

Short abstract
RNAi can be achieved by feeding worms Escherichia coli expressing dousble-stranded RNA corresponding to a specific gene. An optimized feeding method is presented that results in phenotypes at least as strong as those produced by direct injection of RNA for embryonic lethal genes, and stronger for genes with post-embryonic phenotypes.
Abstract
Background
In Caenorhabditis elegans, injection of double-stranded RNA (dsRNA) results in the specific inactivation of genes containing homologous sequences, a technique termed RNA-mediated interference (RNAi). It has previously been shown that RNAi can also be achieved by feeding worms Escherichia coli expressing dsRNA corresponding to a specific gene; this mode of dsRNA introduction is conventionally considered to be less efficient than direct injection, however, and has therefore seen limited use, even though it is considerably less labor-intensive.
Results
Here we present an optimized feeding method that results in phenotypes at least as strong as those produced by direct injection of dsRNA for embryonic lethal genes, and stronger for genes with post-embryonic phenotypes. In addition, the interference effect generated by feeding can be titrated to uncover a series of hypomorphic phenotypes informative about the functions of a given gene. Using this method, we screened 86 random genes on consecutive cosmids and identified functions for 13 new genes. These included two genes producing an uncoordinated phenotype (a previously uncharacterized POU homeodomain gene, ceh-6, and a gene encoding a MADS-box protein) and one gene encoding a novel protein that results in a high-incidence-of-males phenotype.
Conclusions
RNAi by feeding can provide significant information about the functions of an individual gene beyond that provided by injection. Moreover, it can be used for special applications for which injection or the use of mutants is sometimes impracticable (for example, titration, biochemistry and large-scale screening). Thus, RNAi by feeding should make possible new experimental approaches for the use of genomic sequence information.
Background
RNA-mediated interference (RNAi) is the phenomenon first described in the nematode Caenorhabditis elegans in which introduction of double-stranded RNA (dsRNA) results in potent and specific inactivation of the corresponding gene through the degradation of endogenous mRNA [1,2]. This technique rapidly produces gene-specific loss-of-function or hypomorphic phenotypes, and potent interference is also observed in the progeny of the affected animal. Thus, because RNAi results in a robust, specific and durable interference effect, and also because RNAi is the simplest and most efficient method for inactivating genes in C. elegans, it has been rapidly embraced as a reverse-genetics tool for determining the functions of specific genes.
Studies involving RNAi have shown that this mode of interference can function across cell boundaries; that is, the site of injection is not critical for successful gene inactivation [1]. As a result, it is also possible to initiate RNAi either by soaking worms in a solution of dsRNA or by feeding worms with Escherichia coli expressing target gene dsRNA, as RNA can be absorbed through the gut and distributed to somatic tissues and the germ line [3,4]. However, these other delivery systems have seen limited use in published studies as the observed efficiency of gene inhibition is significantly lower than with microinjection of adult hermaphrodite worms [5].
Nevertheless, RNAi by feeding has several distinct advantages over microinjection. First, because feeding is far less labor-intensive than injection, it is extremely convenient for performing RNAi on a large number of worms. In this regard, RNAi by feeding has proved particularly useful in genetic screens to identify C. elegans genes involved in the RNAi mechanism [6]. Second, for the same reason, feeding is useful for performing RNAi on large numbers of genes. And third, feeding is considerably less expensive than injection and results in a durable reagent (a bacterial strain expressing dsRNA corresponding to a gene of interest) which can be reused to reproduce an RNAi phenotype easily and inexpensively.
For these reasons, we explored feeding as a means of delivering dsRNA for RNAi. We have developed an optimized protocol for feeding which is of similar sensitivity to injection and results in phenotypes at least as strong as those produced by injection. Furthermore, the interference effect produced by feeding can be titrated, resulting in the ability to generate a range of strong and hypomorphic phenotypes analogous to an allelic series of mutants. Thus, this method establishes RNAi by feeding as a viable or even preferable alternative to RNAi by injection in C. elegans.
Results
Establishment of optimal RNAi feeding conditions
Timmons and Fire [4] first described a method for RNAi in which bacteria expressing dsRNA are fed to C. elegans. A fragment corresponding to the gene of interest is cloned into a feeding vector (L4440) between two T7 promoters in inverted orientation and is transformed into a bacterial strain carrying IPTG-inducible expression of T7 polymerase [4]. Recently, Timmons and Fire also showed that use of an E. coli strain (HT115(DE3)), which lacks double-strand-specific RNase III, improves the ability to produce RNAi phenotypes by feeding (L. Timmons and A. Fire, personal communication; and Figure 1).
Figure 1.
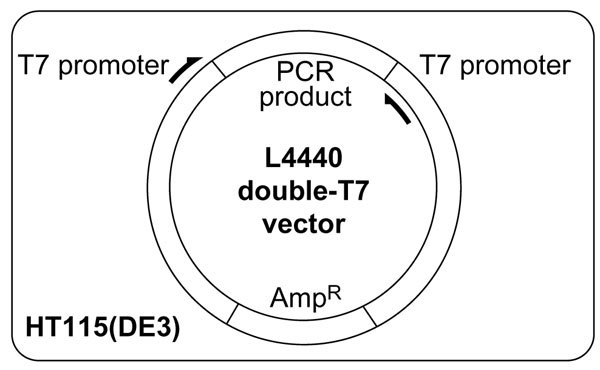
L4440 double-T7 vector inside HT115 RNase-deficient E. coli. A fragment from the gene of interest is amplified by PCR and cloned into the L4440 double-T7 vector, which has two T7 promoters in inverted orientation flanking the multiple cloning site [4]. Cloned plasmids are transformed into HT115(DE3), an RNase III-deficient E. coli strain with IPTG-inducible expression of T7 polymerase (L. Timmons and A. Fire, personal communication).
