

Abstract
Recent studies have shown that the intersection between transcription and proteins involved in the ubiquitin–proteasome pathway encompasses both proteolytic and nonproteolytic functions. Examples of the latter type include evidence that monoubiquitylation of some transcriptional activators stimulates their activity. In addition, the proteasomal ATPases are recruited to many active promoters through binding to activators and play an important, nonproteolytic role in promoter escape and elongation. In this study, we report the discovery of a new nonproteolytic activity of the proteasome (specifically the proteasomal ATPases): the active destabilization of activator–promoter complexes. This reaction depends on the presence of an activation domain and ATP. Destabilization is inhibited in vitro and in vivo if the protein is monoubiquitylated or if ubiquitin is genetically fused to the activator. The fact that monoubiquitylated activator is resistant to the “stripping” activity of the proteasomal ATPases may explain, in part, why some activators require this modification in order to function efficiently.
Keywords: Proteasome, Gal4, transcription, promoter occupancy, Sug1
The 26S proteasome is (Baumeister et al. 1998) comprised of a 20S core complex with the proteolytic active sites sequestered inside the barrel-like cavity, which has small openings on both the top and bottom of the complex. Capping the ends of the 20S core is the 19S regulatory particle (Glickman et al. 1998), which introduces substrates into the core. The substrate is unfolded by the action of six AAA class ATPases (Rpt1–6) and fed into the maw of the proteasome. The proteasome is directly or indirectly involved in many cellular processes, including many nuclear functions such as DNA repair (Russell et al. 1999a) and RNA polymerase II transcription (Collins and Tansey 2006).
The connections between the ubiquitin–proteasome pathway and transcription have been shown to encompass both proteolytic and nonproteolytic events. For example, the levels of many transcriptional activators are kept low by polyubiquitylation and subsequent proteasome-mediated proteolysis, which conspires to keep the expression of the activator’s target genes low. A reduction in polyubiquitylation and/or proteolysis in response to some signaling pathway or a change in cellular conditions can thus serve as the activating event for expression of such genes. Conversely, some activators require proteasome activity in order to function (Lonard et al. 2000; Lipford et al. 2005). In the case of Estrogen Receptor-α (ER-α), it has been shown that proteasome inhibition abrogates cycling of the activator (Reid et al. 2003) and many general transcription factors and cofactors (Métivier et al. 2003) on and off the promoter, suggesting that polyubiquitylation and proteolysis are important for clearing various factors from the promoter after a certain number of transcription events.
The potency of some activators is stimulated by monoubiquitylation (Salghetti et al. 2001; Bres et al. 2003; Greer et al. 2003), which does not signal proteasome-mediated proteolysis (a chain of at least four K48-linked ubiquitins is required for degradation) (Thrower et al. 2000). The mechanistic basis of this effect is not well understood, although monoubiquitylation of LexA-VP16 has been shown to stimulate the ability of the activator to recruit PTEF-b (Kurosu and Peterlin 2004) and thus make elongation more efficient. Ubiquitylation might also stimulate post-transcriptional events such as RNA processing (Muratani et al. 2005). The discovery of a stimulatory role of activator monoubiquitylation, combined with the aforementioned requirement of some activators to be turned over by the proteasome, presumably explains why E3 ubiquitin ligases have been shown to be important for the efficient function of some activators (Salghetti et al. 2001; Kim et al. 2003; von der Lehr et al. 2003).
These discoveries suggest that the ubiquitylation state of an activator may be at the heart of how cells govern their duration of function (Tansey 2001; Conaway et al. 2002; Gonzalez et al. 2002; Muratani and Tansey 2003). The idea is that monoubiquitylation constitutes a “licensing event” for activator function. But once a single ubiquitin is added to a lysine side chain, subsequent chain growth can occur, eventually resulting in a chain with four or more ubiquitins that then attracts the proteasome and results in activator destruction. This model would provide an elegant explanation for how cells keep at least some activators under control and couple their continued activity to continued signaling (for a review, see Kodadek et al. 2006).
Another surprising development in this area was the discovery that the ATPases, particularly Sug1/Rpt6 and Sug2/Rpt4, stimulate promoter escape and elongation in a nonproteolytic fashion (Ferdous et al. 2001) after being recruited to promoters through direct interactions with acidic activation domains (Gonzalez et al. 2002; Archer et al. 2005). Subsequently, it was shown that the proteasomal ATPases play a role in SAGA recruitment to promoters during early stages of the transcription cycle (Lee et al. 2005). Finally, these proteins were also shown to be important in linking histone H2B monoubiquitylation and methylation of histone H3, the latter mark being associated with recently transcribed genes (Ezhkova and Tansey 2004). Recently published studies in yeast show that physical and functional interactions between the proteasomal ATPases and chromatin are quite widespread (Auld et al. 2006; Sikder et al. 2006; Sulahian et al. 2006)
Ironically, none of these interesting insights into the nonproteolytic roles of the proteasomal ATPases in transcription would appear to explain the original observation of a proteasome–transcription connection, which was that specific mutations in the SUG1 and SUG2 genes supress the “no growth on galactose” phenotype of gal4D, which expresses a truncated activator lacking about two-thirds of the activation domain (Swaffield et al. 1992). The sug1-1 and sug2-1 alleles were recessive mutations, suggesting that suppression resulted from loss of some antagonistic function of the wild-type ATPases on Gal4D. This is not easily explained in the context of stimulatory roles in elongation, SAGA recruitment, or histone methylation. Thus, we have remained interested in discovering the putative inhibitory activity of the proteasomal ATPases suggested by the genetic data. Here we describe such an activity. It is demonstrated that purified 19S regulatory particle or 26S proteasome has a general, nonproteolytic activity that can destabilize an activator–DNA complex. This activity is dependent on an activation domain and ATP hydrolysis. In a HeLa nuclear extract (NE) the model activator Gal4-VP16 is monoubiquitylated when bound to DNA, and this modified form of the activator is resistant to 19S-mediated destabilization. The physiological relevance of these findings is demonstrated through the study of a previously identified Gal4 derivative called Gap71 (Corton and Johnston 1989; Corton et al. 1998), which carries mutations in the DNA-binding domain that do not compromise the intrinsic DNA-binding activity of the protein, but that nonetheless cripple it in vivo. It is shown here that the defect in Gap71 lies in its inability to stably occupy promoters under inducing conditions, although stable association is observed when the activation domain is masked by Gal80 under noninducing conditions. We show that the mutations in Gap71 prevent monoubiquitylation of the DNA-binding domain, and that this is linked both in vitro and in vivo with hypersensitivity to the destabilizing effect of the proteasomal ATPases. Genetic fusion of ubiquitin to this mutant form of Gal4 significantly restores its DNA-binding activity even in the presence of the proteasome. These results point to a novel and previously unrecognized nonproteolytic activity of the 19S regulatory particle of the proteasome, and suggest that modulating this destabilization of activator–DNA complexes is at least one mechanism by which monoubiquitylation increases activator potency.
