

Abstract
Telomere erosion and subsequent dysfunction limits the proliferation of normal human cells by a process termed replicative senescence. Replicative senescence is thought to suppress tumorigenesis by establishing an essentially irreversible growth arrest that requires activities of the p53 and pRB tumor suppressor proteins. We show that, depending on expression of the pRB regulator p16, replicative senescence is not necessarily irreversible. We used lentiviruses to express specific viral and cellular proteins in senescent human fibroblasts and mammary epithelial cells. Expression of telomerase did not reverse the senescence arrest. However, cells with low levels of p16 at senescence resumed robust growth upon p53 inactivation, and limited growth upon expression of oncogenic RAS. In contrast, cells with high levels of p16 at senescence failed to proliferate upon p53 inactivation or RAS expression, although they re-entered the cell cycle without growth after pRB inactivation. Our results indicate that the senescence response to telomere dysfunction is reversible and is maintained primarily by p53. However, p16 provides a dominant second barrier to the unlimited growth of human cells.
Keywords: cyclin-dependent kinase/pRB/RAS/senescence/telomerase
Introduction
Normal cells do not divide indefinitely due to a process termed replicative senescence. One important mechanism responsible for the replicative senescence of human cells is the erosion and eventual dysfunction of telomeres (Harley et al., 1990; de Lange, 2001). Telomeres are the DNA sequence and associated proteins that cap and stabilize the ends of linear chromosomes, preventing their degradation or fusion by DNA repair systems. Owing to the biochemistry of DNA replication, several dozen base pairs of telomeric DNA are lost with each cell cycle. Thus, proliferating cells experience progressive telomere shortening, unless they express the enzyme telomerase, which can add the telomeric sequence to chromosome ends de novo. Most human cells do not express this enzyme, and hence can acquire telomeres that are critically short and dysfunctional. Dysfunctional telomeres signal normal cells to cease proliferation with a characteristic senescent phenotype (Blackburn, 2001; Shay and Wright, 2001; Kim et al., 2002).
Replicative senescence is an example of a more general process, herein termed cellular senescence, which arrests the growth of cells in response to many stimuli. These stimuli include dysfunctional telomeres, DNA damage, disrupted chromatin organization, and certain oncogenes, such as activated RAS (Campisi et al., 2001; Serrano and Blasco, 2001). They have in common the potential to initiate or promote neoplastic transformation. Cellular and replicative senescence require activities of the p53 and pRB tumor suppressor proteins, which regulate pathways that suffer mutations in most, if not all, mammalian cancers. Human cells that lose p53 and pRB function are generally refractory to multiple senescence-inducing stimuli (Serrano et al., 1997; Dimri et al., 2000). These and other lines of evidence suggest that the senescence response suppresses the development of cancer in mammals (Reddel, 2000; Campisi et al., 2001; Wright and Shay, 2001).
Although p53 and pRB are clearly critical for establishing the senescence growth arrest, their precise roles in this process are incompletely understood. p53 is presumed to sense dysfunctional telomeres as damaged DNA, whereupon it elicits the senescence response at least in part by increasing expression of the p21 cyclin-dependent kinase inhibitor (CDKI); p21 in turn prevents the phosphorylation and inactivation of pRB (Sherr and Roberts, 1999). However, inactivation of either p53 or pRB (e.g. by viral oncoproteins or anti-sense oligonucleotides) independently extends the replicative lifespan of many human cells, allowing them to proliferate despite short telomeres (Hara et al., 1991; Shay et al., 1991). Thus, although the p53 and pRB pathways interact, they may also act separately to establish the senescence arrest. Indeed, senescent cells have been reported to upregulate another CDKI, p16, which also controls pRB activity (Alcorta et al., 1996; Hara et al., 1996a; Stein et al., 1999). p16 may limit cell proliferation by a mechanism distinct from that utilized by p53, since some human epithelial cells (e.g. initial outgrowths from mammary tissue explants) senesce with relatively long telomeres and high p16 expression (Kiyono et al., 1998; Ramirez et al., 2001). Moreover, ectopic expression of telomerase does not protect such cells from replicative senescence, suggesting that p16 expression and function are independent of telomere status (Kiyono et al., 1998; Rheinwald et al., 2002).
p53 and pRB are also important for maintaining the senescence growth arrest, which, in human cells, is thought to be irreversible. Senescent human cells arrest growth with a G1 DNA content, and cannot be stimulated to divide by physiological mitogens. Moreover, although the potent viral oncoprotein SV-40 T-antigen stimulates DNA replication in senescent human fibroblasts, it does not stimulate cell proliferation (Ide et al., 1983; Gorman and Cristofalo, 1985). T-antigen binds and inactivates both p53 and pRB, and mutants defective in either p53 or pRB binding fail to stimulate DNA synthesis in senescent cells (Sakamoto et al., 1993; Hara et al., 1996b). One interpretation of these findings is that p53 and pRB cooperatively prevent senescent cells from initiating S phase, but another activity prevents completion of the cell cycle. On the other hand, p53 antibodies, when microinjected into senescent cells, were shown to stimulate DNA synthesis and limited proliferation (Gire and Wynford-Thomas, 1998). Thus, requirements for maintaining the senescence arrest are less clear than the requirements for establishing it.
We recently showed that human fibroblasts differ in their sensitivity to BMI-1, an oncogene that extends the replicative lifespan of fibroblasts by repressing p16, apparently because they differ in the level of p16 they express at senescence (Itahana et al., 2003). This finding raises the possibility that human cell strains also differ in the mechanisms that maintain the senescence state. To explore this possibility, and understand the mechanisms that maintain the senescence arrest, we used lentiviruses to express viral and cellular proteins in replicatively senescent human fibroblasts. Our results suggest that the senescence arrest caused by telomere dysfunction is reversible, being maintained primarily by p53 and reversed by p53 inactivation. In some human cells, however, p16 provides a dominant, apparently irreversible, second barrier to cell proliferation, which cannot be completely overcome by subsequent inactivation of pRB.
Results
Telomerase does not reverse the senescence growth arrest
The first candidate we tested for ability to reverse the senescence growth arrest was hTERT, the catalytic subunit and rate-limiting component of telomerase (Bodnar et al., 1998; Vaziri and Benchimol, 1998). For these and subsequent experiments, we used two human fibroblast strains: WI-38 (WI) from fetal lung and BJ (from neonatal foreskin). Neither strain expresses the endogenous TERT gene, and both are devoid of detectable telomerase activity, as determined by the telomere repeat amplification protocol (TRAP) assay.