To determine feeding conditions that maximize observable phenotypes, we started with the existing L4440 vector and strain HT115(DE3) and varied a number of parameters that could affect the efficiency of RNAi. We chose two initial test genes that were easy to assay: gpb-1, for which mutants are embryonic lethal, and unc-22, which results in a post-embryonic uncoordinated phenotype (Unc), as determined by deletion mutants and by RNAi [4,7]. We first tested different methods of induction with isopropylthiogalactoside (IPTG) to see if this would affect the RNAi phenotypes observed. Uninduced bacteria produced no phenotypes, but, somewhat unexpectedly, each presumably stronger method of induction resulted in a lower penetrance of phenotypes, culminating in 0% phenotype from overnight induction in culture (Table 1). The best induction method was to grow bacteria in culture without induction, to seed these bacteria onto plates containing IPTG, and then to incubate overnight at room temperature; with this method, gpb-1 produced 100% dead embryos and unc-22 produced 99% Uncs. To further test this new induction method, we fed two more genes, par-1 and par-3, mutations in either gene result in embryonic lethality [8,9]. Using our optimized induction conditions, feeding par-1 and par-3 resulted in 100% and 96% dead embryos, respectively, similar to the results obtained with null mutants (Table 1).
Table 1.
Induction methods for RNAi by feeding
| Non-Ind | Ind (1) | Ind (2) | Ind (3) | Ind (4) | ||||||
| Test gene | n | % Phe | n | % Phe | n | % Phe | n | % Phe | n | % Phe |
| gpb-1 | 546 | 0 | 530 | 100 | 309 | 84 | 442 | 97 | 346 | 0 |
| unc-22 | 422 | 0 | 255 | 99 | 179 | 80 | ND | ND | ND | ND |
| par-1 | ND | ND | 313 | 100 | 263 | 100 | ND | ND | ND | ND |
| par-3 | ND | ND | 391 | 96 | 325 | 11 | ND | ND | ND | ND |
Four different methods were compared to determine optimal induction conditions for RNAi; non-induced (Non-Ind) bacteria were also included for comparison. Induction conditions (Ind) were as follows: (1) Bacteria were induced on plates with IPTG at room temperature overnight; (2) bacteria were induced in culture at 37°C for 2 h; (3) bacteria were induced on plates with IPTG at 37°C overnight; (4) bacteria were induced in culture at 37°C overnight (see the Materials and methods section for detailed protocols). gpb-1, par-1 and par-3 were scored for percentage of dead embryos, unc-22 was scored for percentage of worms with an uncoordinated phenotype. Data shown represent the progeny of three fed worms. ND, not done; n is the number of worms or embryos scored; %Phe, percentage of worms or embryos with phenotype.
To determine the optimal concentration of IPTG for producing RNAi phenotypes, we titrated the IPTG concentration from 10 mM to 1 pM plus no IPTG. We found that 1 mM IPTG gave us the highest penetrance of phenotypes (Table 2). In addition, we tested the effect of seeded bacterial density and growth phase on the ability to generate phenotypes; for both par-1 and unc-22, saturated cultures produced phenotypes as well as log-phase bacteria (see the Materials and methods section). We also compared feeding at 15°C versus 22°C and found that although there is some gene-specific variation in RNAi effectiveness between these two temperatures, there is no generalizable difference (data not shown).
Table 2.
Hypomorphic RNAi phenotypes produced by titration of IPTG concentration
| Concentration of IPTG | |||||||
| Test gene | Experimental phenotype | 0 | 1 pM | 1 nM | 1 μM | 1 mM | 10 mM |
| unc-37 | Emb | 0% | 4% | 11% | 48% | 100% | 77% |
| Unc | 0% | 10% | 10% | 100% | NA | 69% | |
| hlh-2 | Emb | 0% | 8% | 20% | 97% | 100% | 86% |
| Unc | 0% | 13% | 9% | 100% | NA | 100% | |
| mei-1 | Emb | 0% | 7% | 16% | 71% | 100% | 71% |
| Male | 0% | 0% | 6% | 8% | NA | 3% | |
| rba-2 | Emb | 56% | 100% | 100% | 100% | 100% | 100% |
Four genes were tested to determine whether reducing the concentration of IPTG used to induce the bacteria could elicit hypomorphic phenotypes from worms escaping embryonic lethality. The percentage of embryonic lethality (Emb) was determined from the total number of progeny, whereas the percentage of uncoordinated (Unc) or male worms was determined from escapers only. High concentrations of IPTG (for example 10 mM) overinduce and presumably kill the bacteria, thus leading to a lower penetrance of strong phenotypes. Data shown represent the progeny of three fed worms. NA, not applicable.
Finally, we tested the effect of length of feeding time on the penetrance of RNAi phenotypes observed. Many genes were not effectively silenced after 24 hours at 22°C, but were after 48 hours (see the Materials and methods section and Figure 2). In general, it appears that allowing worms to ingest dsRNA-expressing bacteria for longer periods of time increases the efficiency of RNAi by feeding. However, feeding worms beginning earlier than the L4 stage did not improve the penetrance of RNAi in the progeny (data not shown). After 48 hours, L4-stage hermaphrodites become older adults, and many stop laying fertilized eggs; thus we have routinely used the longest feeding time possible (36-40 hours at 22°C), which ensures that these worms will continue to lay fertilized eggs during the time window from which progeny are assayed (the subsequent 24 hours). We found that 72 hours at 15°C gives a similar level of RNAi inhibition for most genes (data not shown).
Figure 2.
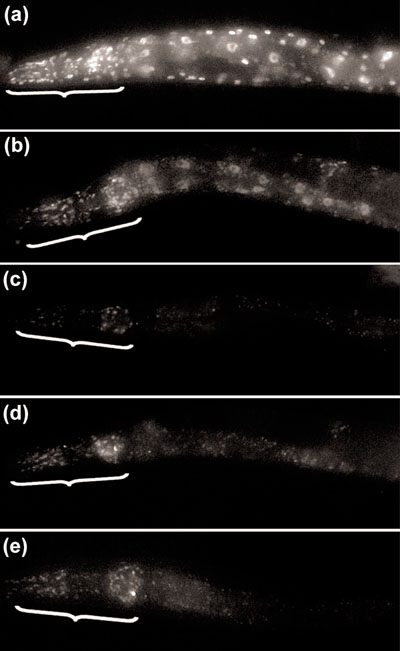
Tissue susceptibility to RNAi by feeding. Worms with a transgenic GFP reporter gene expressed in all somatic tissues (egl-27::gfp) were fed (a) non-dsRNA expressing bacteria or (b-e) bacteria expressing gfp dsRNA for (b) 24 h, (c) 48 h, (d) 72 h, or (e) 96 h. After being fed for 24 h, GFP expression was markedly reduced (b) compared to similarly treated unfed worms (a). (c) Of worms fed for 48 h, none had any visible non-neural GFP expression, and 20/22 (91%) had reduced levels of neural GFP. (d) After 72 h, 93% had no non-neural somatic GFP expression (the remaining 7% had very weak expression), and 28/30 (93%) had reduced neural GFP. (e) Finally, after being fed for 96 h, no worms had any non-neural GFP expression, and 26/27 (96%) also had reduced levels of neural GFP compared with similarly treated unfed worms. The head of each worm (from the nose to the posterior pharynx), which contains the majority of the neurons in the animal, is indicated with a bracket.