Results
Destabilization of an activator–DNA complex by the proteasome without proteolysis
Since the first and essential step in the transcription process involves binding of the activator to DNA, we first assessed whether the 19S affected this activity. GST-tagged Gal4(1–147)-VP16 (hereafter called GST-Gal4-VP16) was bound to a biotinylated duplex DNA containing five Gal4-binding sites linked to streptavidin-coated beads. After the addition of highly purified 19S or 26S complex (Verma et al. 2000; Ferdous et al. 2001) in the presence of ATP, the amount of GST-Gal4-VP16 remaining on the DNA was determined. Neither 26S nor 19S had an effect on the steady state level of GST-Gal4-VP16 occupancy (Fig. 1A). We then performed the same experiment in the presence of a 10-fold excess of nonbiotinylated, soluble DNA containing five tandem repeats of the Gal4-binding sites as a trap for dissociated GST-Gal4-VP16. In the presence of the competitor, the GST-Gal4-VP16 occupancy was reduced ∼70% (Fig. 1B, lanes 1,2). These results indicate that the 19S is capable of reducing GST-Gal4-VP16 occupancy, but since this is only apparent in the presence of competing DNA, GST-Gal4-VP16 must cycle quickly on and off DNA. The loss of the protein–DNA complex was unaffected by the addition of lactacystin (LC) to a level that inhibits ∼95% of the protease activity (Fig. 1B, lanes 4,5) (Ferdous et al. 2001, 2002). These data imply that the removal of GST-Gal4-VP16 by the 19S does not involve the permanent denaturation or degradation of the protein.
Figure 1.

Destabilization of the activator–DNA complex by proteasome is not dependent on its proteolytic activity. (A) The activator–DNA complex is not destabilized by the proteasome without competitor DNA. Activator–DNA complexes of GST-Gal4-VP16 and the DNA template, shown at the top of the gel, were isolated after 30 min of incubation at 30°C and then incubated with ATP and purified 19S (0.5 μg) or 26S (1 μg). After 15 min of incubation at 30°C, the amount of protein retained on the DNA was analyzed by Western blotting. (B) Destabilization of the activator–DNA complex in the presence of competitor DNA by the proteasome is independent of proteolysis. Destabilization reactions were carried out as described in A, except that a 10-fold excess of competitor DNA (containing five Gal4-binding sites only) was added. Preincubation of 19S and 26S with anti-Sug1 antibodies and 200 μM LC is indicated. (C) Effect of competitor DNA on the activator–DNA complex destabilization by 26S proteasome. Destabilization reactions were carried out as described in A, except that activator–DNA complexes were incubated in the presence or absence of 26S proteasome and the indicated fold excess of specific and nonspecific (NS) competitor DNA (C-DNA). The amount of protein retained on the DNA was analyzed by Western blotting with anti-Gal4DBD antibodies.
Addition of antibody raised against Sug1 completely inhibited the destabilization reaction (Fig. 1B, cf. lanes 2,4 and 3,6, respectively). These results demonstrate that this effect was indeed due to 19S and not a contaminant. In order to test for the specificity of the competitor DNA effect, the same experiment was performed in the presence of DNA lacking the Gal4-binding site. As shown in Figure 1C, in order to observe net dissociation of the GST-Gal4-VP16 protein from the immobilized DNA, Gal4-binding sites in the soluble competitor were necessary (cf. lanes 5 and 6).
We conclude that the 19S and 26S complexes have the ability to reversibly disrupt activator–DNA complexes, and that this activity requires the function of Sug1 but not proteolysis.
Destabilization of activator–DNA complexes requires an activation domain and ATP hydrolysis
Given that the proteasomal ATPases act to unfold substrates during proteasome-mediated proteolysis, a reasonable model for the activity described above is that the 19S complex operates on GST-Gal4-VP16 in this fashion, but that this unfolding process is uncoupled from proteolysis. If so, then one would expect that the unfolding reaction would require direct binding of the activator (substrate) to the 19S complex and that ATP would be required for turnover. The Gal4 activation domain binds directly to Sug1 and Sug2 and the activation domain of VP16 (Lee et al. 1995) interacts directly with the Sug1 protein. Therefore, we tested if this physical interaction is critical for the destabilization of the GST-Gal4-VP16–DNA complex.
Figure 2A compares the amount of GST-Gal4, without (lanes 1–3) and with (lanes 4–6) the VP16 activation domain, left on DNA after exposure to 19S. In the presence of ATP, GST-Gal4-VP16 is removed efficiently from the immobilized DNA, whereas the complex lacking the activation domain is unaffected by exposure to the regulatory particle (Fig. 2A, cf. lanes 2 and 5). This suggests that, as expected, the destabilization reaction requires direct substrate–19S contacts. ATP hydrolysis appears to be required. Even the GST-Gal4-VP16-containing complex was stable in the presence of the 19S complex and the nonhydrolyzable analog ADP and AMP-PNP did not support full activity (Fig. 2B, cf. lanes 1,3,4,5).
Figure 2.

Destabilization of the protein–DNA complex by 19S is dependent on ATP and the activation domain. (A) The influence of ATP and the activation domain in protein–DNA complex destabilization by 19S. Isolation and destabilization of the indicated protein–DNA complex by 19S was carried out in the presence or absence of ATP as described in Figure 1B. (B) ATP hydrolysis is required for efficient destabilization of the activator–DNA complex by 26S proteasome. Destabilization of the activator–DNA complex by 26S proteasome was analyzed in the presence of a 10-fold excess of specific competitor DNA as described in Figure 1B, except that 500 μM of ATP and the indicated ATP analogs were added. (C) Destabilization of the GST-Pho4p/DNA complex by the proteasome. Destabilization of Pho4p–DNA complexes by 26S proteasome was performed as described in Figure 1B, except that the DNA template contained two Pho4-binding sites and the proteasome was preincubated with MG132.
To address the generality of this activity, we tested the stability of the DNA complex of Pho4, which also interacts with 19S proteasome (Sun et al. 2002). As is evident in Figure 2C, the GST-Pho4–DNA complex was also destabilized by the addition of 26S in a dose-dependent fashion (lanes 1–3). As with GST-Gal4-VP16, this destabilization was not affected by the inhibition of proteolysis (MG132) (Fig. 2C, cf. lanes 3 and 4). The fact that the destabilization activity is observed using two different protein–DNA complexes suggests that it may be general, at least for acidic activators that bind the 19S complex.
Monoubiquitylation of Gal4-VP16 impedes the destabilizing activity of 19S
The results presented in Figures 1 and 2, while interesting, provide something of a conundrum. The proteasome concentration in the nucleus of a cell is very high (Russell et al. 1999b). Given the potent destabilization activity observed here, how do activators resist this activity in order to function efficiently? One possibility is that the activator is modified post-translationally in such a way that it acquires the ability to resist this destabilization process. Alternatively, there may be other nuclear proteins that modulate this process through binding to the proteasome or the activator.
To begin to probe this issue, immobilized DNA and GST-Gal4-VP16 were incubated at 30°C with HeLa NE, which contains high levels of active proteasome (data not shown), in the presence or absence of ATP. The DNA-bound activator was isolated after 30 min and quantified by SDS-PAGE and Western blotting. As shown in Figure 3A, significant GST-Gal4-VP16 remained bound to the bead-attached DNA even in the presence of ATP, suggesting that there is indeed some nuclear activity that can antagonize the ATP-dependent destabilization activity of the 19S regulatory particle on the activator–DNA complex.
Figure 3.