We passaged pre-senescent (early passage) cultures (P-WI, P-BJ) until replicative senescence. Unless noted otherwise, senescent cultures (S-WI, S-BJ) contained >99.9% non-dividing cells, as determined by no increase in cell number over >4 wks and <1% [3H]thymidine-labeled nuclei after a 3-day labeling interval (% LN). To express hTERT and other proteins, we used lentiviruses, which efficiently infect and stably express genes in non-dividing cells (Bukrinsky et al., 1993). We verified the infection efficiency by infecting parallel cultures with an equivalent titer (see Materials and methods) of virus expressing green fluorescent protein (GFP), and, where possible, immunostaining for the virally expressed proteins. At the titers employed, the lentiviruses transduced >95% of cells in both pre-senescent and senescent cultures.
The hTERT-expressing lentivirus (lenti-hTERT) conferred robust telomerase activity on S-WI cells, whereas S-WI cells infected with lenti–GFP were devoid of telomerase activity (Figure 1A). By contrast, pBabe-hTERT, which requires cell proliferation for integration and expression, failed to confer telomerase activity on S-WI cells, although it conferred robust activity on P-WI cells (Figure 1A).

Fig. 1. hTERT does not reverse the senescent phenotype. (A) Lenti-hTERT confers telomerase activity. Senescent (S) or pre-senescent (P) WI-38 (WI) cells were infected with lenti-hTERT, lenti–GFP or pBABE-hTERT, and telomerase activity was measured using the TRAP assay, as described in Materials and methods. ‘+’ is a positive TRAP control, and ‘–’ and ‘Heat inactivated’ are negative controls. (B) Lenti-hTERT alters telomere length in pre-senescent, but not senescent cells. Terminal restriction fragment (TRF) lengths in P- or S-WI cells infected with lenti–GFP (GFP) or lenti-hTERT (hTERT) were determined as described in Materials and methods. (C) Lenti-hTERT does not alter senescent morphology. S-WI cells were infected with lenti–GFP (+GFP) or lenti-hTERT (+hTERT), and viewed and photographed 7 days later.
S-WI cells infected with lenti-hTERT did not proliferate (Figure 2A), despite expressing high levels of telomerase. Moreover, they did not lose the senescent morphology (Figure 1C) or senescence-associated β-galactosidase (SA-Bgal) expression (Dimri et al., 1995) (not shown). Identical results were obtained when S-BJ cells were infected with lenti-hTERT (Figure 2B, and results not shown). Lenti-hTERT did not alter average telomere length in S-WI cells, even when tested up to 6 weeks after infection; however, the same virus elongated telomeres in P-WI cells (Figure 1B), indicating that the virus expressed a functional hTERT protein.

Fig. 2. p53 inactivation reverses senescence of BJ, but not WI-38 fibroblasts. (A) S-WI cells synthesize DNA, but do not proliferate. S-WI cells were infected with lentiviruses expressing GFP, hTERT, GSE, LgT, LgTK1 and CDK4m as indicated; 72 h later, DNA synthesis was determined by % LN, and percentage growth monitored, as described in the text. (B) S-BJ cells synthesize DNA and proliferate. S-BJ cells were infected and monitored, as described in (A). (C) Morphology of control and rescued S-BJ cells. S-BJ cells infected with GFP or GSE-expressing lentivirus were photographed 6 days later. (D) Lifespan assays. S-BJ cells were infected with lentiviruses expressing the indicated proteins, serially passaged, and cell number determined at each passage, as described in Materials and methods.
These results lead to two important conclusions. First, human telomeres cannot be modified by telomerase in the absence of cell proliferation. Secondly, telomerase cannot reverse the growth arrest or senescent phenotype of replicatively senescent cells.
p53 inactivation reverses senescence of BJ, but not WI-38, cells
Inactivation of either the p53 or pRB pathway is known to postpone, but not prevent, the replicative senescence of human fibroblasts (Wright and Shay, 2001). Inactivation of both pathways, for example by SV-40 T-antigen (LgT), causes crisis, a state characterized by genomic instability, cell death, and the eventual emergence of rare replicatively immortal variants (Shay et al., 1993; Wei and Sedivy, 1999).
To explore the roles of p53 and pRB in maintaining the senescence arrest, we used lentiviruses to express the following proteins in human cells: (i) LgT, which binds and inactivates both p53 and pRb (Fanning, 1992); (ii) LgT-K1, a LgT mutant that binds p53 but not pRB or pRB family members (DeCaprio et al., 1988); (iii) GSE-22, a peptide that inactivates p53 function in a dominant negative fashion (Gudkov et al., 1993); and (iv) CDK4m, a cyclin-dependent kinase 4 mutant that cannot bind p16, and hence constitutively inactivates pRB (Wolfel et al., 1995), although it may also have other modes of action.
Consistent with results from SV-40 infection and plasmid microinjection experiments (Gorman and Cristofalo, 1985; Sakamoto et al., 1993; Hara et al., 1996b) (Figure 2A), LgT stimulated a substantial fraction (60–70%) of S-WI cells to synthesize DNA. CDK4m also stimulated DNA synthesis in S-WI, albeit to a lesser extent (35–40%). By contrast, GSE-22 and LgT-K1 were essentially inactive (<5%). These data suggest that S-WI cells can re-enter the cell cycle upon inactivation of the pRB pathway (by LgT or CDK4m), but inactivation of the p53 pathway alone (by LgT-K1 or GSE) has no effect in these cells. However, regardless of ability to stimulate DNA synthesis, none of the lentiviruses, alone or in combination, efficiently stimulated S-WI cells to proliferate (Figure 2A). We conclude that although S-WI cells enter S-phase upon inactivation of the pRB pathway, they cannot complete the cell cycle and proliferate.
In contrast, a substantial fraction of S-BJ cells initiated DNA synthesis in response to each of the four lenti-expressed proteins (LgT, LgT-K1, GSE-22 and CDK4m) (Figure 2B). Moreover, GSE and LgT-K1 were as effective as LgT, each stimulating 70–90% of the cells (Figure 2B). CDK4m was less effective (25–30%) (Figure 2B). Most striking, all four lentiviruses each stimulated S-BJ cells to complete the cell cycle and proliferate. The extent of proliferation was approximately equal to the extent of DNA synthesis. Proliferation was assessed by the formation of colonies (>50 cells) (Figure 2B) and loss of senescent morphology (Figure 2C). Thus, in contrast to S-WI cells, the growth arrest of replicatively senescent BJ fibroblasts was completely reversible, and p53 inactivation was sufficient to induce both DNA synthesis and proliferation.
With regard to efficacy, GSE-22 was more efficient than LgT or LgT-K1 at reversing the senescence arrest of S-BJ cells (Figure 2B), consistent with reports that LgT and LgT-K1 do not completely inactivate p53 (Deppert et al., 1987). Moreover, failure of GSE-22 to stimulate S-WI cells was not due to an inability to inactivate p53 in these cells. GSE-22 increased p53 levels in both S-BJ and S-WI cells, as expected from its ability to enhance p53 stabilization (Gudkov et al., 1993); moreover, GSE-22 markedly reduced p21 levels in both cell strains, as expected for loss of p53 function (Supplementary figure 1, available at The EMBO Journal Online). Interestingly, CDK4m stimulated DNA synthesis in both S-WI and S-BJ cells (25–35%), despite their different requirements for cell cycle re-entry after senescence. CDK4m may also act indirectly on the p53 block by sequestering p21 (Sherr and Roberts, 1999). Consistent with p21 sequestration accounting for the ability of CDK4m to stimulate S-BJ cells, most of the p21 in S-BJ cells co-immunoprecipitated with CDK4 after infection with lenti-CDK4m (Supplementary figure 2).