Titration of RNAi phenotypes by feeding
Many genes have pleiotropic effects in vivo but have one dominant mutant phenotype that masks other informative phenotypes; thus, we decided to see if we could elicit such masked phenotypes by titrating the concentration of IPTG and, by extension, the degree of RNAi. We titrated the IPTG concentration from 1 pM to 1 mM in three log increments; for comparison, we also tested no IPTG (uninduced) and 10 mM IPTG. We analyzed four genes that could possibly be titrated to other distinct phenotypes: unc-37, which has a known allelic series with strong alleles resulting in embryonic lethality and weak alleles resulting in an uncoordinated (Unc) phenotype [10]; hlh-2, which is embryonic lethal by RNAi injection and is expressed in some neural precursors which eventually form the ventral nerve cord ([11] and M. Krause, personal communication); mei-1, which is required for meiotic spindle formation and is embryonic lethal by RNAi injection, but for which a weak mutant allele produces a high-incidence-of-males (Him) phenotype [12,13]; and rba-2, a very strong embryonic lethal gene by RNAi injection which is also involved in repression of vulval cell fates [14].
All four genes were 100% embryonic lethal at 1 mM IPTG (Table 2). For the first three genes, decreasing the IPTG concentration to 1 μM reduced the embryonic lethality sufficiently to expose high levels of a secondary phenotype. For unc-37 and hlh-2, 100% of the surviving worms were Unc. For mei-1, a Him phenotype was seen: 8% of the surviving worms were male, which is significantly greater than the 0.5% which normally arise by non-disjunction in a wild type hermaphrodite culture. As we further decreased the IPTG concentration to 1 pM, these levels of embryonic lethality tapered off, as did the penetrance of secondary phenotypes. With no IPTG, all the worms fed these genes had wild-type progeny. For rba-2, however, embryonic lethality was observed at all concentrations of IPTG, including 56% lethality without any IPTG. Therefore, these bacteria are likely to express a low level of T7 polymerase in the absence of induction, which is sufficient to produce an RNAi phenotype for some genes. From these data, we conclude that it is possible to titrate RNAi phenotypes by feeding bacteria induced with different concentrations of IPTG, which results in a series of hypomorphic alleles analogous to an allelic series of mutants.
RNAi by feeding multiple genes
We also tested RNAi by feeding for two genes simultaneously. When we fed one embryonic lethal gene and one non-lethal gene, we found a reduced penetrance of embryonic lethality compared to feeding the lethal gene alone: feeding gpb-1 and unc-22 together reduced the embryonic lethality to 50% (versus 100% for gpb-1 alone) and feeding par-3 and unc-22 together reduced the lethality to 85% (versus 96% for par-3 alone). Furthermore, in both cases the resulting progeny also failed to display the unc-22 phenotype (data not shown). Thus, it appears that feeding two genes greatly reduces the strength of phenotype produced by either.
In a separate experiment, we diluted the unc-37 HT115 strain 1:1 with E. coli that does not produce dsRNA (OP50, a strain commonly used for growing C. elegans). In this case, the phenotype changed dramatically from 100% to 0% embryonic lethality, with 100% of progeny displaying post-embryonic phenotypes (uncoordinated, rolling, and body morphology defects), which is similar to the phenotypes obtained by inducing the bacteria with 1 fM IPTG (data not shown). Thus, diluting feeding bacteria with bacteria not expressing the dsRNA can also be used to generate weak hypomorphic phenotypes.
Target RNA expression in RNAi-treated hermaphrodites
To determine which tissues are susceptible to RNAi by feeding, we fed bacteria expressing green fluorescent protein (GFP) dsRNA to hermaphrodite worms with a transgenic GFP reporter gene expressed in all somatic tissues (egl-27::gfp; Figure 2a) [15]. After being fed for 24 hours at 15°C, GFP expression was markedly reduced compared to similarly treated unfed worms (n = 28; compare Figure 2b and a). After 48 hours, with the exception of the nervous system, GFP was not detectable in somatic tissues; furthermore, neural GFP was dramatically reduced in 91% of worms (n = 22; Figure 2c). A similar level of inhibition of GFP expression was observed after 72 and 96 hours of feeding (Figure 2d,e). Although GFP fluorescence in fed worms was abolished or severely reduced, GFP was sometimes expressed at high levels in late-stage embryos derived from these worms (data not shown). This suggests that some zygotic embryonic transcripts may be difficult to silence, possibly because a continuous supply of dsRNA cannot be provided through the eggshell. In summary, RNAi by feeding efficiently silences genes in most C. elegans somatic tissues; however, the nervous system has a delayed and somewhat less robust response to RNAi compared to other tissues.
