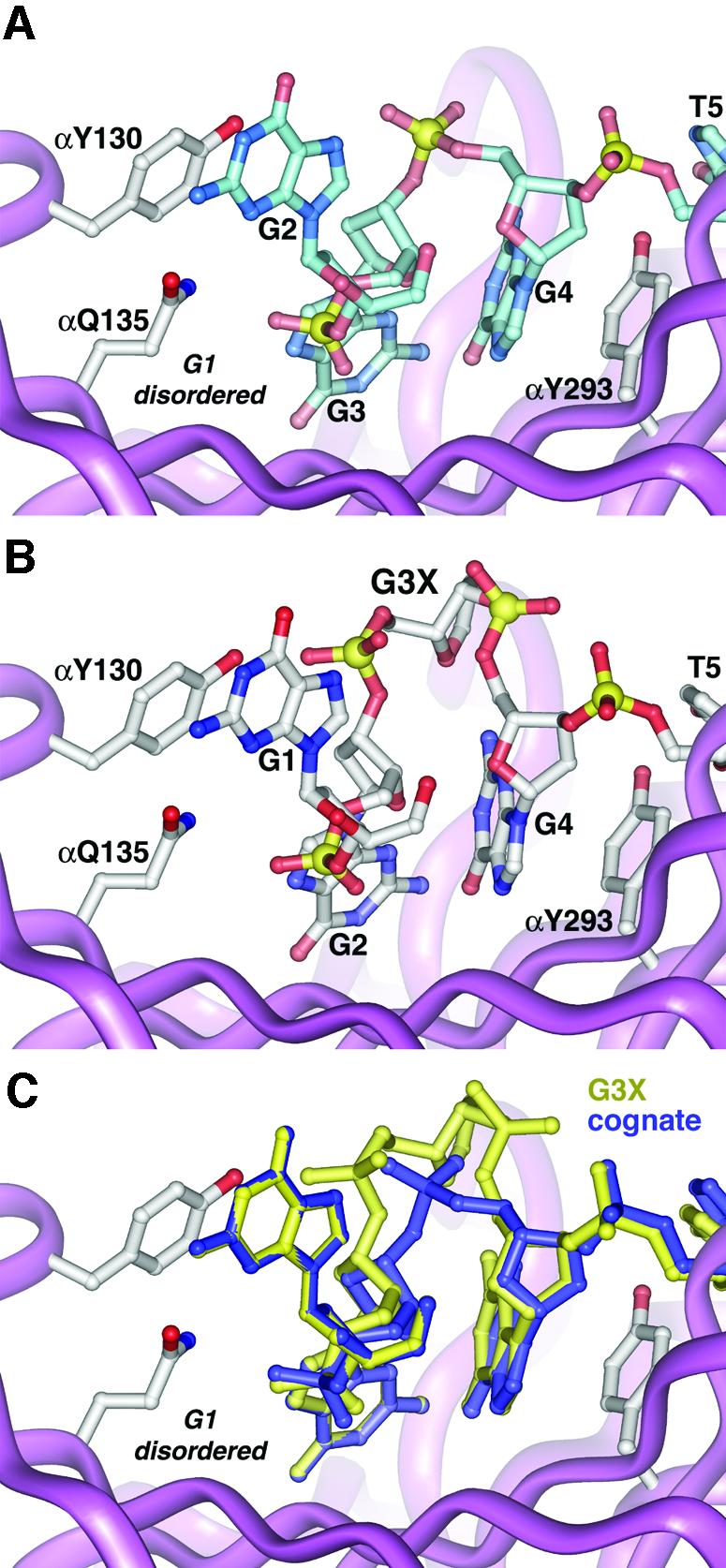
Fig. 4. G3X complex. (A) Cognate ssDNA conformation near the G3 position. G1 at the very 5′ end is disordered in the cognate crystals. (B) The non-cognate ssDNA conformation in the G3X complex. Via a register shift, G1 replaces G2, and G2 in turn replaces G3, leaving the cognate base contacts with the protein unperturbed. The abasic G3X nucleotide is swung away from the protein into solution. (C) Superposition of the non-cognate G3X complex with the cognate complex. Despite introduction of an extra nucleotide, identical protein–ssDNA contacts are made in both complexes.
If the non-cognate G3X crystal structure were not available, the most parsimonious explanation for our thermodynamic results would be that the energetic cost of the G3X mutation (ΔΔGrel = 10 kJ/mol) is due to loss of the various protein contacts with the G3 base. However, the observed difference in binding affinities does not result from contacts perturbed at the G3 binding site, since all contacts there are identical to the cognate complex. Apparently, ΔΔGrel results from the alternate conformation of the G3X phosphodiester backbone and from protein contacts perturbed at G1. This energetic difference is surprisingly small given the extent of the conformational rearrangement of the G3X ssDNA backbone.
Interactions with G10
We chose to investigate G10 because it occupies a pivotal position in the complex, bound between the α- and β-subunits, making an intriguing stacking interaction with the guanidium portion of an arginine side chain (Figure 5A). The G10 base is sandwiched between αY239 and βR140, the latter of which packs flat against the base and interacts electrostatically with the phosphodiester group on the 5′ side of this nucleotide. Two direct hydrogen bonds are made between G10 and the protein: N7 to βK145, and the exocyclic amino group to the backbone oxygen of βS102. The exocyclic oxygen participates in a water-mediated interaction with the backbone amide from βA110 (omitted from Figure 5 for clarity). Interestingly, G10 adopts the syn conformation about the C1′–N glycosyl bond.