GST-Gal4-VP16 bound to DNA is monoubiquitylated in HeLa NE. (A) ATP-dependent modification of DNA-bound GST-Gal4-VP16 in NE. GST-Gal4-VP16 and immobilized DNA were incubated with NE in the presence (+) or absence (−) of ATP, and the DNA-bound activator was affinity-purified after 30 min and analyzed by Western blotting. Numbers on the left show the molecular weight markers (in kilodaltons). (B) DNA-bound GST-Gal4-VP16 is ubiquitylated. HeLa NE and immobilized DNA were incubated in the presence (+) or absence (−) of GST-Gal4-VP16 and ATP for 30 min at 30°C. The DNA-bound activator was isolated and analyzed first with anti-GST antibodies (lanes 1–3) and then reprobed with anti-ubiquitin antibodies (lanes 4–6). (C) Monoubiquitylation of GST-Gal4-VP16 in NE. Gal4-VP16 and DNA were incubated with NE in the absence (−) or presence of wild-type (WT) or mutant (K48R) His6-tagged ubiquitin (His6-Ub) for 30 min at 30°C. An aliquot (5 μL) of the 30-μL reaction was removed to analyze activator levels (Total), and activators, ubiquitylated with tagged-Ub, were affinity-purified under denaturing conditions on Talon beads (Isolate). Proteins were detected by Western blotting using anti-Gal4DBD antibodies. Arrows indicate the ubiquitylated and nonubiquitylated (Gal4, as input [IN]) GST-Gal4-VP16.
A strong clue as to the nature of this “protective” activity was immediately apparent in that the mobility of the DNA-bound GST-Gal4-VP16 in the presence of ATP was retarded, consistent with an ∼7–8 kDa increase in the apparent molecular mass of the GST-Gal4-VP16 protein (Fig. 3A, cf. lanes 2 and 1,3). This shift was not observed in the ATP-depleted extract (Fig. 3A, lane 3). Since this is approximately the shift expected from the addition of one ubiquitin molecule, we probed the membrane with the anti-ubiquitin as well as anti-GST antibody. As is evident in Figure 3B, the DNA-bound GST-Gal4-VP16 from the extract is recognized by the anti-ubiquitin antibody, whereas the input protein is not (lanes 4,6). This suggests that DNA-bound GST-Gal4-VP16 exposed to NE is monoubiquitylated. To test this more rigorously, we performed an assay where GST-Gal4-VP16 was incubated with an excess of His6- tagged ubiquitin or a His6 derivative of a mutant ubiquitin that cannot be chain extended through Lys 48 (K48R). Ubiquitin-tagged proteins were affinity purified under denaturing conditions by incubation with Ni-NTA-agarose resin and analyzed by Western blotting using antibody raised against the Gal4 DNA-binding domain (Gal4DBD). As shown in Figure 3C, both wild-type ubiquitin and the K48R derivative are linked to GST-Gal4-VP16 in the NE in the presence of ATP. This is evidenced by the fact that only in the presence of these His6-tagged proteins does an anti-Gal4DBD-reactive band appear. Furthermore, these bands are shifted with respect to the input protein by an amount consistent with monoubiquitylation. Finally, the fact that incubation with both wild-type His6-ubiquitin and His6-ubiquitin (K48R) produce bands of the same apparent mass argues that this band does not represent a polyubiquitylated product (Fig. 3C, cf. lanes 2,3 and input).
Ubiquitylation occurs within the Gal4 DNA-binding domain
The VP16 activation domain lacks lysine residues. Therefore, the monoubiquitylation of GST-Gal4-VP16 almost certainly occurs within the Gal4DBD or on GST. To identify the ubiquitylated domain, we took advantage of the presence of a TEV protease site located between GST and the Gal4DBD in this construct to remove the GST fusion and purify Gal4-VP16, which was then incubated with HeLa NE in the presence or absence of ATP and an excess of His6-tagged ubiquitin. Ubiquitin-tagged proteins were affinity purified under denaturing conditions and analyzed by Western blotting with antibodies raised against the Gal4DBD. As is evident in Figure 4A, Gal4-VP16 lacking the GST portion is still monoubiquitylated in the presence of ATP, but not in its absence (lanes 2,3). No ubiquitylated band was detected when GST alone was employed in the same assay (see Fig. 5; data not shown).
Figure 4.
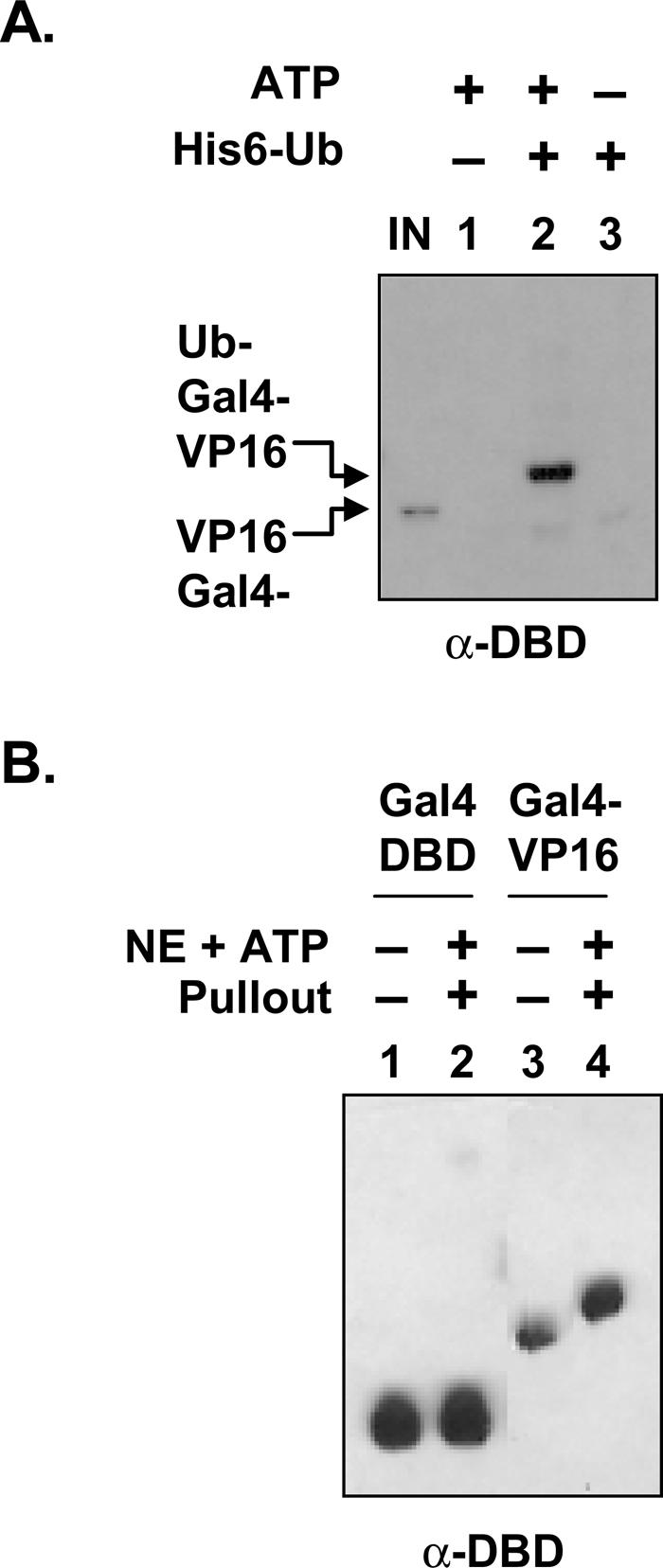
Ubiquitylation of Gal4DBD is dependent on the VP16 activation domain. (A) The Gal4DBD is ubiquitylated in vitro. DNA (150 ng) and TEV-treated Gal4-VP16 (100 ng) were incubated for 30 min at 30°C in HeLa NE in the presence (+) or absence (−) of ATP and His6-tagged ubiquitin (His6-Ub). Proteins conjugated with His6-Ub were affinity-purified as described in Figure 3B. Ubiquitylated proteins were detected by Western blotting with anti-Gal4DBD antibodies. Arrows indicate the ubiquitylated (Ub-Gal4-VP16) and purified Gal4-VP16 (IN). (B) The activation domain is necessary for ubiquitylation of the DNA-bound GST-Gal4-VP16. Immobilized DNA, GST-Gal4DBD (Gal4DBD), and GST-Gal4-VP16 (Gal4-VP16) were incubated with (+) or without (−) HeLa NE and ATP. After 30 min of incubation at 30°C, DNA-bound protein was isolated (+ pullout) and separated on an SDS gel along with the respective purified proteins (− pullout) and detected with anti-Gal4DBD antibodies.