Comparison of RNAi by feeding and injection for maternal-effect lethal genes
To test the strength of RNAi by feeding using the above optimized protocol, we compared it to RNAi by injection. We first tested a set of 14 known maternal-effect embryonic lethal genes (gpb-1, par-1, par-2, par-3, par-6, cyk-1, skn-1, dnc-1, bir-1, pal-1, dif-1, plk-1, dhc-1, and mex-3) to compare the lethality obtained with both methods (see [16,17,18] for a review of maternal-effect genes). All genes tested were 100% embryonic lethal by both feeding and injection except for par-3, which resulted in 97% dead embryos by feeding but 100% by injection, and cyk-1, which resulted in 55% dead embryos by feeding but 100% by injection (Table 3). For some genes, for example par-1, RNAi by feeding (n = 32 fed worms) and by injection (n = 9) both resulted in 100% embryonic lethality of the progeny in all cases. Whereas RNAi of par-3 resulted in 100% embryonic lethality for all injected worms (n = 9), worms subjected to RNAi of par-3 by feeding resulted in 100% dead embryos in 16/24 cases but lower levels of lethality (on average 88%) in 8/24 cases (data not shown). The overall comparison shows that RNAi by feeding is of similar strength to RNAi by injection for maternal-effect embryonic lethal genes. For some genes, however, RNAi by feeding appears to be somewhat more variable than RNAi by injection.
Table 3.
Strength of RNAi by feeding versus injection
| Feeding | Injection | |||
| Test gene | n | % Phe | n | % Phe |
| gpb-1 | 506 | 100 | 175 | 100 |
| par-1 | 262 | 100 | 293 | 100 |
| par-2 | 257 | 100 | 288 | 100 |
| par-3 | 241 | 97 | 540 | 100 |
| par-6 | 361 | 100 | 348 | 100 |
| cyk-1 | 308 | 55 | 275 | 100 |
| skn-1 | 355 | 100 | 311 | 100 |
| dnc-1 | 343 | 100 | 309 | 100 |
| bir-1 | 391 | 100 | 403 | 100 |
| pal-1 | 393 | 100 | 396 | 100 |
| dif-1 | 391 | 100 | 244 | 100 |
| plk-1 | 402 | 100 | 239 | 100 |
| dhc-1 | 269 | 100 | 173 | 100 |
| mex-3 | 200 | 100 | 256 | 100 |
Fourteen known maternal-effect embryonic lethal genes were tested to determine the efficiency of RNAi by feeding relative to injection for producing embryonic lethality. Data shown represent the progeny of three fed worms. n, number of embryos scored; %Phe, percentage of embryos with embryonic lethality.
To further confirm that RNAi by feeding is comparable in effectiveness to injection for analyzing maternal-effect genes, we made four-dimensional time-lapse video recordings of developing embryos whose mothers were fed with dsRNA corresponding to the 10 above genes with a mutant phenotype detectable by the third cell division (gpb-1, par-1, par-2, par-3, par-6, cyk-1, dnc-1, bir-1, plk-1, and dhc-1,). In each case, the known null phenotypes were obtained (Figure 3, and data not shown; for cyk-1, one of two embryos recorded had the known phenotype and the other survived). For example, par-2(RNAi) by either method yielded spindle orientation defects, and bir-1(RNAi) by either method yielded embryos with cytokinesis defects (Figure 3) [19,20].
Figure 3.
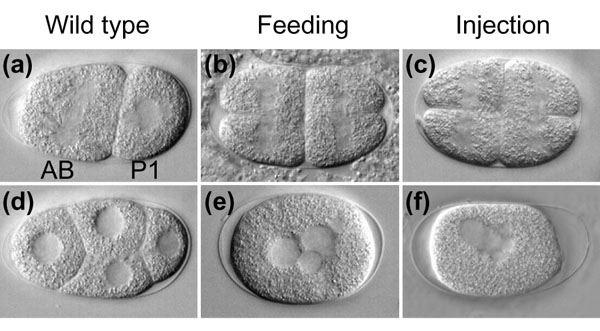
Embryonic phenotypes from RNAi by feeding versus injection. (a) Wild-type N2 embryos divide with the AB (anterior cell) spindle oriented along the dorso-ventral axis and the P1 (posterior cell) spindle along the AP axis to create a four-cell embryo (d). In wild-type embryos, AB is larger and divides slightly before P1. For par-2, RNAi by both (b) feeding and (c) injection results in embryos in which AB and P1 are of equal size and divide synchronously, with both spindles oriented along the dorso-ventral axis. Embryos in (a-c) are undergoing the second mitotic divisions. For bir-1, RNAi by both (e) feeding and (f) injection results in embryos that do not complete cytokinesis and thus form a single multinucleate cell. Embryos in (d-f) have undergone two rounds of mitosis. Anterior is to the left in all panels.
Quantitative analysis of RNAi by feeding versus injection
To compare the sensitivity of feeding versus injection in detecting phenotypes from randomly selected genes, we tested 86 genes from the middle of C. elegans chromosome I on consecutive cosmids from K04G2 to R05D11. Of these genes, 12 yielded a phenotype by injection and 13 by feeding (Table 4). These data suggest that feeding and injection are similarly sensitive for detecting genes that give an RNAi phenotype. The number of phenotypes detected in this experiment is, however, too small to allow a fair comparison of the strengths of the two methods.
Table 4.
Sensitivity of RNAi by feeding versus injection
| Gene(s) | Feeding phenotype | Injection phenotype | Gene information |
| K04G2.8a | Unc, Bmd, Lvl | Emb | apr-1 (APC-related) |
| F18C12.2a | Emb, Unc, Mlt, Bmd | Emb, Unc, Mlt, Lvl | DNA-J domain |
| ZK265.5,6* | Gro | Gro | G-protein-coupled receptor/unknown function |
| T01G9.4 | Emb, Gro, Clr | Emb, Gro | kup-2 (unknown function) |
| T01G9.5 | Emb | Emb | mei-1 (meiotic spindle formation) |
| T01G9.6a | Emb, Gro, Pvl | Emb | kin-10 (CKII-beta subunit) |
| F52B5.6 | Emb, Ste | Emb, Ste | Ribosomal protein L25 |
| T19A6.2a | Gro | Gro | Ynr053p-like protein |
| D1081.2 | Unc, Prz | Unc | MADS domain |
| D1081.8 | Emb | Emb | Myb-like DNA-binding domain |
| K02B12.1 | Unc, Mlt | Gro, Mlt, Lvl | ceh-6 (POU homeodomain protein) |
| K02B12.3 | Ste, Gro | Ste | WD domains |
| K02B12.8 | Him | None | Unknown function |
From chromosome 1, 86 random genes from consecutive cosmids were selected to test the sensitivity of feeding versus injection for detecting RNAi phenotypes. Of these, 13 gave a phenotype by either method, 13 by feeding and 12 by injection; 11 of these genes have no previously described mutant or RNAi phenotype. Genes were determined to have a given phenotype if at least 10% of progeny had that phenotype, except for sterility, which required an average brood size of less than ten, and the Him phenotype, which required at least two out of three fed worms to have >5% male progeny. Bmd, body morphology defect; Clr, clear; Emb, embryonic lethal; Gro, slow growth; Him, high incidence of males; Lvl, larval lethal (death at any larval stage); Mlt, molting defect (old cuticle remains attached); Prz, paralyzed; Pvl, protruding vulva; Ste, sterile; Unc, uncoordinated. *The fragment used overlaps two predicted genes: ZK265.5 and ZK265.6.