Fig. 5. Complexes with G10 nucleotide substitutions. Hydrogen bonds are shown as solid yellow sticks. Water molecules are shown as small turquoise spheres. The backbone amide of βA110, mentioned in the text, has been omitted in all images for clarity. (A) Detail of the cognate protein–ssDNA interactions at the G10 position. G10, in the syn conformation, is bound between αY239 and βR140, with two hydrogen bonds shown between the base and the protein. (B) G10I: note the absence of a hydrogen bond with the backbone carbonyl of βS102. (C) G10X: the protein remains in a conformation similar to cognate. An exception is αY239, which is rotated into a preferred rotamer conformation. A water molecule replaces the G10 guanine and interacts with βK145. (D) G10A: no DNA–protein hydrogen bonds are observed. The adenine is in the anti conformation, flipped 180° relative to the guanine. (E) G10C: two fewer hydrogen bonds, relative to the cognate complex, result in a similar protein conformation, maintaining all stacking interactions. (F) G10T: One hydrogen bond is similar to the cognate complex (between the base and βK145), while a novel hydrogen bond is made between the thymine O2 and αT240.
G10I complex (Krel = 0.5). When G10 is replaced with inosine, which lacks only the exocyclic amino group, only slight changes in the complex are observed (Figure 5B). Relative to the cognate complex, the direct hydrogen bond between the backbone carbonyl group of residue βS102 to the exocyclic amino group of G10 cannot form in the non-cognate G10I complex. All other structural features remain identical. Reflecting the lack of structural rearrangement, the G10I substitution results in a statistically insignificant energetic perturbation. The βS102–G10:N2 hydrogen bond makes no measurable contribution to specific recognition.
G10X complex (Krel = 100). As shown in Figure 5C, complete removal of the base at position G10 of the DNA (G10X) results in larger, although still modest, changes in the structure. βR140, which stacks on the cognate guanine, remains locked in position, while αY239 and αE238 have both reoriented slightly to preferred rotamer conformations. In total, there is a loss of two direct hydrogen bonds between the ssDNA and the protein (guanine N2 to the backbone carbonyl of βS102, and guanine N7 to the ε-amino group of βK145), two stacking interactions with aromatic amino acid residues (βR140 and αY239) and one water-mediated interaction (guanine O6 to the backbone amide of βA110). The sole new interaction is from an ordered water that has entered the cavity vacated by the missing base, where it satisfies the hydrogen bond previously made to βK145 from the cognate guanine. In total, the removal of an entire base at position G10 results in the loss of 111 Å2 of contact surface area relative to the cognate complex.
As might be expected, the abasic modification results in the largest energetic loss at this position, yet this loss is only 11 kJ/mol. The novel water interaction in the G10X complex, combined with the slight repositioning of two amino acid side chains to favorable rotamers, may partially compensate for the loss of interactions with the guanine base.
G10A complex (Krel = 35). In the G10A complex, the conformation of the adenine base is flipped nearly 180° about the C1′–N glycosyl bond relative to the cognate guanine, and it sits in an anti conformation (Figure 5D). Despite this 180° rotation of the base, the adenine remains stacked between βR140 and αY239 with essentially no change in the positions of the side chains. Two direct hydrogen bonds are lost (G10:N2 to the βS102 backbone carbonyl and G10:N7 to βK145), and the adenine lacks any direct hydrogen-bond interactions with the protein. An indirect water-mediated interaction with βA110 is maintained in a manner similar to that of the cognate complex.
The G10A substitution results in a loss of 8.8 kJ/mol of binding energy. Guanine is distinguished from the other three DNA bases by its preference for the syn conformation. The rather small loss of energy due to this substitution may result from a combination of altered stacking, loss of hydrogen bond formation and the anti versus syn preference of the adenosine nucleotide.
G10C complex (Krel = 37). As illustrated in Figure 5E, with G10C a pyrimidine substitutes for a purine with an overall molecular orientation similar to the cognate complex. The N3 of the cytosine hydrogen bonds with βK145, just as the guanine N7 does in the cognate complex. The indirect water-mediated interaction between the cognate guanine O6 carbonyl oxygen and the backbone amide of βA110 is replaced by a similar water-mediated interaction between the exocyclic amino group (N4) of the cytosine and the same backbone amide of βA110. This nucleotide substitution results in only two major differences: (1) the loss of a single direct hydrogen bond at the protein–DNA interface (cognate G10:N2 to the βS102 backbone carbonyl), and (2) the introduction of a small cavity between the ssDNA and the protein where the guanine N1, N2 and O6 are positioned in the cognate complex. The βR140 and α239 side chains are unchanged and stack flat against the cytosine base.
Like G10A, the G10C substitution results in a loss of ∼9.0 kJ/mol of binding energy, likely due to the altered stacking energies and the loss of close packing for cytosine versus guanine.
G10T complex (Krel = 12). Similar to G10C, the thymidine base replaces the cognate guanine with relatively little change (Figure 5F). In the cognate complex, the guanine N7 group hydrogen bonds to βK145; this hydrogen bond is maintained in the non-cognate complex in which the O2 hydrogen bonds to βK145. The indirect water-mediated interaction seen in the cognate complex is also maintained. The G10T substitution results in two noteworthy differences: (i) the loss of a single direct hydrogen bond at the protein–DNA interface (guanine N2 to the βS102 backbone carbonyl), and (ii) the introduction of a novel interaction between the DNA and protein (thymine O2 to the αT240 hydroxyl group). The thymidine methyl group is exposed to solvent.