Figure 5.

Ubiquitylation of Gal4-VP16 impedes the destabilization activity of the 19S. (A) Effect of ubiquitylation on the activator–DNA complex destabilization. GST-Gal4-VP16, DNA, and NE were first incubated for 30 min at 30°C (1st Inc.) with (+) or without (−) ATP. Activator–DNA complexes were isolated, washed, and analyzed for 19S-dependent destabilization as described in Figure 1B. (B) Ubiquitylation of GST-Gal4-VP16 but not GST-Gap71-VP16 in NE. Ubiquitylation of activator proteins was analyzed as described in Figure 3C, except that the indicated proteins were incubated in the presence (+) or absence (−) of ATP and wild-type (WT) His6-tagged ubiquitin (His6-Ub) for 30 min at 30°C. An arrowhead indicates the ubiquitylated GST-Gal4-VP16.
Is ubiquitylation of the Gal4DBD, dependent on the activation domain? To address this, the GST-Gal4DBD with and without the VP16 activation domain was incubated with HeLa NE and ATP, and then isolated as described in Figure 3A. As is clear in Figure 4B (lanes 1,2), the DNA-binding domain itself was not ubiquitylated, as is evident by a lack of mobility shift. In contrast, Gal4 with the VP16 activation domain was shifted, and that mobility shift (Fig. 4B, lane 4) was approximately the same as that of recombinant GST-ubiquitin–Gal4-VP16, where a single ubiquitin is genetically fused to mutant Gal4-VP16 (data not shown). We conclude that DNA-bound Gal4-VP16 is monoubiquitylated and that ubiquitylation of Gal4DBD is dependent on the presence of the activation domain.
Monoubiquitylation of the activator correlates with resistance to 19S-mediated dissociation of the protein–DNA complex
We next tested if the monoubiquitylated form of the activator is resistant to destabilization in the more defined system using purified proteasome using the protocol outlined in Figure 5A. GST-Gal4-VP16 and DNA were first incubated in NE with or without ATP. Since, as shown in Figure 3A, the ubiquitylation is ATP-dependent, this allowed the production of ubiquitylated and nonubiquitylated forms of Gal4-VP16 bound to the DNA. The bead-attached GST-Gal4-VP16–DNA complex was isolated, washed, and then incubated with or without purified 19S in the presence of ATP and excess competing DNA. As shown in Figure 5A, exposure of the nonubiquitylated form of GST-Gal4-VP16 to 19S and ATP destabilized the complex relative to the amount on the DNA in the absence of the 19S (lanes 3,4). However, the ubiquitylated form of GST-Gal4-VP16 was quite stable in the presence of 19S (Fig. 5A, lanes 1,2). These results, along with the data in Figure 4A, indicate that monoubiquitylation of Gal4-VP16 leads to higher Gal4-VP16 occupancy in the presence of 19S.
To investigate the significance of this ubiquitylation and 19S-mediated destabilization further, we took advantage of a puzzling mutation in Gal4 that had been noted earlier. It had been reported that three mutations in the Gal4DBD (S22D, K23Q, and K25F), providing a Gal4 derivative called Gap71, had no effect on intrinsic DNA-binding activity in vitro, but resulted in a protein with little activity to activate transcription in both yeast and mammalian cells (Corton and Johnston 1989; Corton et al. 1998), an odd result given the general separability of the DNA-binding and activation domains of the protein. To test if these mutations affected ubiquitylation, both GST-Gal4-VP16 and GST-Gap71-VP16 were incubated in HeLa NE with or without an excess of His-tagged ubiquitin and ATP. Ubiquitin-tagged proteins were then purified, separated on an SDS gel, and then probed with the antibody to GST as described in Figure 3C. While ubiquitylated GST-Gal4-VP16 was readily isolated, there was no evidence of ubiquitylated GST-Gap71-VP16 (Fig. 5B, lanes 3,6).
Ubiquitylation of Gal4-VP16 is necessary for its stable DNA occupancy and transcriptional activity in vitro
To investigate the potential connection between ubiquitylation of GST-Gap71-VP16 and its transcriptional defect, we performed in vitro transcription using HeLa NE. The Gap71 variant of Gal4-VP16 is indeed less active compared with wild-type GST-Gal4-VP16 in stimulating transcription in vitro (Fig. 6A, cf. lanes 1,2,5 and 6–9). Given the ATP-dependent destabilization of activator–DNA complex by purified proteasome (Figs. 1, 2, 3), we asked whether the failure to activate transcription in vitro was due to the inability of GST-Gap71-VP16 to occupy DNA or to it being rapidly degraded in NE. Both the wild-type and mutant activators were preincubated in NE in the presence or absence of the immobilized template, NTPs were added, and proteins were affinity-purified by virtue of the immobilized template and separated on an SDS gel. Consistent with the previous results, GST-Gal4-VP16 is retained on the bead-bound DNA, and all of it (∼2% of input) migrates as the mono- ubiquitylated form (Fig. 6B, lanes 1,3). However, GST-Gap71-VP16, although stable in the extract (Fig. 6B, lane 5), does not effectively bind the DNA template (Fig. 6B, lane 6). The addition of LC does not stabilize the preformed GST-Gap71-VP16–DNA complex in NE (Fig. 6C, lane 5). However, the GST-Gap71-VP16–DNA complex was stable in NE preincubated with hexokinase to deplete ATP levels (Fig. 6C, lane 2), or with the addition of the anti-human Sug1 (hSug1) antibody (Fig. 6C, lane 4), but not anti-TNP antibody (Fig. 6C, lane 6). This highlights the fact that the GST-Gap71-VP16 protein has a normal intrinsic DNA-binding capacity, but is much more sensitive to destabilization by the 19S complex in the presence of ATP. We conclude that ubiquitylation of Gal4DBD is essential to block ATP-dependent destabilization of the activator–DNA complex by the 19S.
Figure 6.

Ubiquitylation of Gal4-VP16 is essential for DNA binding in the presence of NE and ATP. (A) GST-Gap71-VP16 does not activate transcription in vitro. In vitro transcription reactions with HeLa NE were carried out essentially as described (Ferdous et al. 2002) except that increased amounts of indicated activators were added. Control buffer was added in lane 1. (B) GST-Gap71-VP16 cannot bind to the promoter under in vitro transcription conditions. In vitro DNA-binding reactions of indicated activators were processed as described in Figure 3A, except that isolated complexes were analyzed along with an aliquot from the reaction without DNA (lanes 2,5) and purified proteins (lanes 1,4). (C) ATP-dependent destabilization of the preformed activator–DNA complex by 19S. Preformed GST-Gap71-VP16–DNA complexes were incubated in the presence (+) or absence (−) of NE, NTP, and anti-TNP (negative control) antibodies. Preincubation (PI) of NE with buffer (−), hexokinase (HK), lactacystin (LC), and antibodies against Trip1 (hSug1) or TNP is indicated. The amount of DNA-bound activator after 30 min of incubation in the absence of competitor DNA was analyzed. (D) Genetic fusion of ubiquitin restores DNA binding of Gap71-VP16 in NE. DNA-binding activity of the indicated proteins without a GST tag was analyzed in the presence (+) or absence (−) of NE and ATP. DNA-bound protein was detected by Western blotting with anti-Gal4DBD antibodies.