To obtain a quantitative measure of the efficiency of feeding versus injection for RNAi, we compared the two methods on a larger data set. Fraser et al. have constructed an RNAi feeding library for C. elegans chromosome I [21]. After performing RNAi by feeding on the first 1,200 predicted genes on chromosome I, phenotypes were identified for 168 predicted genes. We then performed RNAi by injection on these genes and compared the phenotypes to those obtained by feeding (Figure 4; see the Materials and methods section for scoring criteria) We reasoned that using a set of genes initially identified by feeding for this comparison would be valid, as in our previous comparison of feeding and injection, feeding successfully detected all those genes with RNAi phenotypes detected by injection.
Figure 4.

Large-scale comparison of RNAi by feeding versus injection. The first 1,200 predicted genes from chromosome I were screened by feeding [21]. Those genes with a phenotype were subjected to RNAi by injection, and results obtained by the two methods were compared according to phenotypic class - embryonic lethal (Emb), sterile (Ste), or post-embryonic (PostEmb; see the Materials and methods section for scoring criteria). Data shown compare results from three worms subjected to RNAi by feeding or injection.
Of the 168 genes, 123 were determined to be embryonic lethal by either method, of which feeding detected 97% and injection 91%. The embryonic lethality was of equal penetrance using either method for 77% of these genes, was stronger by feeding for 15% of the genes, and was stronger by injection for 8% of the genes. Of 52 genes giving a sterile phenotype by either method, 96% were identified by feeding and 48% by injection; 44% were detected by both methods, 52% only by feeding, and 4% only by injection. And, finally, of 154 genes giving a post-embryonic phenotype by either method, 96% were identified by feeding and 64% by injection; 60% were detected by both methods, 36% only by feeding, and 4% only by injection. In addition to the above, we also injected 30 random genes that gave no phenotype by feeding, and none gave a detectable phenotype by injection (data not shown). Thus, from this expanded data set, we conclude that RNAi by feeding is roughly equivalent to injection for detecting embryonic lethality, and is somewhat better for detecting genes causing sterility or other post-embryonic phenotypes.
Discussion
We present a method by which RNAi by feeding is as strong and as sensitive as RNAi by injection for detecting embryonic phenotypes, and, furthermore, is more sensitive for detecting sterility and post-embryonic phenotypes. The fact that feeding is superior to injection for detecting sterile phenotypes is most likely due to the fact that with feeding the RNA interference effect is begun at L4 stage, giving it more time to affect the germline or gonad, whereas young adults are usually injected; nevertheless, this does not diminish the utility of feeding for detecting sterility as L4-stage hermaphrodites are considerably more difficult to inject. Similarly, the fact that feeding is better than injection for producing post-embryonic phenotypes could be due to the fact that both fed mothers and their progeny are constantly exposed to dsRNA, whereas by injection only the mothers are subjected to a single dose. Again, this confers an inherent advantage on feeding in producing post-embryonic phenotypes that can be exploited by those studying genes important later in development. It should be noted, however, that there are gene-specific differences between RNAi by feeding and injection. We found that some genes are more sensitive to RNAi by injection and others to RNAi by feeding (Figure 4). In addition, for some genes, RNAi by feeding is more variable than RNAi by injection. Thus feeding is a useful tool to complement, rather than replace, RNAi by injection.
We also showed that by titrating the IPTG concentration, RNAi by feeding can generate a range of strong and hypomorphic phenotypes. Because many embryonic lethal genes, for example, have informative post-embryonic RNAi phenotypes, it is extremely useful to be able to elicit such phenotypes for specific genes. Hypomorphic phenotypes were also seen by analyzing the progeny of fed or injected worms immediately following exposure to dsRNA (during the time when RNAi was taking effect); however, these phenotypes were not as reproducible or as robust as those generated by titrating the IPTG concentration. Furthermore, titration allows a low level of RNAi to be consistently applied to a large number of worms, increasing the possibility of detecting low-penetrance phenotypes.
We also tested other variables that might have affected the strength and utility of feeding. Interestingly, the current feeding vector does not have any transcriptional terminators, and thus we would expect transcripts to vary in size and also to contain sequences from the vector backbone. As a result, we tested a modified vector containing T7 terminators just outside the T7 promoters and discovered that inclusion of such terminators greatly diminished the effectiveness of RNAi (data not shown). Because it is also useful to concomitantly inhibit the functions of two genes, we tested the ability to feed two dsRNAs and learned that this greatly reduces the ability of each gene to independently produce a phenotype. Other methods could be tested for this purpose, however, including co-transforming two feeding vectors into the same bacteria or inserting two gene fragments into the same vector, either as single fragments or as inverted repeats.
Our analysis of 86 genes on chromosome I by feeding versus injection identified 13 with an RNAi phenotype. Among these 13 genes were two, apr-1 and mei-1, previously reported to have loss-of-function phenotypes. apr-1 encodes a protein similar to the APC (adenomatous polyposis coli) protein; it is involved in Wnt signaling and has previously been shown to control endoderm induction in the embryo [22]. Although we failed to obtain embryonic lethality for apr-1 by feeding, we did identify reproducible and specific phenotypes - uncoordinated movement, body morphology defects, and larval lethality - consistent with the expression pattern and previously demonstrated roles for this gene [23]. Both feeding and injection produced strong embryonic lethality for mei-1, a regulator of meiosis [12]. Furthermore, by titrating the IPTG concentration, we were able to phenocopy the Him phenotype of a weak mei-1 mutant, which is indicative of X-chromosomal non-disjunction during meiosis [13].