Intriguingly, the G10T substitution results in a slightly smaller yet statistically significant energetic loss (ΔΔGrel = 6.2 kJ/mol; P < 0.05) than either the G10A or the G10C substitution. It is perhaps noteworthy that the G10T substitution maintains two direct hydrogen bonds with the protein, in contrast with the G10A and G10C complexes which both have fewer hydrogen bonds.
Interactions with G11
The G11 was chosen because it is bound deep within a markedly hydrophobic pocket between the two OB folds of the α-subunit in an anti conformation (Figure 6A), features that may contribute to specificity for a guanine base. The two faces of the G11 base pack against several aliphatic and aromatic protein side chains. A direct hydrogen bond is made between the backbone carbonyl of αK261 and the exocyclic amino group of G11, which in turn participates in a water-mediated interaction with a ring nitrogen of αH114.

Fig. 6. G11 nucleotide substitutions. (A) View of the cognate protein–ssDNA interactions at the G11 position. G11 is bound in a deep cleft between two domains of the α subunit, surrounded by hydrophobic residues. A direct hydrogen bond is made between the G11 base and αK261. The base and αH114 interact via a bound water molecule. (B) G11C and (C) G11T: a water molecule is involved in a three-way indirect interaction between the bases, αK261 and αH114. The pyrimidines are tilted 15° relative to the cognate guanine.
G11C complex (Krel = 81). As with the G10 substitutions, the G11C substitution is accommodated by the ternary complex with only minor structural changes. The protein side-chain conformations are indistinguishable from those of the cognate complex. As shown in Figure 6B, the plane of the cytosine base has rotated about the glycosidic bond ∼15° with respect to the plane of the cognate guanine, resulting in a more perpendicular orientation of the base with respect to the attached ribose ring.
The cytosine O2 makes a water-mediated contact with a ring nitrogen of αH114. This water molecule is well ordered in the G11C crystal structure and interacts with the backbone carbonyl of αK261 to form a three-way interaction at the protein–DNA interface. This water-mediated interaction has replaced a direct hydrogen bond that previously existed between the exocyclic amino group of the cognate G11 guanine and the backbone carbonyl of αK261. Therefore a water molecule appears to help adapt this site for interaction with a non-cognate cytidine nucleotide. The G11C substitution results in a larger reduction in binding free energy (ΔΔGrel = 11 kJ/mol) than the equivalent substitution at G10, perhaps due to the greater hydrophobic character of the G11 binding pocket.
G11T complex (Krel = 44). The ternary complex also accommodates the G11T substitution with minor structural changes (Figure 6C). Virtually identical to the G11C complex, the thymine base has rotated about the glycosidic bond by ∼15°, while the thymine O2 is involved in a three-way water-mediated interaction at the protein–DNA interface.
G11T also results in a somewhat larger reduction in binding free energy (ΔΔGrel = 9.4 kJ/mol) than the corresponding substitution at the G10 position, which again may reflect the greater hydrophobic nature of the G11 binding site.
Discussion
We have explored the molecular basis of cognate telomeric ssDNA recognition via structural and thermodynamic comparisons of 10 different non-cognate OnTEBP complexes, two of which include ssDNAs that correspond to sequences found during the process of telomeric polymerization. This analysis has revealed an unanticipated plasticity in accommodating non-cognate sequences, including conformational rearrangements involving shifts in the ssDNA register of up to nine nucleotides, a phenomenon that we have termed nucleotide shuffling. The conformational perturbations that result from individual base changes in the cognate sequence are accompanied by significant, yet relatively modest, reductions in binding affinity. Without the structural data, these energetic differences might have been misinterpreted as reflecting loss of binding interactions in an otherwise unchanged ssDNA register. Instead, we show that the complex often finds a more energetically favorable solution for dealing with the altered nucleotide sequence, often involving contacts far removed from the mutated site.
Relative to dsDNA, single-stranded nucleic acids are very flexible and the ssDNA phosphodiester backbone can explore a large conformational space. Via various conformational rearrangements that exploit this flexibility, a non-cognate ssDNA can adjust its sequence register to preserve cognate-like interactions and maximize the energetics of favorable contacts. By using the different techniques discussed below, the strict requirement for a specific linear sequence of bases can be eliminated, often without substantial energetic penalty.
In some structures, such as the G3X, Tel+T and Tel-3 complexes, a specific base has been substituted, deleted or modified, yet all cognate interactions at the corresponding binding site are maintained because a neighboring base is able to replace the non-cognate base. In these three non-cognate structures, a large fraction of the energetic effects of the ssDNA base substitutions arise from structural perturbations that are distant from the cognate mutated site. Therefore structural data is essential for correctly interpreting the physical basis of our thermodynamic perturbation studies. On the other hand, the magnitude of structural change observed in the ssDNA and protein does not necessarily correlate with a similar change in binding affinity, a fact that underscores the importance of binding studies in dissecting the energetic importance of interactions observed in high-resolution structures.
Local sequence context is critical for determining the effects of a particular base substitution. For example, a register shift and base expulsion is conceivable in the G10X complex, as is seen in the G3X complex. However, the interaction energy contributed by adjacent bases (e.g. T8, G9, G11 and G12) appears to outweigh the effects of removing G10 and thereby prevents a shift in G10X DNA register. In complexes where the register does not shift, base stacking is always maintained (both with other ssDNA bases and with aromatic protein side chains), likely reflecting the energetic importance of stacking interactions in protein–ssDNA complexes.