As a direct test of this conclusion we asked if the genetic fusion of ubiquitin to Gap71-VP16 restores its DNA occupancy. Genetic fusions of ubiquitin to activators have been shown previously to at least partially substitute for monoubiquitin attached naturally to a lysine side chain (Salghetti et al. 2001; Bres et al. 2003). As shown in Figure 6D, N-terminal addition of ubiquitin to Gap71-VP16 lacking the GST tag indeed stabilizes its DNA occupancy in HeLa NE in the presence of ATP.
Gap71–promoter complexes are hypersensitive to destabilization in vivo under inducing conditions
We next used chromatin immunoprecipitation (ChIP) assays in yeast to determine if the in vitro phenomena we describe above are evident in vivo. We had established earlier using this assay that wild-type Gal4 protein occupies the GAL1/10 promoter both in noninducing (raffinose) and inducing (galactose) conditions (Gonzalez et al. 2002). When we performed the ChIP assay on a strain expressing Gap71 in place of Gal4 we find that it is pres- ent in noninducing conditions (Fig. 7A; raffinose, R). This confirms that the intrinsic DNA-binding activity of this protein is intact. In striking contrast however, no ChIP signal reflecting Gap71 occupancy of the promoter is observed in galactose (G), whereas the wild-type Gal4 protein provides clear evidence of occupancy (Fig. 7A). The failure of Gap71 to occupy the promoter in galactose is not due to its proteolysis, as there is as much Gap71 protein in raffinose as in galactose medium as detected by Western blotting for a T7 tag epitope tag included in the Gap71 construct (Fig. 7B).
Figure 7.

Gap71 dissociates from its DNA-binding sites on activation of the GAL1 gene. (A) Gap71 is not bound to the GAL1 promoter on induction of gene expression. Promoter occupancy of T7-tagged Gap71 and Gal4 in raffinose (R) and after 60 min of induction with galactose (G) was analyzed by ChIP assay as described (Gonzalez et al. 2002). Totals indicate the PCR reaction from each chromatin solution before immunoprecipitation by anti-Gal4 antibodies. (B) Steady-state protein levels of Gap71 do not change after induction. Gal4 and Gap71 were immunoprecipitated before and after 30 min of induction with anti-Gal4 antibodies and then analyzed for protein level by Western blotting with anti-T7 antibodies. (C) Kinetics of Gap71 occupancy on the promoter after induction. ChIP assays were done essentially as described in A, except that formaldehyde was added after the indicated time points (ΔT min) of galactose addition. Preimmune (PI) serum was used as negative control. (D) Modulation of Gap71 promoter occupancy by 19S. Yeast strains expressing either wild-type or mutant Gal4p and Sug1p were grown in raffinose-containing medium at 25°C. At OD600 ∼ 0.5, growth continued for another 2 h at 25°C or 37°C, formaldehyde was added after the indicated time of induction by galactose, and samples were processed for ChIP assay as above.
We then examined the kinetics of Gap71 loss from the GAL1/10 promoter upon addition of galactose to raffinose media using the ChIP assay. As seen in Figure 7C, while the Gal4 stably occupies the promoter before and after induction, the Gap71 protein is lost rapidly, with a half-life of between 1 and 2 min. It is well known that galactose induction results in rearrangement or disruption of the Gal4–Gal80 complex and exposes the activation domain of the activator. Thus, these ChIP results argue that destabilization of the activator–promoter complex is triggered by activation domain exposure.
If the destabilization of the Gap71–promoter complex in vivo involves the 19S, as is the case in vitro, we would predict that mutations in the 19S might affect the lifetime of the Gap71–promoter complex upon induction. To test this, we used yeast strains containing the sug1-20 mutation. This mutation results in a highly thermolabile protein that is inactivated rapidly upon shifting the temperature to 37°C (Ferdous et al. 2001; Sulahian et al. 2006). A sug1-20 strain, containing either wild-type Gal4 or Gap71 was grown to mid-log phase at 25°C, then shifted to 37°C. After inactivation of the Sug1-20 protein, galactose was added. As controls, the same experiment was done using strains that carried a wild-type SUG1 gene, and SUG1 and sug1-20 strains expressing wild-type Gal4 were also examined. The results are shown in Figure 7D. As expected, the temperature shift had no effect on wild-type Gal4 occupancy of the promoter in either strain. In contrast, the Gap71–GAL1/10 promoter complex lifetime was increased at least twofold in the sug1-20 strain as compared with the SUG1 strain. This result may underestimate the magnitude of the effect of inactivation of Sug1-20 on the complex. At 25°C, the lifetime of the Gap71–promoter complex is much shorter than in the wild-type strain after induction, suggesting that the sug1-20 mutation has the unanticipated effect of stimulating the destabilization reaction at the permissive temperature. Thus, the more relevant and striking comparison is of the Gap71–promoter complex lifetimes in the sug1-20 strain at 25°C and 37°C, which shows clearly that inactivation of Sug1-20 has a strong stabilizing effect on the complex. We conclude that that the stability of the Gap71 protein–promoter complex is affected strongly by the19S proteasome in vivo, but only after exposure of the activation domain. This is precisely the result that would have been predicted from the in vitro results discussed above, arguing that they indeed reflect events of physiological relevance.
Discussion
This study reports two seminal findings. The first is a new, nonproteolytic activity of the 19S regulatory particle of the proteasome, which is the ability to destabilize activator–DNA complexes in an ATP-dependent fashion. The second is that activators are protected from this potentially repressive activity by monoubiquitylation.
The destabilization process does not involve degradation or irreversible denaturing of the protein (Fig. 1). It does require an activation domain and ATP (Fig. 2). The activity is not peculiar to Gal4 as a substrate, as the same is true for the Pho4 protein (Fig. 2C). The DNA-bound form of Gal4-VP16 is monoubiquitylated somewhere in the DNA-binding domain when exposed to a HeLa NE (Fig. 4), and this modification blocks its removal from the DNA by the 19S (Figs. 3, 4). Gap71, a Gal4 derivative with three mutations in the DNA-binding domain (two of which convert lysines to other residues), is not mono- ubiquitylated in vitro, and this protein is rapidly removed from DNA by the 19S (Figs. 4, 5, 6). In vivo Gap71 is bound to the promoter in the uninduced state when its activation domain is bound by Gal80, but is quickly removed from DNA when Gal80 is removed upon induction (Fig. 7). Finally, a mutation in the Sug1 protein of the 19S can partially suppress the hypersensitivity of the Gap71–promoter complex to destabilization in vivo (Fig. 7D). We conclude that we have discovered a previously unknown activity of the proteasomal ATPases, that this activity can be blocked by ubiquitylation, and that this activity is operable in vivo.
ATP- and activation domain-dependent destabilization of activator–DNA complexes by proteasome
This study provides evidence that the 19S regulatory subunit of the proteasome destabilizes activator–DNA complexes both in vitro and in vivo. Others have reported that the 19S subcomplex exhibits chaperone-like activity (Braun et al. 1999; Strickland et al. 2000). However, this is the first example of the 19S or 26S acting on DNA–protein complexes. ATP hydrolysis is required for the 19S to destabilize a protein–DNA complex (Figs. 2, 6). Although nonhydrolyzable analogs (ADP and AMP-PNP) supported some destabilization, it was not as efficient as ATP (Fig. 2). Of note is that in the absence of ATP, a low-level destabilization was also observed with the purified 19S (Fig. 2). It is possible that this is due to residual ATP contamination in purified proteasome since it was purified in the presence of high ATP concentration (Verma et al. 2000).