To determine the extent to which GSE, LgT, LgT-K1 and CDK4m stimulated S-BJ cell proliferation, we determined the growth of mass cultures after infection. GSE stimulated >20 additional population doublings (PDs) (Figure 2D). Towards the end of this extended growth, the fraction of cells that synthesized DNA (% LN) gradually declined (not shown). However, the % LN did not decline below 20–25%, even after there was no net increase in cell number. This phenotype (high labeling index without net proliferation) is characteristic of cultures in crisis (Wei and Sedivy, 1999). LgT and LgT-K1 each stimulated 10–11 additional PDs (Figure 2D), also culminating in crisis. Cultures driven to crisis by GSE-22 showed fewer apoptotic cells than cultures driven by LgT or LgT-K1 (not shown), which likely explains why GSE-22-expressing cultures completed more PDs than LgT- or LgT-K1-expressing cultures. In contrast to the other proteins, CDK4m stimulated very limited proliferation of S-BJ cells (2–3 PDs) (Figure 2D). This finding suggests that although p21 binding by CDK4m may partially relieve the p53 block (Figure 2B and Supplementary figure 2), it is not equivalent to p53 inactivation.
Together, the results indicate that p53 maintains the senescence growth arrest of S-BJ cells, and that inactivation of p53 alone is sufficient to reset their replicative lifespan. A third human fibroblast strain (82-6, from skin) displayed an intermediate response to p53 inactivation: 20–25% of senescent 82-6 cells initiated DNA synthesis in response to GSE-22 (not shown). Thus, some human fibroblast strains have phenotypes intermediate between WI-38 and BJ with respect to reversibility of the senescence growth arrest by p53 inactivation.
p16 prevents reversal of senescence by p53 inactivation
Why do S-BJ cells resume growth upon p53 inactivation, without pRB inactivation, whereas S-WI cells fail to proliferate (despite undergoing DNA synthesis) even when both pRB and p53 are inactivated? One possibility might be intrinsic differences in the ability to induce p16 at senescence. WI-38, like several human epithelial cells, appear to undergo replicative senescence prior to critical telomere shortening owing to induction of p16 by as yet unidentified factors (Wright and Shay, 2001; Itahana et al., 2003). Thus, p16 may impose a proliferative block that cannot be overcome by p53 inactivation. Consistent with this idea, WI-38 cells consistently expressed higher levels of p16 than BJ cells, whether pre-senescent or senescent (Figure 3A and B). Moreover, S-BJ cells that were rescued from senescence by GSE-22 ceased proliferation (after >20 additional PDs; Figure 2D) with low but significant p16 expression (Figure 3C), and could not be rescued from this second growth arrest by LgT (not shown). Finally, senescent 82-6 fibroblasts expressed p16 at levels intermediate between S-WI and S-BJ cells; immediately after rescue by GSE-22, the cells had substantially less p16 than the starting population (not shown), suggesting that GSE rescued only those cells that expressed little or no p16. Thus, p16 may prevent reversal of the senescence arrest by p53 inactivation.

Fig. 3. Senescence reversal correlates inversely with p16 expression. (A) p16 expression. p16 and actin (control) protein levels were assessed in pre-senescent (P) and senescent (S) BJ and WI-38 (WI) cells by western blotting (WB). The labeling index of the cultures is shown below the blot (% LN). (B) p16 immunostaining. Pre-senescent (P) and senescent (S) BJ and WI-38 cells were immunostained for p16, and nuclei stained with DAPI, as described in Materials and methods. HeLa cells served as a positive control. (C) p16 and p21 levels after rescue from senescence by GSE-22. S-BJ cells were infected with control (GFP) or GSE-22-expressing (rescued GSE) lentivirus. Rescued cells were harvested while proliferating (16 PDs) or after proliferation ceased (23 PDs), and analyzed for p21, p16 and actin (control) by western blotting. (D) Senescence reversal in HMECs. Post-selection HMECs were infected with lentiviruses expressing the indicated proteins, and monitored for growth, as described in Materials and methods. Shown are cells 72 h following infection.
Results with human mammary epithelial cells (HMECs), a culture system in which p16 expression is well characterized, support this idea. HMECs that proliferate from tissue explants generally do so for only 10–25 PDs before undergoing a senescence arrest with relatively long telomeres and high p16 expression. However, variants that spontaneously silence p16 by methylation can emerge (self selection) (Hammond et al., 1984). Post-selection HMECs proliferate for an additional 50–75 PDs before senescing with short telomeres and genomic instability, a crisis-like state termed agonescence (Romanov et al., 2001). Agonescent HMECs resumed proliferation upon expression of LgT or GSE, but not hTERT (Figure 3D), and thus resembled S-BJ fibroblasts. However, pre-selection HMECs that ceased growth with high p16 levels were not rescued from the growth arrest by LgT (not shown), suggesting that the senescence arrest is reversible only in p16-negative cells.
Suppression of p16 confers sensitivity to senescence reversal by p53 inactivation
To evaluate more critically the role of p16 in the senescence arrest, we manipulated p16 expression in human fibroblasts. First, we used RNA interference (Paddison et al., 2002) to stably suppress p16 expression in WI38 cells. A short hairpin RNA (shRNA) capable of suppressing p16 expression (Narita et al., 2003) was introduced into proliferating (P-WI) cells using a retroviral vector. The p16 shRNA modestly extended the replicative lifespan of P-WI cells (3–4 PDs) (Supplementary figure 3), and the cells senesced with significantly reduced p16 levels (Figure 4A). S-WI cells with suppressed p16 senesced with elevated levels of p21 (Figure 4A), suggesting that they now senesced primarily due to activation of the p53 pathway, similar to the phenotype of BJ fibroblasts. Indeed, subsequent infection with lenti-GSE-22 completely reversed the senescence arrest of p16-suppressed S-WI cells (Figure 4B), stimulating both DNA synthesis and cell proliferation (>70%). Thus, GSE-22 acted similarly in S-BJ and p16-suppressed S-WI cells, in contrast to its effects in S-WI cells that express high p16 (compare Figure 2A and B with Figure 4B). Next, we ectopically expressed p16 in P-WI cells using a lentivirus (lenti-p16). Western blotting (Figure 4C) and immunofluorescence (Supplementary figure 4) showed that lenti-p16 markedly elevated p16 expression in P-WI cells. As expected (McConnell et al., 1998), lenti-p16 infected P-WI cells arrested growth with a senescent morphology and SA-Bgal expression (not shown). Subsequent infection with lenti-GSE-22 failed to stimulate DNA synthesis or cell proliferation (Supplementary figure 5), similar to its effect in S-WI cells.