Water molecules and hydrogen bonds are used creatively in these complexes to accommodate various base changes. Water-mediated interactions can substitute for direct protein–ssDNA hydrogen-bond interactions (e.g. G11C and G11T complexes). In the abasic G10X complex, water fills the cavity left by removal of the base. Novel and seemingly specific hydrogen-bond interactions can be formed in non-cognate complexes (such as seen with G10T). In other cases, cognate-like hydrogen-bond interactions are preserved despite the substitution of a non-cognate base.
Nucleotide shuffling has important consequences for protein recognition. In double-stranded DNA–protein complexes, single base-pair changes from the cognate sequence frequently result in very large reductions in binding affinity. Because one base pair can account for nearly the entire binding energy in dsDNA systems, the energetic contribution of single nucleotides to the overall specific binding affinity is not additive but highly cooperative (Lesser et al., 1990; Takeda et al., 1992; Engler et al., 1997; Frank et al., 1997). In contrast, we observe that single base changes in the cognate ssDNA result in modest decreases in binding affinity that are consistent with additivity. Detailed binding studies with OnTEBP have confirmed this observation (Gray, 1992; Classen et al., 2003), and similar results have been seen in several other ssDNA– and ssRNA–protein complexes, including Cdc13, PTB, SF1, U2AF65 and Sex-lethal (Berglund et al., 1997; Perez et al., 1997; Singh et al., 2000; Anderson et al., 2003). Therefore, unlike what is commonly observed with sequence-specific dsDNA–protein complexes, low cooperativity between cognate nucleotides may be a general characteristic of sequence-specific recognition of single-stranded nucleic acids. In light of our crystal structures with non-cognate ssDNAs, this feature can be straightforwardly interpreted as the consequence of multiple near-isoenergetic conformations available to many similar ssDNA sequences.
The generality of this principle is suggested by three protein–RNA complexes, two involving hairpins with short RNA stretches comparable to a single-stranded nucleic acid. Alternate nucleic acid conformations, albeit less extensive than those described here, are also seen with an unnatural nucleotide in the RNA tetraloop of the MS2 coat binding protein complex (Grahn et al., 2000) and in the Nova KH domain complexed with a stem loop SELEX RNA involving nucleotides, outside the core UCAC recognition motif, with no or few protein contacts (Lewis et al., 2000). A subtle conformational rearrangement has also been observed in the ssRNA ‘spacer’ region of the TRAP–ssRNA complex (involving an inserted base lacking direct protein contacts) (Hopcroft et al., 2002).
These observations emphasize the fundamental differences between protein recognition of single-stranded versus double-stranded nucleic acids. Cognate complexes with double-stranded nucleic acids have a high degree of complementarity with few waters at the protein/nucleic acid interface (Garner and Rau, 1995; Sidorova and Rau, 1996) and involve local folding of the protein (Spolar and Record, 1994). Non-cognate complexes in these systems have quite different structures, such as poor interface complementarity with a large solvent content and an absence of local folding (Record et al., 1991; Engler et al., 1997; Oda et al., 1998). In contrast, both the cognate OnTEBP–ssDNA complex and the non-cognate complexes described here have extensive comparable shape complementarity at the protein–ssDNA interface with few water-mediated contacts. Similarly, there is no obvious pathway for the ssDNAs to enter the protein binding site if the OnTEBP protein surface is preformed. Thus both cognate and non-cognate OnTEBP complex formation appears to proceed by a cofolding pathway (Horvath et al., 1998), in which protein and nucleic acid fold together to form the final complex.
Telomeric end protection is a critical function of proteins that specifically bind to the telomeric single-stranded overhang, including OnTEBP, Schizosaccharomyces pombe Pot1, and Saccharomyces cerevisiae Cdc13 (Gottschling and Zakian, 1986; Nugent et al., 1996; Baumann and Cech, 2001). The combination of sequence, structural and functional similarity among these divergent eukaryotic proteins indicates that mechanisms of telomeric end protection are broadly conserved throughout evolution (Mitton-Fry et al., 2002; Theobald et al., 2003b). Interestingly, two of the ssDNAs studied in this work, Tel+T and Tel-3, correspond to intermediates generated during extension of the telomere by the enzyme telomerase. Although it is unclear whether OnTEBP is present at the telomere in vivo when telomerase adds repeats, OnTEBP binding to the telomere inhibits telomerase activity in vitro, indicating a role for OnTEBP in telomere length regulation (Froelich-Ammon et al., 1998). Despite the limited in vitro tolerance of OnTEBP for non-cognate sequences, the length and sequence identity of O.nova telomeric ssDNA is closely regulated in vivo (Klobutcher et al., 1981). Taken together, these observations suggest that the widespread biological preference for specific telomeric repeats and guanosine-rich telomeres largely results from factors other than these telomeric end-binding proteins, with telomerase a likely candidate.
Nucleotide shuffling may also explain how a variety of other proteins recognize heterogeneous single-stranded sequences. For example, the yeast telomeric proteins Cdc13 and Pot1 bind irregular G-rich ssDNA repeats, and splicing factors such as PTB, U2AF and Sex-lethal recognize heterogeneous ssRNA sequences (Singh et al., 2000). Several known in vivo PTB binding sites (e.g. human α-tropomyosin IVS2 and IVS3 and rat β-tropomyosin IVS6) differ from the PTB consensus sequence by a single-base insertion. This puzzling feature could result from the ability of the pre-mRNA to shift register and flip an unfavorable base into solution. Similarly, much of the variability in U2AF binding sites can be readily explained by invoking nucleotide shuffling, especially the single-base differences observed in the conserved internal CCC tract.