This activity requires an activation domain. The Gal4DBD without the VP16 activation domain was not removed from DNA (Fig. 2). This is consistent with our previous observations that the activation domains of Gal4 (Swaffield et al. 1995; Chang et al. 2001; Gonzalez et al. 2002; Archer et al. 2005) and VP16 (Lee et al. 1995) interact directly with Sug1 and Sug2 proteins. We also found that these activation domains apparently disassociate the 26S, so that only the base (APIS) of the 19S is retained by the activation domain (Gonzalez et al. 2002). Apparently this is also true in vivo, as upon induction we find that only the base components are recruited to the GAL promoter (Gonzalez et al. 2002; Sulahian et al. 2006), although others have reported association of 20S components as well (Morris et al. 2003; Lee et al. 2005).
This interaction of part of the 19S with the Gal4 activation domain is also important for in vivo destabilization activity (Fig. 7). Results from ChIP assays demonstrate that under noninducing conditions the Gap71 form of Gal4 is bound to the GAL1 promoter, and that binding is rapidly destabilized on induction (Fig. 7). The Gal80 repressor protein binds tightly to the Gal4 activation domain and prevents its interaction with other proteins under noninducing conditions, but either dissociates or rearranges under inducing conditions so as to expose the activation domain (Leuther and Johnston 1992; Peng and Hopper 2000, 2002).
The ability of the proteasome to destabilize activator–DNA complexes clearly proceeds via a nonproteolytic mechanism that primarily involves the ATPases. First, addition of MG132 or LC, potent and selective inhibitors, respectively, of the proteasome-mediated proteolysis, had no effect on in vitro destabilization of the activator–DNA complex by proteasome, even at a concentration where the vast majority of proteasomal peptidase activity was lost (Figs. 1, 2, 6C; Ferdous et al. 2001, 2002). Second, in contrast to proteasome inhibitors, preincubation of purified 19S or 26S proteasome with yeast anti-Sug1 (Fig. 1) or human anti-human Trip1/Sug1, but not with control (Fig. 6) antibodies, almost completely blocked proteasome-mediated destabilization of preformed protein–DNA complexes.
It is of note that Baker and colleagues (Levchenko et al. 1995; Kim et al. 2000) have elegantly demonstrated that the bacterial homolog of the Sug ATPase, ClpX, acts on the stable Mu protein–DNA complex by nonproteolytically destabilizing Mu, which rapidly renatures to occupy its site again, a process quite analogous to the reversible destabilization process reported here. This denaturation requires specific sequences in Mu for ClpX recognition, analogous to the apparent requirement for Sug1/Sug2 binding to the activation domain.
By analogy to the activity of ClpX on Mu–DNA complexes and based on the presumed protein unfolding/unwinding role of the 19S base (specifically the six ATPases) (Braun et al. 1999; Strickland et al. 2000), we propose that the mechanism of the destabilization reaction involves binding of the base of the 19S complex to the activation domains of the activator, followed by ATP-dependent unfolding of the protein, which results in it being stripped from the DNA. At some point, the activator is released from the ATPases, and may refold and reassociate with the DNA under appropriate conditions.
Gap71–promoter complexes are unstable in vivo under activating conditions
Inhibition of the destabilization reaction by anti-Trip1 or anti-Sug1 antibody demonstrates an important role for this protein in vitro. That this is also the case in vivo is supported by the fact that heat inactivation of temperature-sensitive Sug1-20 at 37°C in yeast stabilizes Gap71 promoter binding in galactose (Fig. 7) relative to the binding at the unrestricted temperature. We note that in these experiments the Gap71 is gradually stripped from the promoter, whereas the wild-type activator is not. This could be due to another destabilizing activity besides the 19S. For example, a destabilization activity has also been associated with the p23 molecular chaperone that disrupts the receptor protein–DNA complex in vivo and in vitro (Freeman and Yamamoto 2002). It could also be due to incomplete inactivation of Sug1 activity.
The effect of the ubiquitylation state on the destabilization activity
Besides the discovery of an unrecognized activity of the proteasome, the other surprising result was that activator monoubiquitylation blocks this process. When Gal4-VP16 was incubated with a NE, essentially all the DNA bound was monoubiquitylated (Figs. 3, 4, 6). We note, however, that this was only ∼2% of the total activator introduced into the reaction, and an analysis of bulk Gal4-VP16 would have shown only trace ubiquitylation, highlighting the importance of focusing on the active species. Remarkably, while the extract supported polyubiquitylation of this excess Gal4-VP16 (data not shown), none of the polyubiquitylated forms of the activator were retained on the DNA. Clearly, this effect merits further investigation. The ubiquitylation of the DNA-bound activator was dependent on the presence of an activation domain (Fig. 4B).
The E3 ubiquitin ligase that targets the Gal4DBD is unknown
Gap71, lacking two lysines present in Gal4 (K23 and K25), is not ubiquitylated, and is rapidly destabilized by 19S in vitro (Fig. 6) and in vivo (Fig. 7). These results explain the basis of the transcriptional defect of Gap71 (Corton and Johnston 1989; Corton et al. 1998). As direct evidence, we show that genetic fusion of ubiquitin to Gap71-VP16 stabilized the Gap71-VP16–DNA complex in NE in the presence of ATP (Fig. 6). As will be reported elsewhere, analysis of the individual point mutations that together comprise Gap71 has shown that mutation of K25 does not compromise ubiquitylation, but mutation of K23 does (A. Ferdous, M. O’Neal, K. Nalley, D. Sikder, T. Kodadek, and S.A. Johnston, in prep.). Thus, K23 might be the site of this important modification, although we cannot eliminate the possibility that this residue (and perhaps S22) is somehow critical for monoubiquitylation elsewhere in the DBD. The E3 ubiquitin ligase that operates on the Gal4DBD is currently unknown. Tansey and coworkers (Muratani et al. 2005) have recently identified two F-box-containing E3 ligases, Grr1 and Dsg1, that regulate the stability of different forms of Gal4 in raffinose and galactose. However, deletion of either gene had no effect on Gal4 transcriptional activity. Rather, it was shown that the no-growth phenotype in galactose of the Dsg1 deleted strain was due to a defect in some post-transcription step in GAL1 gene expression (Muratani et al. 2005). Thus, the E3 ligase involved in regulating Gal4 promoter occupancy has yet to be identified.
Biological role of the destabilization activity
What is the physiological role of this activity in transcription? One possibility is that it is a constitutive activity of the proteasomal ATPases that potentially can operate on any activator with an exposed activation domain. This would tend to keep activator–DNA interactions in flux, which might be desirable in providing a mechanism with which to modulate the potency of activation by limiting the number of transcription cycles an activator could drive before forcing the system to “reboot” (Brady et al. 2005; Kodadek et al. 2006). Active destabilization of complexes by other ATPases has been reported to limit the lifetime of certain activator–promoter complexes (Fletcher et al. 2002; Freeman and Yamamoto 2002) and, given the high levels of 19S complex in the nucleus (Russell et al. 1999b), this idea seems reasonable. Proteolytic turnover of some activators has been proposed to serve this need as well (for reviews, see Collins and Tansey 2006; Kodadek et al. 2006).