The remaining 11 genes for which we identified an RNAi phenotype have no previously reported function. RNAi of two of these genes caused an Unc phenotype, suggesting roles in the neuromuscular system: ceh-6 is a POU homeodomain protein, and D1081.2 encodes a MADS-box transcription factor [24,25]. Another gene, K02B12.8, produced a Him RNAi phenotype, suggesting a possible function in meiotic chromosome segregation. RNAi of D1081.8, which encodes a novel protein with a Myb-like DNA-binding domain, resulted in 100% embryonic lethality, as did, unsurprisingly, F52B5.6, which is thought to encode a ribosomal protein [26,27]. K02B12.3 resulted in sterility by RNAi; this gene encodes a WD-domain protein which is weakly similar to TUP1, a general transcriptional repressor in Saccharomyces cerevisiae [28]. Finally, T19A6.2A, which is similar to a human breast cancer autoantigen, resulted in slow growth [29]. The ability of RNAi by feeding to detect a wide range of phenotypes for these genes demonstrates its value for studying gene function in C. elegans.
One major advantage of RNAi by feeding over injection is that it is considerably less labor-intensive. In practice, this means that RNAi can be performed on thousands of worms for little more effort than feeding a single worm. Thus, feeding affords the possibility of using RNAi in ways not practically possible by injection, such as doing large-scale biochemistry on worms with a mutant RNAi phenotype. A second advantage of RNAi by feeding is that once a bacterial strain expressing a specific dsRNA is created, it can be reused indefinitely to repeatedly perform RNAi on a given gene. Thus, feeding is extremely useful for large-scale experiments in which either many worms will be subjected to RNAi, many genes will be used for RNAi, or a few genes will be used for RNAi many times. Indeed, this method has been used by Fraser et al. [21] to efficiently screen roughly 90% (2,500 predicted genes) of C. elegans chromosome I by RNAi.
Materials and methods
Strains and clones
Standard methods were used for culturing C. elegans on NGM (nematode growth medium) [30]. Fragments designated for RNAi were obtained by polymerase chain reaction (PCR) from genomic DNA and were cloned into the L4440 feeding vector (pPD129.36) [4]; all fragments were between 500 and 2,700 base pairs (bp) in length. The resulting plasmids were transformed into the HT115(DE3) RNase Ill-deficient E. coli strain, which was previously shown by Timmons and Fire to be beneficial for RNAi by feeding (L. Timmons and A. Fire, personal communication). The HT115 genotype is as follows: (F-, mcrA, mcrB, IN(rrnD-rrnE)1, lambda-, rnc14::Tn10(DE3 lysogen:lacUV5 promoter-T7 polymerase)). The RNase III gene is disrupted by a Tn10 transposon carrying a tetracycline-resistance marker. Inclusion of tetracycline in feeding plates or in bacterial cultures used for feeding in many cases resulted in a weaker RNAi effect (data not shown), perhaps because the cultures grew very poorly, and thus it was not included in our feeding experiments; however, bacteria were selected on tetracycline plates before feeding.
The following primer pairs were used for PCR amplification: gpb-1 (5'-ATGAGCGAACTTGACCAAC-3' and 5'-TTAATTCCAGATCTTGAGG-3'), par-1 (5'-CAAAGCACGTGATAACCGG-3' and 5'-TTGGTGGCTCAATAAATGGC-3'), par-3 (5'-TTTGGCTTCACTGTGACCG-3' and 5'-TGATGTGCTGTGGATCAGC-3'), par-2 (5'-GCCGTCGCCCACTGTCG-3' and 5'-CCGGCTCCAGAGTGTCC-3'), cyk-1 (5'-GAAGAACAGCTGACCAGCG-3' and 5'-GACGATTCAATGCAATGATGG-3'), skn-1 (5'-CTGCCGAAGAGAATGCTCG-3' and 5'-GTTTGGTACAACTTCTGTTGG-3'), dnc-1 (5'-TCTCCACTTTCTACTACAGC-3' and 5'-TGTTCTTGGAAGCCAGCG-3'), bir-1 (5'-ATGGCACCCGGGACCAA-3' and 5'-TTATTTGCCGCGGCGGC-3'), pal-1 (5'-GGGGTACCCCAATGTCGGTCGATGTCAAGTCG-3' and 5'-CATGCCATGGCATGGTACTTATAGCCGAATCTTCTG-3'), dif-1 (5'-ACGCATTGAAATGTCGGACG-3' and 5'-TTGCAGGGAAAGCACGGAG-3'), and plk-1 (5'-GACAAGGATCGTGGGACC-3' and 5'-AGCACAGCAACTTGGTGG-3'). Primer pairs for mex-3, dhc-1, par-6, unc-37, hlh-2, mei-1 and rba-2, the 88 genes on cosmids K04G2 to R05D11, and the 1,200 genes used for large-scale comparison of feeding and injection were obtained as part of the Research Genetics C. elegans GenePairs collection. Fragments corresponding to unc-22 and GFP were generated from L4440-based vectors containing those inserts (pLT 61.1 and pPD128.110, respectively) [4].
Bacterial induction method tests
Single colonies of HT115 bacteria containing cloned L4440 plasmids were picked and grown in culture in LB with 50 μg/ml ampicillin (Amp), except where indicated. The following methods were used to induce expression of dsRNA:
Non-induced
Bacteria were grown for 8 h, then seeded directly onto NGM plates with 50 μg/ml Amp and incubated at room temperature overnight.
Protocol I (optimal)
Bacteria were grown for 8 h, then seeded directly onto NGM plates with 1 mM IPTG and 50 μg/ml Amp. (Similar results were obtained from bacterial cultures grown for 8-18 h before seeding plates; RNAi results obtained after growth longer than 24 h were sometimes weaker.) Seeded plates were allowed to dry at room temperature and induction was continued at room temperature overnight.