Materials and methods
Nitrocellulose filter-binding assays were used to determine relative KD values for the 10 non-cognate ssDNA sequences (Table I). The ssDNAs used in these filter-binding experiments each had an additional 28 nucleotides (‘Oxy’ = AAGACGACATCGCTCAGCCAGACATTTT) at the 5′ end, included to pre-empt G-quartet side reactions and to facilitate efficient binding of free ssDNA to the bottom filter. Weak and non-specific interaction of the 5′ tail with the protein (Raghuraman et al., 1989) was abrogated by addition of non-specific competitor ssDNA. Analogous binding experiments using an agarose gel system with the shorter ssDNAs, though more difficult, yielded qualitatively similar results. Final binding reactions contained 25 mM HEPES pH 7.5, 150 mM NaCl, 5% glycerol, 0.01% Tween-20, 2 mM dithiothreitol (DTT), 1 mM EDTA, 10 µM non-specific competitor ssDNA (TelRNDM) and 50–200 pM 5′-32P-labeled ssDNA. Reaction mixtures were allowed to equilibrate at room temperature for 24 h and were filtered using a two-membrane Schleicher & Schuell 96-well minifold dotblot apparatus with a nitrocellulose top layer and a Hybond-N+ (Amersham-Pharmacia) bottom layer. The β-subunit contains a 28 kDa core domain (β28), used here, that is sufficient for ternary complex formation and that lacks G-quartet binding activity (Fang and Cech, 1993a, 1993b; Fang et al., 1993).
In this ternary system, the unitless KD is formally defined as
KD = (γα[α]γβ[β28]γDNA[DNA])/(γα-β28-DNA[α-β28-DNA])
where activity coefficients γX are ∼1.0/M at low concentration. Binding constants were fitted to
f = ([α][β28])/([α][β28] + KD)
where f is the fraction of DNA bound. Concentrations of α and β28 were held equivalent, and thus at half-bound DNA, the KD equals the product of [α] and [β28]. All binding experiments were performed in triplicate. For cognate OxyTel, KD = (2.8 ± 0.3 × 10–9)2, consistent within error with previously reported measurements (Raghuraman et al., 1989; Raghuraman and Cech, 1990; Gray, 1992; Fang et al., 1993).
For the Kspec competition assay, a series of binding reactions were made in which the protein and radioactively labeled cognate ssDNA were held at constant concentration and combined with increasing amounts of competitor ssDNA (TelRNDM, final concentrations 10 µM–1 mM). Labeled ssDNA was in slight (∼10%) molar excess over protein (1–2 µM). Reactions were incubated at room temperature for 24–48 h before agarose gel electrophoresis. Results were fitted to the equation
(1 – f)(f0/f – 1) = [TelRNDM]tot/(Kspec·[Tel ssDNA]tot)
where f is the fraction of DNA bound and f0 is the fraction bound with no competitor ssDNA.
The α- and β-protein subunits were expressed in Escherichia coli and purified as previously described (Horvath et al., 1998). Ternary complex with modified ssDNA was crystallized as described previously (Horvath et al., 1998) with one major alteration based on the discovery that intact G-quartets were necessary for crystallization. Briefly, G-quartets of a 13-nucleotide ssDNA (2.86 mM G4T4G4T) were preformed by annealing from 100°C to room temperature, followed by incubation in 100 mM NaCl for 3 weeks. Ternary complex was formed first (200 µM α-subunit with 1.1 to 1.4 molar equivalents of β28 and modified ssDNA), followed by addition of the preformed G-quartets (250 µM final). Ternary complex (∼2 µL) was added to an equal volume of precipitant (30% ethylene glycol, 1–6% polyethylene glycol 4000, 40 mM MES pH 5.5–7.0, 0.02% NaN3, 2 mM DTT) and allowed to equilibrate against a 1 ml volume of precipitant with an additional 50 mM NaCl. Crystal morphology of non-cognate complexes was indistinguishable from cognate complexes (space group P6122 with a = b = 94 Å and c = 425 Å).
Diffraction data were collected at the synchrotron beamline 14BM-C (Argonne APS) from cryoprotected crystals. Images were processed with HKL (Otwinowski and Minor, 1997). All structures were solved by molecular replacement (MR) using CNS (Brunger et al., 1998). Phases were generated from two complementary models derived from the cognate OnTEBP:Tel complex structures (1OTC and 1JB7): an OnTEBP:Tel model containing the ssDNA and one omitting the ssDNA. Rigid body refinement followed by rounds of minimization, simulated annealing and manual refinement reduced Rfree from >0.4 to <0.3. Differences between cognate and non-cognate complexes were clearly revealed in FO–FC maps. As a precaution to eliminate bias, the ssDNAs in the OnTEBP:Tel+T and OnTEBP:Tel-3 models were built independently of the cognate model, using phases calculated from the MR model omitting the Tel ssDNA. No crystal contacts involving the ssDNAs are observed, though there is a close approach to a symmetry-related molecule near G1. Comparison with independent crystal structures of α:ssDNA complexes (1K8G and 1KIX) indicates that at least two adjacent 5′ cognate ssDNA binding sites are unobstructed in the ternary complex crystals, suggesting that it is unlikely this symmetry-related molecule would influence binding of these non-cognate ssDNAs.