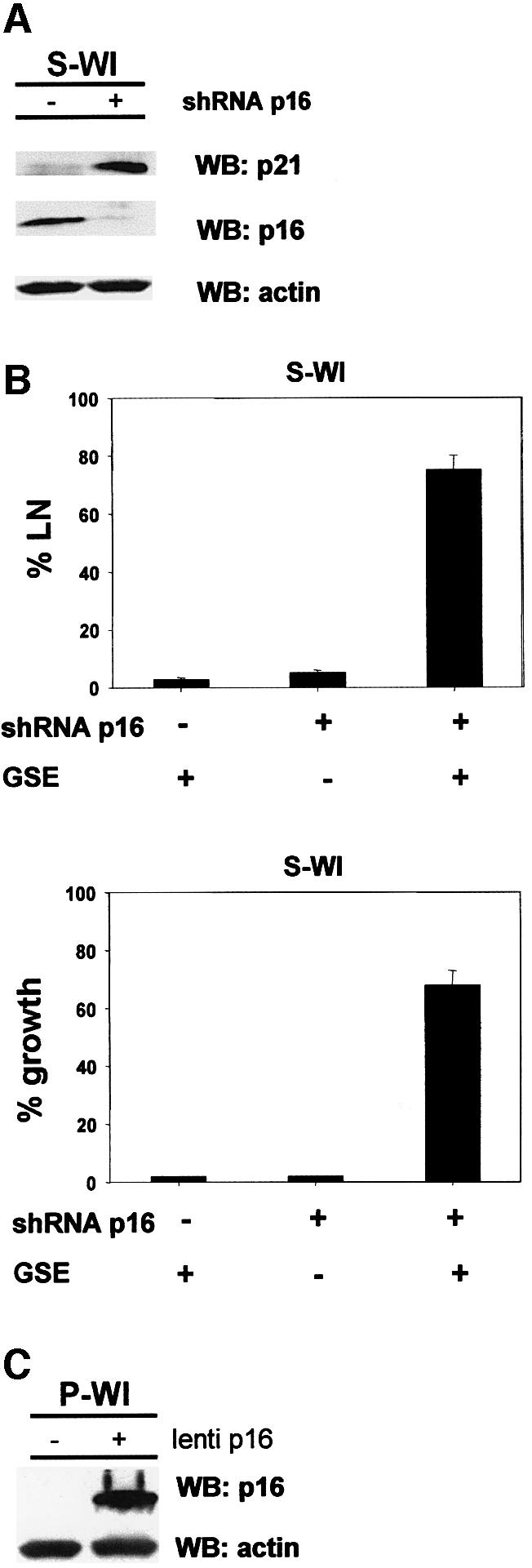
Fig. 4. p16 suppression allows senescence reversal in WI-38 cells. (A) shRNA-p16 suppresses p16 expression in WI-38 cells. P-WI cells were mock infected (–) or infected with shRNA-p16 expressing pMSCV retroviruses, and passaged until replicatively senescent (S-WI; % LN <1%). p16, p21 and actin (control) levels were assessed by western blotting (WB). (B) % LN. P-WI were mock infected (–) or infected with shRNA-p16 expressing retrovirus (+) and cultured until senescent (S-WI). Cells were then monitored for ability to synthesize DNA (% LN) upon p53 inactivation by subsequent infection with lenti-GSE-22 (GSE). Parallel cultures were monitored for proliferation (% growth), as described in Materials and methods. (C) Ectopic p16 expression. P-WI cells were mock infected (–) or infected (+) with lenti-p16; p16 and actin (control) were assessed by western blotting (WB). At the exposure shown, endogenous p16 in mock-infected P-WI cells is undetectable.
Taken together, these results support the idea that p16 imposes a senescence-associated block to cell proliferation that cannot be reversed by p53 inactivation.
Sequential action of p16
p16 is known to exert its effects through pRB, specifically by inhibiting CDKs and thus preventing pRB inactivation by phosphorylation. However, LgT, which inactivates pRB by direct binding, stimulated DNA synthesis but not proliferation in p16-expressing cells (S-WI or pre-selection HMECs). This finding raises the possibility that the block to cell proliferation imposed by p16 can be independent of continual pRB activity. To explore the nature of the p16 block, we infected P-WI cells with lenti-p16, and superinfected with lenti-LgT. LgT stimulated DNA synthesis in ∼40% of the p16-expressing P-WI cells (Figure 5A), but cell proliferation did not occur (Figure 5A). By contrast, P-WI cells that were first infected with lenti-LgT and then superinfected with lenti-p16 continued to synthesize DNA and proliferate (Figure 5B) despite high p16 expression. The presence or absence of LgT did not affect the level of p16 expressed by lenti-p16 (not shown). Similar results were obtained using HMECs. LgT prevents pre-selected HMECs from senescing when introduced prior to upregulation of p16 (Huschtscha et al., 2001). However, LgT did not stimulate cell proliferation when introduced after HMECs had undergone the p16-induced senescence arrest (not shown). Finally, although 70–75% of P-WI cells expressing shRNA p16 and cultured until senescence resumed growth upon p53 inactivation by GSE-22 (Figure 4B), this was not the case when p16 was downregulated in cells that had already undergone senescence with high p16 expression. We constructed a lentivirus to express the shRNA-p16 and used it to downregulate p16 in S-WI cells. We then superinfected the cells with lenti-GSE-22. Although GSE-22 stimulated DNA synthesis, very few cells (∼1–2%) resumed proliferation (Figure 5C). Therefore, once the p16/pRB pathway is engaged, its downregulation is insufficient for DNA synthesis unless p53 is also targeted. Moreover, once the p16/pRB pathway engaged, neither p53 nor pRB inactivation is sufficient to allow cell proliferation.

Fig. 5. Sequential inactivation of p16/Rb determines senescence reversibility. (A) p16 followed by LgT. P-WI cells were infected with lenti-p16, and then mock infected (–) or infected with lenti LgT (+). Cells were monitored for ability to synthesize DNA (% LN) and proliferate (% growth), as described in Materials and methods. (B) LgT followed by p16. P-WI cells were infected with lenti-LgT, followed by mock infection (–) or lenti-p16 infection (+). The infected cells were monitored for DNA synthesis (% LN) and proliferation (% growth). (C) Silencing p16 after senescence. S-WI cells were infected with lenti-shRNA-p16, and then mock infected (–) or infected with lenti-GSE-22 (+). The infected cells were monitored for DNA synthesis (% LN) and proliferation (% growth).
p16 suppresses the response to oncogenic RAS
Oncogenic RAS (Ha-RASv12) (Shih and Weinberg, 1982) delivers a strong mitogenic signal that transforms immortal cells (Land et al., 1983). However, normal human fibroblasts respond to oncogenic RAS by a senescence growth arrest accompanied by upregulation of p16 (Serrano et al., 1997). Because cells differ in their ability to upregulate p16 upon replicative senescence, we asked whether ability to upregulate p16 also influences the senescence response to mitogenic signals, such as those delivered by Ha-RASv12.