Protocol 2
Bacteria were grown to OD595 = 0.4, then IPTG was added to 0.4 mM and bacteria were induced shaking at 37°C for 2 h. After induction, additional IPTG and Amp were added to a total concentration of 0.8 mM and 100 μg/ml, respectively, before seeding onto NGM plates with 50 μg/ml Amp.
Protocol 3
Bacteria were treated as in Protocol 1, but induction was performed on seeded plates at 37°C overnight.
Protocol 4
Bacteria were treated as in Protocol 2, but induction was performed in culture shaking at 37°C overnight.
RNAi by feeding
L4-stage hermaphrodite worms were placed onto NGM plates containing seeded bacteria expressing dsRNA for each gene and were incubated for 36-40 h at 22°C or for 72 h at 15°C. Then, three worms were independently replica plated onto plates seeded with the same bacteria and were allowed to lay eggs for 24 h at 22°C before being removed. Progeny were scored for embryonic lethality after a further 24 h at 22°C, and post-embryonic phenotypes were scored blindly by two independent observers at the end of four successive 12-h intervals. Progeny laid on the first plate were also scored for post-embryonic phenotypes. A gene was found to be positive for a given phenotype if it could be observed in at least two of three worms or their progeny in at least two independent feeding experiments. Feeding times shorter than 36-40 h at 22°C or 72 h at 15°C were not always sufficient to produce a strong RNAi effect; for example, par-2 and par-3 were 53% and 23% embryonic lethal, respectively, after feeding for 24 h but were both 100% embryonic lethal after feeding for at least 36 h at 22°C (our unpublished results).
Titration of hypomorphic phenotypes
Single colonies of HT115 bacteria containing cloned L4440 plasmids with fragments corresponding to unc-37, hlh-2, mei-1 or rba-2 were treated as described in the optimal induction method above, except that for each gene, NGM plates were used with the following IPTG concentrations: 0, 1 pM, 1 nM, 1 μM, 1 mM, and 10 mM. Worms were incubated for 72 h at 15°C before being replica plated and scored.
Tissue susceptibility to RNAi by feeding
Worms containing an extrachromosomal array expressing egl-27::gfp, a ubiquitous somatic GFP reporter, were fed bacteria expressing GFP dsRNA for either 24, 48, 72, or 96 h at 15°C. Those worms, as well as control worms fed bacteria not expressing GFP dsRNA, were photographed under identical conditions. Figure 2 illustrates the GFP expression of a representative worm from each time point. Many worms from the 72 h and 96 h time points had almost no detectable GFP expression in any tissue.
RNA synthesis and microinjection
Injections were performed as in [1]. Templates for dsRNA synthesis were made by PCR on L4440-based feeding constructs using T7 primer (5'-CGTAATACGACTCACTATAG-3'). Sense and antisense RNAs were synthesized in a single reaction in vitro using a T7 polymerase-based kit (Promega). Double-stranding was achieved by incubation at 72°C for 10 min, and the sizes of dsRNA products were verified by electrophoresis. dsRNA was injected at a concentration of 0.5-1.0 mg/ml into one or both gonad arms (we and others have found that injection into one or both gonad arms produces equivalent effects). Injected worms were allowed to recover at 22°C for 24 h post-injection, then were replica plated and allowed to lay eggs for 24 h. Injected worms and their progeny were scored as previously described for RNAi by feeding.
Four-dimensional recordings of developing embryos
Mothers fed dsRNA were dissected in egg buffer (118 mM And, 40 mM KCl, 3 mM CaCl2, 3 mM MgCl2, 5 mM HEPES pH 7.2) on a coverslip to release young embryos. The cover-slip was inverted onto a 3% agar pad, which was then sealed with petroleum jelly. A series of 12 focal planes was recorded every 30 sec for 1 h using Openlab software (Improvision) controlling either a Zeiss Axioplan 2 or Leica DMBRE microscope.
Quantitative comparison of RNAi by feeding versus injection
RNAi was performed by feeding at 15°C using the optimized method described above or by injection. Embryonic lethality was scored by estimating the percentage of dead embryos to the nearest 10% among the offspring of the three worms replica plated for each gene. For embryonic lethality, feeding and injection were considered to be of equal strength if the percentages of dead embryos were within 10%, and one method was considered stronger than the other if the percentage of dead embryos was at least 20% greater than that obtained by the other method. Fed or injected worms were considered sterile if they had fewer than 10 progeny on average, as wild-type worms under similar conditions typically have more than 50 progeny. Post-embryonic phenotypes were scored if more than 10% of the progeny had a given phenotype. For sterility and post-embryonic phenotypes, feeding and injection were considered to be of equal strength if both resulted in that phenotype, and one method was considered stronger if it resulted in that phenotype but the other method did not.
Acknowledgments
Acknowledgements
We thank Andy Fire and Lisa Timmons for kindly providing protocols, feeding vectors, and the HT115(DE3) bacterial strain. We also thank Mike Krause, Lisa Timmons and Andy Fire for sharing unpublished data. Björn Schumacher and Monica Gotta gave us helpful comments on the manuscript. R.S.K. was supported by a Howard Hughes Medical Institute Predoctoral Fellowship, M.M.C. by an EC-TMR Network Grant, P.Z. by a Wellcome Trust Prize Studentship, A.G.F. by a US Army Breast Cancer Research Fellowship, and J.A. by a Wellcome Trust Senior Research Fellowship (No. 054523).