Table III. Crystallographic statistics.
| Complex | Rcrysta (%) | Rfreea (%) | Resolutionb (Å) | Completeness (%) | Rsym (%) | Unique reflections | PDB accession code |
|---|---|---|---|---|---|---|---|
| Tel+T | 23.2 | 26.1 | 2.50 | 96.6 | 10.6 | 37 742 | 1PH1 |
| Tel-3 | 21.7 | 26.9 | 3.10 | 91.1 | 12.6 | 19 176 | 1PH2 |
| G3X | 24.6 | 27.3 | 2.50 | 92.9 | 6.7 | 36 489 | 1PHJ |
| G10I | 23.0 | 26.8 | 2.90 | 96.4 | 8.0 | 24 336 | 1PH7 |
| G10X | 22.8 | 25.8 | 2.30 | 83.5 | 5.3 | 41 698 | 1PH5 |
| G10A | 21.4 | 26.4 | 2.50 | 76.2 | 9.6 | 29 531 | 1PH9 |
| G10C | 22.2 | 25.6 | 2.36 | 83.8 | 9.6 | 38 960 | 1PH8 |
| G10T | 24.2 | 27.6 | 2.10 | 92.5 | 9.8 | 60 505 | 1PH6 |
| G11C | 21.4 | 25.1 | 2.30 | 99.7 | 6.3 | 49 778 | 1PH4 |
| G11T | 23.0 | 26.3 | 2.30 | 99.3 | 5.6 | 49 804 | 1PH3 |
aRcryst = Σ||FO| – |FC||/Σ|FO|; Rfree was calculated with a randomly selected 10% of the total reflections.
bResolution is defined as the shell where the ratio of average reflection intensity to average intensity error is 2.
Acknowledgments
Acknowledgements
We thank Thomas Cech and Deborah Wuttke for critical review of the manuscript and Catherine Theobald for contribution to figure design. The American Cancer Society (RPG-93–011–04-NP) and the NIH (1R01CA81109) provided support.
References
- Anderson E.M., Halsey,W.A. and Wuttke,D.S. (2003) Site-directed mutagenesis reveals the thermodynamic requirements for single-stranded recognition by the telomere-binding protein Cdc13. Biochemistry, 42, 3751–3758. [DOI] [PubMed] [Google Scholar]
- Antson A.A., Dodson,E.J., Dodson,G., Greaves,R.B., Chen,X. and Gollnick,P. (1999) Structure of the trp RNA-binding attenuation protein, TRAP, bound to RNA. Nature, 401, 235–242. [DOI] [PubMed] [Google Scholar]
- Baumann P. and Cech,T.R. (2001) Pot1, the putative telomere end-binding protein in fission yeast and humans. Science, 292, 1171–1175. [DOI] [PubMed] [Google Scholar]
- Berglund J.A., Chua,K., Abovich,N., Reed,R. and Rosbash,M. (1997) The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell, 89, 781–787. [DOI] [PubMed] [Google Scholar]
- Bodnar A.G. et al. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- Bogden C.E., Fass,D., Bergman,N., Nichols,M.D. and Berger,J.M. (1999) The structural basis for terminator recognition by the Rho transcription termination factor. Mol. Cell, 3, 487–493. [DOI] [PubMed] [Google Scholar]
- Braddock D.T., Baber,J.L., Levens,D. and Clore,G.M. (2002) Molecular basis of sequence-specific single-stranded DNA recognition by KH domains: solution structure of a complex between hnRNP K KH3 and single-stranded DNA. EMBO J., 21, 3476–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A.T. et al. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Changela A., DiGate,R.J. and Mondragon,A. (2001) Crystal structure of a complex of a type IA DNA topoisomerase with a single-stranded DNA molecule. Nature, 411, 1077–1081. [DOI] [PubMed] [Google Scholar]
- Classen S., Ruggles,J.A. and Schultz,S.C. (2001) Crystal structure of the N-terminal domain of Oxytricha nova telomere end binding protein α subunit both uncomplexed and complexed with telomeric ssDNA. J. Mol. Biol., 314, 1113–1125. [DOI] [PubMed] [Google Scholar]
- Classen D.S., Lyons,D., Cech,T.R. and Schultz,S.C. (2003) Sequence-specific and 3′-end selective single-strand DNA binding by Oxytricha nova telomere end binding protein (OnTEBP) α subunit. Biochemistry, 42, 9269–9277. [DOI] [PubMed] [Google Scholar]
- Deo R.C., Bonanno,J.B., Sonenberg,N. and Burley,S.K. (1999) Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell, 98, 835–845. [DOI] [PubMed] [Google Scholar]
- Ding J., Hayashi,M.K., Zhang,Y., Manche,L., Krainer,A.R. and Xu,R.M. (1999) Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev., 13, 1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler L.E. (1998) Specificity determinants in the BamHI endonuclease-DNA interaction. Unpublished Ph.D. Thesis, Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA.