A recent study from our laboratory has demonstrated that under inducing conditions the Gal4 protein forms highly stable, long-lived promoter complexes (Nalley et al. 2006), arguing that Gal4–promoter complexes are not subject to constant 19S-mediated destabilization. Given the tight linkage shown in this study between activator monoubiquitylation and the ability to resist the destabilization process, we suggest that native Gal4 is monoubiquitylated, and thus protected from the destabilizing activity of the 19S base under inducing conditions. However, we note that a direct demonstration of Gal4DBD ubiquitylation has proven elusive due to the low level of the activator in yeast cells, ubiquitylation of Gal4 by other E3 ligases (Muratani et al. 2005) and other technical difficulties (K. Nalley and T. Kodadek, unpubl.). If the 19S-mediated destabilizing reaction is indeed a pervasive process that operates on many transactivators, one could imagine that the activator ubiquitylation state, determined by the balance between monoubiquitylation, chain extension, and de ubiquitylation activities, might provide a previously unrecognized mechanism for regulating activator–promoter interactions.
Another potentially enlightening way to consider this newly discovered activity of the base of the 19S ATPases is as an unavoidable side reaction of the basic activity of these proteins, which is to unfold or unwind proteins to which they bind. Several years ago, we showed that the proteasomal ATPases play an important role in promoter escape and elongation (Ferdous et al. 2001, 2002). In yeast extracts, when Sug1-20 is inactivated by a temperature shift, transcripts up to ∼50 nucleotides are formed efficiently, but further extension is crippled. While the mechanism of this effect is unknown, it is reasonable to speculate that the protein unfolding activity of the ATPases may be necessary to aid in remodeling preinitiation complexes into processive elongation complexes, a transition that requires many protein–protein and protein–nucleic acid interactions to be dissociated. Recently, involvement of ubiquitin/19S proteasome machinery in exchanging corepressor/coactivator complex has been proposed for activated transcription by several activators (Perissi et al. 2004), so the idea that this complex is involved in transcription complex remodeling is not new. Both in vitro and in vivo, this activity is manifest only if the ATPases are recruited to the promoter by direct interaction of Sug1 and Sug2 with the activation domain of the transactivator (Ferdous et al. 2001; Gonzalez et al. 2002; Archer et al. 2005). Therefore, one could view the destabilization activity reported here as a necessary consequence of activator binding to an unfolding machine. The activator becomes a substrate. This required the development of a mechanism that can protect the activator from this activator, which involves mono- ubiquitylation. The details of how ubiquitin mediates this protective effect are under investigation.
Materials and methods
Preparation of immobilized DNA templates
A biotinylated DNA template containing five tandem repeats of Gal4-binding sites or two Pho4p-binding sites upstream of the core promoter (TATA) sequence was generated by PCR methods using pG5E4T-550 C2AT (Ferdous et al. 2002) and pPho4E4T-550 C2AT as templates, respectively, and universal primers where the primer at the 3′-end of the each template (i.e., 3′-end of the G-less cassette [C2AT]) was biotinylated. The PCR products were purified by QIAquick column (Qiagen) and then immobilized on Dynabeads M-280 (Dynal) according to the manufacturer’s instructions. Unbiotinylated primers were used to PCR-amplify a DNA fragment that contained only the five Gal4-binding sites or two Pho4p-binding sites and was used as competitor DNA. Plasmid (pPho4E4T-550 C2AT) containing two Pho4p-binding sites was generated as described (Sun et al. 2002), except that EcoR1 and BamH1 sites were used into primers to replace the Gal4-binding sites from pG5E4T-550 C2AT.
Purification of recombinant proteins
An expression vector for His6-tagged ubiquitin protein (pQE-Ub) was constructed by inserting a PCR-generated DNA fragment from UBI1 into pQE-31 (Qiagen) between Sph1 and Kpn1 sites. GST-Gap71-VP16 was generated by site-directed mutagenesis of the pGEXCS-Gal4(1–141)-VP16 (Ferdous et al. 2001) using the Quickchange system (Stratagene). GST-Ub–Gap71-VP16 was constructed by in-frame insertion of a PCR-generated DNA fragment from pQE-Ub into the NcoI site of GST-Gap71-VP16 where Gly76 (GGG) was changed to Ala (GCC). An expression vector for GST-Pho4p was described previously (Sun et al. 2002). His6-tagged ubiquitin and GST-tagged activator proteins were expressed and purified from Escherichia coli as described (Ferdous et al. 2001). A bead-bound activator was treated with TEV (Stratagene) to purify activator proteins (Gal4-VP16, Gap71-VP16, and Ub-Gap71-VP16) without a GST tag. His6-tagged ubiquitin with a Lys 48-to-arginine (K48R) mutation was from Boston Biochem.
Isolation of activator–DNA complex and analysis of its destabilization by 19S and 26S
Immobilized DNA (∼100–150 ng), salmon sperm DNA (2 μg), 20 μg of bovine serum albumin (BSA), and indicated activator proteins (100 ng/reaction) in transcription (TXN) buffer (20 μL) (Ferdous et al. 2002) were incubated for 30 min at 30°C. Activator–DNA complexes were isolated by magnetic particle concentrator, washed, and then analyzed by Western blotting. Activator–DNA complexes were also isolated after 30 min of incubation with or without ATP in hexokinase-treated (all lanes without ATP/NTP) or untreated HeLa NE (40 μg), and the bound proteins were analyzed by Western blotting. For destabilization assays, activator–DNA complexes were aliquotted into different tubes precoated with BSA and incubated in the presence or absence of HeLa NE or highly purified yeast 19S (500 ng), 26S (0.8–1.6 μg) (Ferdous et al. 2001), and ATP (500 μM), antibodies, and 10-fold excess of competitor DNA containing either five Gal4-binding sites or two Pho4p-binding sites, respectively. The amount of DNA-bound activators after 15–30 min of incubation at 30°C were analyzed as above. Preincubation of NE or purified 19S and 26S with buffer, antibodies, hexokinase (Sigma), LC, and MG132 (Calbiochem) for 15 min at 30°C is indicated. In some reactions, Gal4-VP16 and DNA were first incubated in hexokinase-treated or untreated HeLa NE with or without ATP and activator–DNA complexes were then isolated to analyze 19S-mediated destabilization in the presence of ATP and excess competitor DNA. The amount of DNA-bound activator was quantified and the percent of DNA-bound protein has been indicated where the amount of activator in the absence of 19S/26S and with or without ATP was considered 100%.
Antibodies and Western blotting
Mouse monoclonal anti-GST, anti-Gal4DBD and anti-T7 antibodies were from Santa Cruz Biotechnology and anti-TNP antibodies were from PharMingen. Affinity-purified rabbit polyclonal anti-ubiquitin antibodies were from Calbiochem. Mouse monoclonal anti-Trip1/Rpt6 (human Sug1) antibodies were described (Ferdous et al. 2001). Western blotting was conducted as described (Ferdous et al. 2001).
Analysis of activator ubiquitylation
HeLa NE and immobilized DNA were incubated for 30 min at 30°C under in vitro transcription conditions in the presence or absence of GST-Gal4-VP16 and ATP or nucleotide mixture, NTP (100 μM of ATP, GTP, and CTP, and 5 μM of UTP). DNA-bound proteins were isolated for Western analysis with anti-GST antibodies first and then the membrane was reprobed with anti-ubiquitin antibodies. In some reactions, hexokinase-treated or untreated HeLa NE, DNA, and activators were incubated for 30 min at 30°C in the presence or absence of ATP and His6-tagged wild-type or mutated (K48R) ubiquitin (Ub). An aliquot (5 μL) was removed, and proteins conjugated with His-tagged Ub were affinity-purified with Talon beads (Clontech) under denaturing conditions (50 mM sodium phosphate at pH 7.0, 6 M urea, 300 mM NaCl, 0.1% NP40). Ubiquitylated activators were detected by Western blotting as above.