References
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Bass BL. Double-stranded RNA as a template for gene silencing. Cell. 2000;101:235–238. doi: 10.1016/s0092-8674(02)71133-1. [DOI] [PubMed] [Google Scholar]
- Tabara H, Grishok A, Mello CC. RNAi in C. elegans: soaking in the genome sequence. Science. 1998;282:430–431. doi: 10.1126/science.282.5388.430. [DOI] [PubMed] [Google Scholar]
- Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- Hunter CP. Genetics: a touch of elegance with RNAi. Curr Biol. 1999;9:R440–R442. doi: 10.1016/s0960-9822(99)80276-0. [DOI] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;99:123–132. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- Zwaal RR, Ahringer J, van Luenen HG, Rushforth A, Anderson P, Plasterk RH. G proteins are required for spatial orientation of early cell cleavages in C. elegans embryos. Cell. 1996;86:619–629. doi: 10.1016/s0092-8674(00)80135-x. [DOI] [PubMed] [Google Scholar]
- Guo S, Kemphues KJ. par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell. 1995;81:611–620. doi: 10.1016/0092-8674(95)90082-9. [DOI] [PubMed] [Google Scholar]
- Etemad-Moghadam B, Guo S, Kemphues KJ. Asymmetrically distributed PAR-3 protein contributes to cell polarity and spindle alignment in early C. elegans embryos. Cell. 1995;83:743–752. doi: 10.1016/0092-8674(95)90187-6. [DOI] [PubMed] [Google Scholar]
- Pflugrad A, Meir JY, Barnes TM, Miller DM., 3rd The Groucho-like transcription factor UNC-37 functions with the neural specificity gene unc-4 to govern motor neuron identity in C. elegans. Development. 1997;124:1699–1709. doi: 10.1242/dev.124.9.1699. [DOI] [PubMed] [Google Scholar]
- Krause M, Park M, Zhang JM, Yuan J, Harfe B, Xu SQ, Greenwald I, Cole M, Paterson B, Fire A. A C. elegans E/Daughterless bHLH protein marks neuronal but not striated muscle development. Development. 1997;124:2179–2189. doi: 10.1242/dev.124.11.2179. [DOI] [PubMed] [Google Scholar]
- Clark-Maguire S, Mains PE. mei-1, a gene required for meiotic spindle formation in Caenorhabditis elegans, is a member of a family of ATPases. Genetics. 1994;136:533–546. doi: 10.1093/genetics/136.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mains PE, Kemphues KJ, Sprunger SA, Sulston IA, Wood WB. Mutations affecting the meiotic and mitotic divisions of the early Caenorhabditis elegans embryo. Genetics. 1990;126:593–605. doi: 10.1093/genetics/126.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Mello CC. A CBP/p300 homolog specifies multiple differentiation pathways in Caenorhabditis elegans. Genes Dev. 1998;12:943–955. doi: 10.1101/gad.12.7.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solari F, Bateman A, Ahringer J. The Caenorhabditis elegans genes egl-27 and egr-1 are similar to MTA1, a member of a chromatin regulatory complex, and are redundantly required for embryonic patterning. Development. 1999;126:2483–2494. doi: 10.1242/dev.126.11.2483. [DOI] [PubMed] [Google Scholar]
- Bowerman B, Ingram MK, Hunter CP. The maternal par genes and the segregation of cell fate specification activities in early Caenorhabditis elegans embryos. Development. 1997;124:3815–3826. doi: 10.1242/dev.124.19.3815. [DOI] [PubMed] [Google Scholar]
- Rose LS, Kemphues KJ. Early patterning of the C. elegans embryo. Annu Rev Genet. 1998;32:521–545. doi: 10.1146/annurev.genet.32.1.521. [DOI] [PubMed] [Google Scholar]
- Bowerman B. Maternal control of pattern formation in early Caenorhabditis elegans embryos. Curr Top Dev Biol. 1998;39:73–117. doi: 10.1016/s0070-2153(08)60453-6. [DOI] [PubMed] [Google Scholar]
- Levitan DJ, Boyd L, Mello CC, Kemphues KJ, Stinchcomb DT. par-2, a gene required for blastomere asymmetry in Caenorhabditis elegans, encodes zinc-finger and ATP-binding motifs. Proc Natl Acad Sci U S A. 1994;91:6108–6112. doi: 10.1073/pnas.91.13.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser AG, James C, Evan GI, Hengartner MO. Caenorhabditis elegans inhibitor of apoptosis protein (IAP) homologue BIR-1 plays a conserved role in cytokinesis. Curr Biol. 1999;9:292–301. doi: 10.1016/s0960-9822(99)80137-7. [DOI] [PubMed] [Google Scholar]
- Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, Ahringer J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–330. doi: 10.1016/S0370-2693(97)00774-0. [DOI] [PubMed] [Google Scholar]
- Rocheleau CE, Downs WD, Lin R, Wittmann C, Bei Y, Cha YH, Ali M, Priess JR, Mello CC. Wnt signaling and an APC-related gene specify endoderm in early C. elegans embryos. Cell. 1997;90:707–716. doi: 10.1016/s0092-8674(00)80531-0. [DOI] [PubMed] [Google Scholar]
- Hoier EF, Mohler WA, Kim SK, Hajnal A. The Caenorhabditis elegans APC-related gene apr-1 is required for epithelial cell migration and Hox gene expression. Genes Dev. 2000;14:874–886. [PMC free article] [PubMed] [Google Scholar]
- Burglin TR, Finney M, Coulson A, Ruvkun G. Caenorhabditis elegans has scores of homoeobox-containing genes. Nature. 1989;341:239–243. doi: 10.1038/341239a0. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Durbin R, Eddy SR, Finn RD, Sonnhammer EL. Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 1999;27:260–262. doi: 10.1093/nar/27.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi R, Feoktistova A, McCann S, Valentine V, Look AT, Lipsick JS, Gould KL. Myb-related Schizosaccharomyces pombe cdc5p is structurally and functionally conserved in eukaryotes. Mol Cell Biol. 1998;18:4097–4108. doi: 10.1128/mcb.18.7.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervitz SA, Aravind L, Sherlock G, Ball CA, Koonin EV, Dwight SS, Harris MA, Dolinski K, Mohr S, Smith T, et al. Comparison of the complete protein sets of worm and yeast: orthology and divergence. Science. 1998;282:2022–2028. doi: 10.1126/science.282.5396.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keleher CA, Redd MJ, Schultz J, Carlson M, Johnson AD. Ssn6-Tup1 is a general repressor of transcription in yeast. Cell. 1992;68:709–719. doi: 10.1016/0092-8674(92)90146-4. [DOI] [PubMed] [Google Scholar]
- Racevskis J, Dill A, Stockert R, Fineberg SA. Cloning of a novel nucleolar guanosine 5'-triphosphate binding protein autoantigen from a breast tumor. Cell Growth Differ. 1996;7:271–280. [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]