- Engler L.E., Welch,K.K. and Jen-Jacobson,L. (1997) Specific binding by EcoRV endonuclease to its DNA recognition site GATATC. J. Mol. Biol., 269, 82–101. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1999) Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. D, 55, 938–940. [DOI] [PubMed] [Google Scholar]
- Fang G. and Cech,T.R. (1993a) The beta subunit of Oxytricha telomere-binding protein promotes G- quartet formation by telomeric DNA. Cell, 74, 875–885. [DOI] [PubMed] [Google Scholar]
- Fang G. and Cech,T.R. (1993b) Characterization of a G-quartet formation reaction promoted by the beta- subunit of the Oxytricha telomere-binding protein. Biochemistry, 32, 11646–11657. [DOI] [PubMed] [Google Scholar]
- Fang G., Gray,J.T. and Cech,T.R. (1993) Oxytricha telomere-binding protein: separable DNA-binding and dimerization domains of the alpha-subunit. Genes Dev., 7, 870–882. [DOI] [PubMed] [Google Scholar]
- Frank D.E., Saecker,R.M., Bond,J.P., Capp,M.W., Tsodikov,O.V., Melcher,S.E., Levandoski,M.M. and Record,M.T., Jr. (1997) Thermodynamics of the interactions of lac repressor with variants of the symmetric lac operator: effects of converting a consensus site to a non-specific site. J. Mol. Biol., 267, 1186–1206. [DOI] [PubMed] [Google Scholar]
- Froelich-Ammon S.J., Dickinson,B.A., Bevilacqua,J.M., Schultz,S.C. and Cech,T.R. (1998) Modulation of telomerase activity by telomere DNA-binding proteins in Oxytricha. Genes Dev., 12, 1504–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallia G.L., Johnson,E.M. and Khalili,K. (2000) Puralpha: a multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res., 28, 3197–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner M.M. and Rau,D.C. (1995) Water release associated with specific binding of gal repressor. EMBO J., 14, 1257–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior S.L., Olivares,H., Ear,U., Hari,D.M., Weichselbaum,R. and Bishop,D.K. (2001) Assembly of RecA-like recombinases: distinct roles for mediator proteins in mitosis and meiosis. Proc. Natl Acad. Sci. USA, 98, 8411–8418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis-Ruth F.X., Moncalian,G., Perez-Luque,R., Gonzalez,A., Cabezon,E., de la Cruz,F. and Coll,M. (2001) The bacterial conjugation protein TrwB resembles ring helicases and F1-ATPase. Nature, 409, 637–641. [DOI] [PubMed] [Google Scholar]
- Gottschling D.E. and Zakian,V.A. (1986) Telomere proteins: specific recognition and protection of the natural termini of Oxytricha macronuclear DNA. Cell, 47, 195–205. [DOI] [PubMed] [Google Scholar]
- Grahn E., Stonehouse,N.J., Adams,C.J., Fridborg,K., Beigelman,L., Matulic-Adamic,J., Warriner,S.L., Stockley,P.G. and Liljas,L. (2000) Deletion of a single hydrogen bonding atom from the MS2 RNA operator leads to dramatic rearrangements at the RNA-coat protein interface. Nucleic Acids Res., 28, 4611–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J.T. (1992) The telomere protein of Oxytricha nova: expression and characterization of the subunits. PhD thesis, University of Colorado, Boulder, CO.
- Handa N., Nureki,O., Kurimoto,K., Kim,I., Sakamoto,H., Shimura,Y., Muto,Y. and Yokoyama,S. (1999) Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature, 398, 579–585. [DOI] [PubMed] [Google Scholar]
- Hohjoh H. and Singer,M.F. (1997) Sequence-specific single-strand RNA binding protein encoded by the human LINE-1 retrotransposon. EMBO J., 16, 6034–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopcroft N.H., Wendt,A.L., Gollnick,P. and Antson,A.A. (2002) Specificity of TRAP-RNA interactions: crystal structures of two complexes with different RNA sequences. Acta Crystallogr. D., 58, 615–621. [DOI] [PubMed] [Google Scholar]
- Horvath M.P. and Schultz,S.C. (2001) DNA G-quartets in a 1.86 A resolution structure of an Oxytricha nova telomeric protein–DNA complex. J. Mol. Biol., 310, 367–377. [DOI] [PubMed] [Google Scholar]
- Horvath M.P., Schweiker,V.L., Bevilacqua,J.M., Ruggles,J.A. and Schultz,S.C. (1998) Crystal structure of the Oxytricha nova telomere end binding protein complexed with single strand DNA. Cell, 95, 963–974. [DOI] [PubMed] [Google Scholar]
- Klobutcher L.A., Swanton,M.T., Donini,P. and Prescott,D.M. (1981) All gene-sized DNA molecules in four species of hypotrichs have the same terminal sequence and an unusual 3′ terminus. Proc. Natl Acad. Sci. USA, 78, 3015–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Lesser D.R., Kurpiewski,M.R. and Jen-Jacobson,L. (1990) The energetic basis of specificity in the EcoRI endonuclease–DNA interaction. Science, 250, 776–786. [DOI] [PubMed] [Google Scholar]
- Lewis H.A., Musunuru,K., Jensen,K.B., Edo,C., Chen,H., Darnell,R.B. and Burley,S.K. (2000) Sequence-specific RNA binding by a Nova KH domain: implications for paraneoplastic disease and the fragile X syndrome. Cell, 100, 323–332. [DOI] [PubMed] [Google Scholar]
- Liu Z., Luyten,I., Bottomley,M.J., Messias,A.C., Houngninou-Molango,S., Sprangers,R., Zanier,K., Kramer,A. and Sattler,M. (2001) Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science, 294, 1098–1102. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Miller S.D., Moses,K., Jayaraman,L. and Prives,C. (1997) Complex formation between p53 and replication protein A inhibits the sequence-specific DNA binding of p53 and is regulated by single-stranded DNA. Mol. Cell. Biol., 17, 2194–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitton-Fry R.M., Anderson,E.M., Hughes,T.R., Lundblad,V. and Wuttke,D.S. (2002) Conserved structure for single-stranded telomeric DNA recognition. Science, 296, 145–147. [DOI] [PubMed] [Google Scholar]