We infected S-WI and S-BJ cells with a lentivirus expressing Ha-RASv12 (lenti-RAS). Lenti-RAS did not stimulate S-WI cells to initiate DNA synthesis (Figure 6A), in agreement with reports using a different fibroblast strain and plasmid microinjection (Lumpkin et al., 1986). In contrast, lenti-RAS induced >20% of S-BJ cells to synthesize DNA (Figure 6B). Thus, oncogenic RAS did not stimulate DNA synthesis in senescent cells that express high p16, but modestly stimulated senescent cells that express low levels of p16.

Fig. 6. Oncogenic Ras partially reverses p16-independent senescence. (A) S-WI cells do not synthesize DNA in response to oncogenic Ras. S-WI cells were infected with the indicated lentiviruses, and 72 h later assessed for ability to synthesize DNA (% LN) and proliferate (% growth), as described in Materials and methods. (B) S-BJ cells synthesize DNA and undergo limited proliferation in response to oncogenic Ras. S-BJ cells were infected with the indicated lentiviruses, and assessed for % LN and % growth. The asterisk indicates that the cells underwent limited proliferation, amounting to 3 PDs or less. (C) Mitogenic effects of Ras are concentration- and p16-dependent. S-BJ cells were infected with 1× (low Ras) or 3× (high Ras) lenti-Ras virus concentrations (determined by p24 levels, as described in Materials and methods) and % LN was measured. Where indicated, S-BJ cells were infected with lenti-p16 5 days prior to subsequent infection with high lenti-Ras. (D) Ras immunostaining. S-BJ cells were mock infected or infected with lenti-Ras at 1× or 3× virus concentrations, and immunostained for Ras. Nuclei were identified by DAPI staining.
Regardless of the p16 level, oncogenic RAS synergized with CDK4m to stimulate senescent cells to synthesize DNA (Figure 6A and B). CDK4m alone induced only 20–35% of S-WI and S-BJ cells to synthesize DNA. However, CDK4m plus RAS induced DNA synthesis in 85–90% of the cells (Figure 6A and B). Nonetheless, neither oncogenic RAS nor CDK4m, either alone or in combination, stimulated appreciable cell proliferation (Figure 6A). In S-BJ, RAS or CDK4m each induced ∼20%, and the combination induced >90% of cells to undergo a few divisions. However, proliferation was limited to 2–3 PDs (see Figure 2D). Interestingly, these cells arrested growth with low p16 levels, but they were significantly higher than control p16 levels (Figure 6E), indicating that RAS can induce p16 even in cells such as BJ, which do not express p16 upon replicative senescence. Together, these results suggest that oncogenic RAS cannot sustain the growth of senescent cells, even when p16 expression is initially low.
The ability of RAS to stimulate DNA synthesis depended on the level at which it was expressed, and was abrogated by p16. We infected S-BJ cells with lenti-RAS at a 3-fold higher titer than routinely used (high RAS). High RAS did not increase the fraction of infected cells (>90% infectivity for both high and low RAS; Figure 6D), but increased the % LN of S-BJ cells from 20 to 55% (Figure 6C). This DNA synthesis was markedly suppressed by superinfection with lenti-16 (Figure 6C). Taken together, these data indicate that p16 provides a formidable barrier to reversal of the senescence growth arrest by p53 inactivation, as well as by the strong mitogenic signal delivered by oncogenic RAS.
Discussion
It is well established that the p53 and pRB pathways are critical for establishing the replicative senescence of human cells. Much less is known about the requirements for maintaining the senescence growth arrest. Our results support a model in which human fibroblasts establish and maintain the senescence growth arrest by either of two mechanisms, depending on whether p16 is expressed. Both mechanisms impose a growth arrest that cannot be reversed by known physiological signals. However, in the absence of p16 expression, the senescence arrest can be reversed by inactivation of p53. Thus, the replicative senescence of human cells is not necessarily irreversible once established, and p16 plays a critical role in preventing its reversal by p53 inactivation (Figure 7).

Fig. 7. Pathways leading to reversible and essentially irreversible senescence growth arrests in human cells. Proliferating cells (Presenescent) arrest growth with a senescent phenotype in response to telomere erosion, which is p53 dependent, or a combination of telomere erosion and an as yet unidentified stimulus that induces p16. The p53-dependent arrest increases p21 expression, and is reversed by p53 inactivation or oncogenic Ras. p53 inactivation results in extensive proliferation (growth) culminating in crisis, whereas Ras causes limited proliferation. Cells that senesce with high p16 can be stimulated to synthesize DNA (S-phase) upon inactivation of p53 and pRb, or pRb inactivation plus oncogenic Ras, but do not proliferate (no growth).
In agreement with previously proposed models, our results support the idea that telomere-dependent replicative senescence depends primarily on the p53 pathway (Atadja et al., 1995; Gire and Wynford-Thomas, 1998). In some human cell strains, such as BJ, this p53-dependent growth arrest is the predominant mechanism that limits replicative lifespan, and is reversible upon inactivation of p53. Thus, we found that the replicative senescence of S-BJ cells was completely reversed by GSE-22, which inactivates p53 (Gudkov et al., 1993). The arrest of these cells was also reversed by LgT and LgT-K1, which, among other activities, also inactivate p53. This reversal resulted in extensive (>20 PDs) cell proliferation, and, eventually, the reversed S-BJ cultures ceased proliferation with characteristics of crisis.
BJ fibroblasts expressed very low levels of p16 throughout their replicative lifespan. WI-38 cells, in contrast, expressed p16 even when pre-senescent, and showed a progressive increase in expression throughout their lifespan. We suggest that BJ and WI-38 cells represent extremes in a spectrum of p16 expression in human cells, since a third fibroblast strain, 82-6, showed intermediate p16 expression. However, it remains to be seen whether the cell strains we studied here are representative among the many dozens available for study. Nonetheless, our data support the idea that the ability to induce p16 provides a second mechanism for establishing and maintaining the replicative senescence of human fibroblasts. In contrast to the block established by p53, we were unable to reverse the arrest established by p16. Direct inactivation of pRB (by LgT) or suppression of p16 expression by shRNA allowed cells with high p16 (S-WI cells, P-WI + lenti-p16) to enter the S-phase of the cell cycle upon p53 inactivation. However, these cells failed to proliferate, indicating that the p16/pRB pathway, which is known to regulate entry into S-phase, must also act subsequent to the initiation of S-phase to prevent cell division.