- Mossing M.C. and Record,M.T.,Jr. (1985) Thermodynamic origins of specificity in the lac repressor-operator interaction. Adaptability in the recognition of mutant operator sites. J. Mol. Biol., 186, 295–305. [DOI] [PubMed] [Google Scholar]
- Negroni M. and Buc,H. (2001) Mechanisms of retroviral recombination. Annu. Rev. Genet., 35, 275–302. [DOI] [PubMed] [Google Scholar]
- Nugent C.I., Hughes,T.R., Lue,N.F. and Lundblad,V. (1996) Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science, 274, 249–252. [DOI] [PubMed] [Google Scholar]
- Oda M., Furukawa,K., Ogata,K., Sarai,A. and Nakamura,H. (1998) Thermodynamics of specific and non-specific DNA binding by the c-Myb DNA-binding domain. J. Mol. Biol., 276, 571–590. [DOI] [PubMed] [Google Scholar]
- Osugi T., Ding,Y., Tanaka,H., Kuo,C.H., Do,E., Irie,Y. and Miki,N. (1996) Involvement of a single-stranded DNA binding protein, ssCRE-BP/Pur alpha, in morphine dependence. FEBS Lett., 391, 11–16. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. In Carter,R.E.Jr and Sweet,R.M. (eds) Macromolecular Crystallography, Part A. Academic Press, San Diego, CA. [DOI] [PubMed] [Google Scholar]
- Peersen O.B., Ruggles,J.A. and Schultz,S.C. (2002) Dimeric structure of the Oxytricha nova telomere end-binding protein α-subunit bound to ssDNA. Nat. Struct. Biol., 9, 182–187. [DOI] [PubMed] [Google Scholar]
- Pennock E., Buckley,K. and Lundblad,V. (2001) Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell, 104, 387–396. [DOI] [PubMed] [Google Scholar]
- Perez I., Lin,C.H., McAfee,J.G. and Patton,J.G. (1997) Mutation of PTB binding sites causes misregulation of alternative 3′ splice site selection in vivo. RNA, 3, 764–778. [PMC free article] [PubMed] [Google Scholar]
- Price C.M. and Cech,T.R. (1987) Telomeric DNA–protein interactions of Oxytricha macronuclear DNA. Genes Dev., 1, 783–793. [DOI] [PubMed] [Google Scholar]
- Raghuraman M.K. and Cech,T.R. (1990) Effect of monovalent cation-induced telomeric DNA structure on the binding of Oxytricha telomeric protein. Nucleic Acids Res., 18, 4543–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman M.K., Dunn,C.J., Hicke,B.J. and Cech,T.R. (1989) Oxytricha telomeric nucleoprotein complexes reconstituted with synthetic DNA. Nucleic Acids Res., 17, 4235–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Record M.T. Jr, Ha,J.H. and Fisher,M.A. (1991) Analysis of equilibrium and kinetic measurements to determine thermodynamic origins of stability and specificity and mechanism of formation of site-specific complexes between proteins and helical DNA. Methods Enzymol., 208, 291–343. [DOI] [PubMed] [Google Scholar]
- Sidorova N.Y. and Rau,D.C. (1996) Differences in water release for the binding of EcoRI to specific and nonspecific DNA sequences. Proc. Natl Acad. Sci. USA, 93, 12272–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Banerjee,H. and Green,M.R. (2000) Differential recognition of the polypyrimidine-tract by the general splicing factor U2AF65 and the splicing repressor Sex-lethal. RNA, 6, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spolar R.S. and Record,M.T.,Jr. (1994) Coupling of local folding to site-specific binding of proteins to DNA. Science, 263, 777–784. [DOI] [PubMed] [Google Scholar]
- Stevens S.Y. and Glick,G.D. (1999) Evidence for sequence-specific recognition of DNA by anti-single-stranded DNA autoantibodies. Biochemistry, 38, 560–568. [DOI] [PubMed] [Google Scholar]
- Takeda Y., Ross,P.D. and Mudd,C.P. (1992) Thermodynamics of Cro protein–DNA interactions. Proc. Natl Acad. Sci. USA, 89, 8180–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi S., Sakuraba,Y., Masuyama,S., Inoue,H. and Yamaizumi,M. (2000) Dysfunction of human Rad18 results in defective postreplication repair and hypersensitivity to multiple mutagens. Proc. Natl Acad. Sci. USA, 97, 7927–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theobald D.L., Cervantes,R.B., Lundblad,V. and Wuttke,D.S. (2003a) Homology among telomeric end-protection proteins. Structure, in press. [DOI] [PubMed] [Google Scholar]
- Theobald D.L., Mitton-Fry,R.M. and Wuttke,D.S. (2003b) Nucleic acid recognition by OB-fold proteins. Annu. Rev. Biophys Biomol. Struct., 32, 115–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H., Schachter,F., Uchida,I., Wei,L., Zhu,X., Effros,R., Cohen,D. and Harley,C.B. (1993) Loss of telomeric DNA during aging of normal and trisomy 21 human lymphocytes. Am. J. Hum. Genet., 52, 661–667. [PMC free article] [PubMed] [Google Scholar]
- vonHippel P.H. (1994) Protein-DNA recognition: new perspectives and underlying themes. Science, 263, 769–770. [DOI] [PubMed] [Google Scholar]
- Waga S. and Stillman,B. (1998) The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem., 67, 721–751. [DOI] [PubMed] [Google Scholar]
- Yang H. et al. (2002) BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science, 297, 1837. [DOI] [PubMed] [Google Scholar]
- Zahler A.M. and Prescott,D.M. (1988) Telomere terminal transferase activity in the hypotrichous ciliate Oxytricha nova and a model for replication of the ends of linear DNA molecules. Nucleic Acids Res., 16, 6953–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]