Of particular interest, the ability of p16 to prevent LgT-stimulated cell proliferation depended on the order of expression. Thus, LgT failed to stimulate the growth of p16-expressing cells, but ectopic p16 expression did not inhibit the proliferation of LgT-expressing cells. Similarly, inactivation of both p16 and p53 (by ShRNA and GSE-22) failed to stimulate the growth of S-WI, but the same combination was very effective at reversing the growth arrest if the shRNA-p16 was expressed before WI38 cells reached replicative senescence. These findings suggest that irreversible, presumably epigenetic, changes can determine the extent to which cells are susceptible to growth stimulation by p53 inactivation. We hypothesize that once p16 is expressed, unphosphorylated pRB establishes an essentially irreversible repressive chromatin state. This repressive chromatin may then persist, even if pRB is subsequently inactivated (e.g. by LgT binding) or even if p16 itself is subsequently suppressed (for example, by shRNA) (Brehm and Kouzarides, 1999; Narita et al., 2003). This model is supported by the recent finding that some senescent cells acquire heterochromatic domains that depend on pRB activity (Narita et al., 2003).
In contrast to p53 inactivation, hTERT expression had no effect on the proliferation of replicatively senescent cells, regardless of p16 expression. This result indicates that cell proliferation is required for telomerase to extend the replicative lifespan or to immortalize human cells. Consistent with this idea, hTERT immortalized S-BJ cells only after they had been stimulated to proliferate by LgT or GSE-22 (data not shown). Moreover, hTERT did not alter telomere length in senescent cells, suggesting that DNA synthesis is needed for telomerase to extend the telomeres of human cells. Studies in yeast similarly suggested that an S-phase is required for telomerase to act at the telomeres (Wellinger et al., 1993).
Interestingly, Rasv12 stimulated limited proliferation in S-BJ, but was completely inactive in S-WI, or S-BJ cells that ectopically expressed p16. Earlier microinjection studies indicated that oncogenic RAS was incapable of stimulating senescent human fibroblasts to synthesize DNA (Lumpkin et al., 1986). Presumably, the cells used in this study expressed high levels of p16. Our results suggest that oncogenic RAS can overcome the growth-inhibitory effects of p53 and p21, but not p16. However, oncogenic RAS did not induce robust proliferation even in cells that express low p16, presumably because RAS itself eventually induces a senescent-like arrest (Serrano et al., 1997), and, at least in BJ cells, eventually increased p16 expression. RAS also synergized with CDK4m to induce cell cycle progression, although its mechanism in this regard is not clear, given that CDK4m probably acts on both the p16/pRB and p53 pathways. Nonetheless, our results suggest that transformation by oncogenic RAS may require complete or partial inactivation of the INK4a locus.
Our results raise several important questions regarding how cells respond to senescence-inducing stimuli and the role of p16 in this response. Little is known about what determines whether, and to what extent, cells express p16. Fibroblast strains clearly differ in their propensity to upregulate p16. However, it is not clear whether this difference reflects individual-to-individual variation, or selection for spontaneous silencing of p16 in some cell strains, as appears to be the case for HMEC. In addition, little is known about the signals that increase p16 expression. p16 induction has been proposed to be a response to the stress of standard culture conditions (Sherr and DePinho, 2000; Wright and Shay, 2000), although the nature of the culture stress is not known. One possibility is that DNA replication puts cells at risk for inaccurate re-establishment of repressive chromatin, which can result in p16 expression, and cells may differ in the efficiency with which they maintain chromatin organization.
Cellular senescence is thought to be important for preventing unregulated growth and malignant transformation in mammalian cells (Reddel, 2000; Campisi et al., 2001). Moreover, the ability to undergo a senescence response may determine the efficacy of cancer therapy (Schmitt et al., 2002; te Poele et al., 2002). Our data indicate that p16 is crucial for ensuring the irreversibility of the senescence arrest, consistent with its important role in tumor suppression.
Materials and methods
Cells and cell culture
Human WI-38 lung, BJ foreskin and 82-6 skin fibroblasts were obtained and grown as described previously (Dimri et al., 2000; Itahana et al., 2001). Subconfluent cells were passaged in 20% oxygen until senescence, as determined by % LN, as described previously (Dimri et al., 2000). Cultures with >75% LN were considered pre-senescent, while those with <1% LN were considered senescent. To achieve <1% LN, it was critical to maintain cultures at subconfluent densities. Senescence reversibility assays were performed on cultures that had ceased proliferation (<1% LN) for 1–3 months. Where indicated, a fixed titer (see below) of lentivirus was added simultaneously with [3H]thymidine. PDs were calculated from the cumulative cell number at each passage. When senescent cells proliferated >5–6 PDs, percentage growth was determined by clonogenic assays. When growth comprised <5–6 PDs, percentage growth was determined by counting the total number of cells per culture, and, where the senescent morphology was reversed, by counting the fraction of cells with senescent versus pre-senescent morphology per field. Clonogenic assays were performed by seeding 0.5–1 × 103 cells per 35 mm dish and counting the number of colonies with >50 cells 14–18 days later. HMECs (M.Stampfer, Lawrence Berkeley National Laboratory) were grown as described previously (Stampfer and Bartley, 1985). In our hands, the cell strains used in this study never spontaneously gave rise to replicatively immortal variants.
Vectors and viral infections
pBABE-hTERT (Kim et al., 1999) was used to produce infectious retrovirus using PT67 packaging cells (Clontech). The following DNA fragments were obtained by restriction digestion or PCR: GSE-22 (from pBabe-GSE, encoding an interfering p53 fragment) (Gudkov et al., 1993), LgT and LgTK1 [from pCMV(T) and pCMV (TK1), neither of which encodes small t-antigen] (Hara et al., 1996b), p16 (cDNA from E.Hara, University of Manchester), CDK4R24C (CDK4m; cDNA from W.Hahn, Dana-Farber Cancer Institute), EGFP cDNA (Clontech), hTERT cDNA (Counter et al., 1998), Ha-RASv12 cDNA (from pBABE-RAS) (Serrano et al., 1997), and a p16 short-hairpin RNA (shRNA) expressed from the U6 promoter in MSCV-shp16 as described by Narita et al. (2003). DNA fragments were subcloned into the pRRL.SIN-18 lentivector, which places inserted DNA under the control of the CMV promoter (Dull et al., 1998). Infectious virus was produced by transiently transfecting lentivector and packaging vectors into 293T cells as described previously (Naldini et al., 1996). Viral supernatants were concentrated by ultracentrifugation, and titers determined by ELISA for p24 (viral capsid protein) using a commercial kit (Perkin-Elmer). Cells were infected with a minimum of 40 (up to 100) ng/ml p24 equivalents (1–2 ng/1000 cells) in the presence of 6 µg/ml polybrene. Test infections using lenti–GFP showed the infection efficiency was >95% using 30 ng/ml p24 equivalents. Infection efficiencies were confirmed by immunofluorescence, except for hTERT, for which antibodies suitable for immunofluorescence were not available.
Telomerase activity and telomere length determinations
Telomerase activity was determined by TRAP, using a commercial kit (Intergen, Purchase, NY), and telomere length was assessed by Southern blot analysis, as described previously (Kim et al., 1999).
Western blotting and immunofluorescence
Western analysis was performed as described previously (Dimri et al., 2000). Primary antibodies were p21 (BD Biosciences 556430), Ras (BD Biosciences 610001), LgT (Santa Cruz 147), p53 (Santa Cruz 6243), actin (Chemicon MAB1501), p16 (JC8, gift of J.Koh, University of Vermont) and CDK4 (NeoMarker MS-864-P1). For immunofluorescence, we seeded cells in four-well chamber slides, fixed them with 3.7% formaldehyde, permeabilized them with 0.1% Triton X-100 in phosphate-buffered saline (5 min), treated them with ice-cold methanol (20 min) and blocked with 5% goat serum (1 h). Cells were incubated with primary antibodies (2–16 h) and secondary antibodies plus DAPI (1 h) in blocking solution.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank J.Garbe for senescent HMEC, and E.Hara, W.Hahn, G.Dimri and J.Koh for valuable reagents. This work was supported by grants from the US National Institutes of Health (grant Nos AG09909 and AG16379 to to J.C. and S.W.L.), US Department of Defense (grant Nos DAMD17-00-0308 and DAMD17-01-1-0209 to to P.Y. and M.N.), California Breast Cancer Research Program (grant No. 8KB-0100 to A.K.), the Canadian Institute of Health Research to C.M.B., Jose Carreras International Leukemia Foundation to F.G. and Uehara Memorial Foundation to M.N.
References
- Alcorta D.A., Xiong,Y., Phelps,D., Hannon,G., Beach,D. and Barrett,J.C. (1996) Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl Acad. Sci. USA, 93, 13742–13747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atadja P., Wong,H., Garkavtsev,I., Veillette,C. and Riabowol,K. (1995) Increased activity of p53 in senescing fibroblasts. Proc. Natl Acad. Sci. USA, 92, 8348–8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn E.H. (2001) Switching and signaling at the telomere. Cell, 106, 661–673. [DOI] [PubMed] [Google Scholar]
- Bodnar A.G. et al. (1998) Extension of life span by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- Brehm A. and Kouzarides,T. (1999) Retinoblastoma protein meets chromatin. Trends Biochem. Sci., 24, 142–145. [DOI] [PubMed] [Google Scholar]
- Bukrinsky M.I. et al. (1993) A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature, 365, 666–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J., Kim,S., Lim,C. and Rubio,M. (2001) Cellular senescence, cancer and aging: The telomere connection. Exp. Gerontol., 36, 1619–1637. [DOI] [PubMed] [Google Scholar]
- Counter C.M., Hahn,W.C., Wei,W., Caddle,S.D., Beijersbergen,R.L., Lansdorp,P.M., Sedivy,J.M. and Weinberg,R.A. (1998) Dissociation among in vitro telomerase activity, telomere maintenance and cellular immortalization. Proc. Natl Acad. Sci. USA, 95, 14723–14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. (2001) Telomere capping—one strand fits all. Science, 292, 1075–1076. [DOI] [PubMed] [Google Scholar]
- DeCaprio J.A., Ludlow,J.W., Figge,J., Shew,J.Y., Huang,C.M., Lee,W.H., Marsilio,E., Paucha,E. and Livingston,D.M. (1988) SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell, 54, 275–283. [DOI] [PubMed] [Google Scholar]
- Deppert W., Haug,M. and Steinmayer,T. (1987) Modulation of p53 protein expression during cellular transformation with simian virus 40. Mol. Cell. Biol., 7, 4453–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri G.P. et al. (1995) A novel biomarker identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA, 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri G.P., Itahana,K., Acosta,M. and Campisi,J. (2000) Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14/ARF tumor suppressor. Mol. Cell. Biol., 20, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T., Zufferey,R., Kelly,M., Mandel,R.J., Nguyen,M., Trono,D. and Naldini,L. (1998) A third-generation lentivirus vector with a conditional packaging system. J. Virol., 72, 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning E. (1992) Structure and function of simian virus 40 large tumor antigen. Annu. Rev. Biochem., 61, 55–85. [DOI] [PubMed] [Google Scholar]
- Gire V. and Wynford-Thomas,D. (1998) Reinitiation of DNA synthesis and cell division in senescent human fibroblasts by microinjection of anti-p53 antibodies. Mol. Cell. Biol., 18, 1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman S.D. and Cristofalo,V.J. (1985) Reinitiation of cellular DNA synthesis in BrdU-selected nondividing senescent WI38 cells by simian virus 40 infection. J. Cell. Physiol., 125, 122–126. [DOI] [PubMed] [Google Scholar]
- Gudkov A.V., Zelnick,C.R., Kazarov,A.R., Thimmapaya,R., Suttle,D.P., Beck,W.T. and Roninson,I.B. (1993) Isolation of genetic suppressor elements, inducing resistance to topoisomerase II-interactive cytotoxic drugs, from human topoisomerase II cDNA. Proc. Natl Acad. Sci. USA, 90, 3231–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond S.L., Ham,R.G. and Stampfer,M.R. (1984) Serum-free growth of human mammary epithelial cells: rapid clonal growth in defined medium and extended serial passage with pituitary extract. Proc. Natl Acad. Sci. USA, 81, 5435–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara E., Tsuri,H., Shinozaki,S. and Oda,K. (1991) Cooperative effect of antisense-Rb and antisense-p53 oligomers on the extension of lifespan in human diploid fibroblasts, TIG-1. Biochem. Biophys. Res. Commun., 179, 528–534. [DOI] [PubMed] [Google Scholar]
- Hara E., Smith,R., Parry,D., Tahara,H., Stone,S. and Peters,G. (1996a) Regulation of p16/CDKN2 expression and its implications for cell immortalization and senescence. Mol. Cell. Biol., 16, 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara E., Uzman,J.A., Dimri,G.P., Nehlin,J.O., Testori,A. and Campisi,J. (1996b) The helix-loop-helix protein Id-1 and a retinoblastoma protein binding mutant of SV40 T antigen synergize to reactivate DNA synthesis in senescent human fibroblasts. Dev. Genet., 18, 161–172. [DOI] [PubMed] [Google Scholar]
- Harley C.B., Futcher,A.B. and Greider,C.W. (1990) Telomeres shorten during aging of human fibroblasts. Nature, 345, 458–460. [DOI] [PubMed] [Google Scholar]
- Huschtscha L.I., Neumann,A.A., Noble,J.R. and Reddel,R.R. (2001) Effects of simian virus 40 T-antigens on normal human mammary epithelial cells reveal evidence for spontaneous alterations in addition to loss of p16(INK4a) expression. Exp. Cell Res., 265, 125–134. [DOI] [PubMed] [Google Scholar]
- Ide T., Tsuji,Y., Ishibashi,S. and Mitsui,Y. (1983) Reinitiation of host DNA synthesis in senescent human diploid cells by infection with simian virus 40. Exp. Cell Res., 143, 343–349. [DOI] [PubMed] [Google Scholar]
- Itahana K., Dimri,G. and Campisi,J. (2001) Regulation of cellular senescence by p53. Eur. J. Biochem., 268, 2784–2791. [DOI] [PubMed] [Google Scholar]
- Itahana K. et al. (2003) Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol. Cell. Biol., 23, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.H., Kaminker,P. and Campisi,J. (1999) TIN2, a new regulator of telomere length in human cells. Nat. Genet., 23, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.H., Kaminker,P.G. and Campisi,J. (2002) Telomeres, cancer and aging: in search of a happy ending. Oncogene, 21, 503–511. [DOI] [PubMed] [Google Scholar]
- Kiyono T., Foster,S.A., Koop,J.I., McDougall,J.K., Galloway,D.A. and Klingelhutz,A.J. (1998) Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature, 396, 84–88. [DOI] [PubMed] [Google Scholar]
- Land H., Parada,L.F. and Weinberg,R.A. (1983) Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature, 304, 596–602. [DOI] [PubMed] [Google Scholar]
- Lumpkin C.K., Knepper,J.E., Butel,J.S., Smith,J.R. and Pereira-Smith,O.M. (1986) Mitogenic effects of the proto-oncogene and oncogene forms of c-H-ras DNA in human diploid fibroblasts. Mol. Cell. Biol., 6, 2990–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell B.B., Starborg,M., Brookes,S. and Peters,G. (1998) Inhibitors of cyclin- dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr. Biol., 8, 351–354. [DOI] [PubMed] [Google Scholar]
- Naldini L., Blomer,U., Gage,F.H., Trono,D. and Verma,I.M. (1996) Efficient transfer, integration and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl Acad. Sci. USA, 93, 11382–11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M., Nunez,S., Heard,E., Narita,M., Lin,A.W., Hearn,S.A., Spector,D.L., Hannon,G.J. and Lowe,S.W. (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell, 113, 703–716. [DOI] [PubMed] [Google Scholar]
- Paddison P.J., Caudy,A.A., Bernstein,E., Hannon,G.J. and Conklin,D.S. (2002) Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev., 16, 948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez R.D., Morales,C.P., Herbert,B.S., Rohde,J.M., Passons,C., Shay,J.W. and Wright,W.E. (2001) Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev., 15, 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddel R.R. (2000) The role of senescence and immortalization in carcinogenesis. Carcinogen, 21, 477–484. [DOI] [PubMed] [Google Scholar]
- Rheinwald J.G. et al. (2002) A two-stage, p16(INK4A)- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Mol. Cell. Biol., 22, 5157–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov S.R., Kozakiewicz,B.K., Holst,C.R., Stampfer,M.R., Haupt,L.M. and Tlsty,T.D. (2001) Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature, 409, 633–637. [DOI] [PubMed] [Google Scholar]
- Sakamoto K., Howard,T., Ogryzko,V., Xu,N.Z., Corsico,C.C., Jones,D.H. and Howard,B. (1993) Relative mitogenic activities of wild-type and retinoblastoma binding defective SV40 T antigens in serum deprived and senescent human fibroblasts. Oncogene, 8, 1887–1893. [PubMed] [Google Scholar]
- Schmitt C.A., Fridman,J.S., Yang,M., Lee,S., Baranov,E., Hoffman,R.M. and Lowe,S.W. (2002) A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell, 109, 335–346. [DOI] [PubMed] [Google Scholar]
- Serrano M. and Blasco,M.A. (2001) Putting the stress on senescence. Curr. Opin. Cell Biol., 13, 748–753. [DOI] [PubMed] [Google Scholar]
- Serrano M., Lin,A.W., McCurrach,M.E., Beach,D. and Lowe,S.W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell, 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Shay J.W. and Wright,W.E. (2001) Aging and cancer: the telomere and telomerase connection. Novartis Found. Symp., 235, 116–125. [PubMed] [Google Scholar]
- Shay J.W., Pereira-Smith,O.M. and Wright,W.E. (1991) A role for both Rb and p53 in the regulation of human cellular senescence. Exp. Cell Res., 196, 33–39. [DOI] [PubMed] [Google Scholar]
- Shay J.W., Van Der Haegen,B.A., Ying,Y. and Wright,W.E. (1993) The frequency of immortalization of human fibroblasts and mammary epithelial cells transfected with SV40 large T-antigen. Exp. Cell Res., 209, 45–52. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and DePinho,R.A. (2000) Cellular senescence: Mitotic clock or culture shock? Cell, 102, 407–410. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev., 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Shih C. and Weinberg,R.A. (1982) Isolation of a transforming sequence from a human bladder carcinoma cell line. Cell, 29, 161–169. [DOI] [PubMed] [Google Scholar]
- Stampfer M.R. and Bartley,J.C. (1985) Induction of transformation and continuous cell lines from normal human mammary epithelial cells after exposure to benzo[a]pyrene. Proc. Natl Acad. Sci. USA, 82, 2394–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein G.H., Drullinger,L.F., Soulard,A. and Dulic,V. (1999) Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol., 19, 2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Poele R.H., Okorokov,A.L., Jardine,L., Cummings,J. and Joel,S.P. (2002) DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res., 62, 1876–1883. [PubMed] [Google Scholar]
- Vaziri H. and Benchimol,S. (1998) Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr. Biol., 8, 279–282. [DOI] [PubMed] [Google Scholar]
- Wei W. and Sedivy,J.M. (1999) Differentiation between senescence (M1) and crisis (M2) in human fibroblast cultures. Exp. Cell Res., 253, 519–522. [DOI] [PubMed] [Google Scholar]
- Wellinger R.J., Wolf,A.J. and Zakian,V.A. (1993) Saccharomyces telomeres acquire single-strand TG1-3 tails late in S phase. Cell, 72, 51–60. [DOI] [PubMed] [Google Scholar]
- Wolfel T. et al. (1995) A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science, 269, 1281–1284. [DOI] [PubMed] [Google Scholar]
- Wright W.E. and Shay,J.W. (2000) Telomere dynamics in cancer progression and prevention: fundamental differences in human and mouse telomere biology. Nat. Med., 6, 849–851. [DOI] [PubMed] [Google Scholar]
- Wright W.E. and Shay,J.W. (2001) Cellular senescence as a tumor-protection mechanism: The essential role of counting. Curr. Opin. Genet. Dev., 11, 98–103. [DOI] [PubMed] [Google Scholar]