In vitro transcription assay
Kinetically synchronized transcription assays were carried out essentially as described (Ferdous et al. 2002), except that 125 ng of linear template, 25–200 ng of indicated activators, and 40 μg of HeLa NE (Promega) were used.
ChIP assay
ChIP assays were performed according to the protocol described (Gonzalez et al. 2002). T7-tagged wild-type Gal4 or Gap71 were transfected into a gal4-deleted strain expressing either wild-type Sug1 (Sc748) (Gonzalez et al. 2002) or sug1-20 (Sc 733) (Russell and Johnston 2001). Cells grown in raffinose-containing medium at 25°C or 30°C were treated with galactose to 2% to induce the GAL genes. Induction was carried out as indicated (0–60 min) prior to addition of formaldehyde. In some assays, cells grown in raffinose-containing medium at 25°C were shifted to 37°C for 2 h and then induced for the indicated time points prior to the addition of formaldehyde. Immunoprecipitations were carried out using anti-Gal4 C-terminal antibody and promoter occupancy of wild-type and mutant Gal4 was analyzed by amplifying the DNA fragment corresponding to three upstream Gal4 sites as described (Gonzalez et al. 2002). Before formaldehyde addition, an aliquot of the culture was removed to analyze the protein levels by Western blotting with anti-T7 antibodies after immunoprecipitation of Gal4 with anti-Gal4 antibodies.
Acknowledgments
We thank Dr. Liping Sun for initial investigations of the destabilization activity, Dr. Carillee Denison for constructing the His6-tagged Ub expression vector, Eunice Webb for technical assistance, and members of the Johnston and Kodadek laboratories for kind cooperation and valuable input. We also thank Klaus B. Hendil for anti-Trip1 antibodies. This work was supported by the NIH (GM066380).
Footnotes
Present address: Center for Innovations in Medicine, Biodesign Institute, Arizona State University, P.O. Box 875001, Tempe, AZ 85287, USA.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.1493207
References
- Archer C., Burdine L., Kodadek T., Burdine L., Kodadek T., Kodadek T. Identification of transactivation domain-binding proteins in the 26S proteasome by periodate-triggered cross-linking. Mol. BioSyst. 2005;1:366–372. doi: 10.1039/b510019d. [DOI] [PubMed] [Google Scholar]
- Auld K.L., Brown C.R., Casolari J.M., Komilli S., Silver P.A., Brown C.R., Casolari J.M., Komilli S., Silver P.A., Casolari J.M., Komilli S., Silver P.A., Komilli S., Silver P.A., Silver P.A. Genomic association of the proteasome demonstrates overlapping gene regulatory activity with transcription factor substrates. Mol. Cell. 2006;21:861–871. doi: 10.1016/j.molcel.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Baumeister W., Walz J., Zuhl F., Seemuller E., Walz J., Zuhl F., Seemuller E., Zuhl F., Seemuller E., Seemuller E. The proteasome: Paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- Brady K.L., Ponnampalam S.N., Bumbulis M.J., Setzer D.R., Ponnampalam S.N., Bumbulis M.J., Setzer D.R., Bumbulis M.J., Setzer D.R., Setzer D.R. Mutations in TFIIIA that increase the stability of the TFIIIA–5S r RNA gene complex. J. Biol. Chem. 2005;280:26743–26750. doi: 10.1074/jbc.M502677200. [DOI] [PubMed] [Google Scholar]
- Braun B.C., Glickman M., Kraft R., Dahlmann B., Kloetzel P.-M., Finley D., Schmidt M., Glickman M., Kraft R., Dahlmann B., Kloetzel P.-M., Finley D., Schmidt M., Kraft R., Dahlmann B., Kloetzel P.-M., Finley D., Schmidt M., Dahlmann B., Kloetzel P.-M., Finley D., Schmidt M., Kloetzel P.-M., Finley D., Schmidt M., Finley D., Schmidt M., Schmidt M. The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat. Cell Biol. 1999;1:221–226. doi: 10.1038/12043. [DOI] [PubMed] [Google Scholar]
- Bres V., Kiernan R.E., Linares L.K., Chable-Bessia C., Plechakova O., Treand C., Emiliani S., Peloponese J.-M., Jeang K.-T., Coux O., Kiernan R.E., Linares L.K., Chable-Bessia C., Plechakova O., Treand C., Emiliani S., Peloponese J.-M., Jeang K.-T., Coux O., Linares L.K., Chable-Bessia C., Plechakova O., Treand C., Emiliani S., Peloponese J.-M., Jeang K.-T., Coux O., Chable-Bessia C., Plechakova O., Treand C., Emiliani S., Peloponese J.-M., Jeang K.-T., Coux O., Plechakova O., Treand C., Emiliani S., Peloponese J.-M., Jeang K.-T., Coux O., Treand C., Emiliani S., Peloponese J.-M., Jeang K.-T., Coux O., Emiliani S., Peloponese J.-M., Jeang K.-T., Coux O., Peloponese J.-M., Jeang K.-T., Coux O., Jeang K.-T., Coux O., Coux O., et al. A non-proteolytic role for ubiquitin in tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 2003;5:754–761. doi: 10.1038/ncb1023. [DOI] [PubMed] [Google Scholar]
- Chang C., Gonzalez F., Rothermel B., Sun L., Johnston S.A., Kodadek T., Gonzalez F., Rothermel B., Sun L., Johnston S.A., Kodadek T., Rothermel B., Sun L., Johnston S.A., Kodadek T., Sun L., Johnston S.A., Kodadek T., Johnston S.A., Kodadek T., Kodadek T. The Gal4 activation domain binds Sug2 protein, a proteasome component, in vivo and in vitro. J. Biol. Chem. 2001;276:30956–30963. doi: 10.1074/jbc.M102254200. [DOI] [PubMed] [Google Scholar]
- Collins G.A., Tansey W.P., Tansey W.P. The proteasome: A utility tool for transcription? Curr. Opin. Genet. Dev. 2006;16:197–202. doi: 10.1016/j.gde.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Conaway R.C., Brower C.S., Conaway J.W., Brower C.S., Conaway J.W., Conaway J.W. Emerging roles of ubiquitin in transcriptional regulation. Science. 2002;296:1254–1258. doi: 10.1126/science.1067466. [DOI] [PubMed] [Google Scholar]
- Corton J.C., Johnston S.A., Johnston S.A. Altering DNA-binding specificity of GAL4 requires sequences adjacent to the zinc finger. Nature. 1989;340:724–727. doi: 10.1038/340724a0. [DOI] [PubMed] [Google Scholar]
- Corton J.C., Moreno E., Johnston S.A., Moreno E., Johnston S.A., Johnston S.A. Alterations in the GAL4 DNA-binding domain can affect transcriptional activation independent of DNA-binding. J. Biol. Chem. 1998;273:13776–13780. doi: 10.1074/jbc.273.22.13776. [DOI] [PubMed] [Google Scholar]
- Ezhkova E., Tansey W.P., Tansey W.P. Proteasomal ATPases link ubiquitylation of histone H2B to methylation of histone H3. Mol. Cell. 2004;13:435–442. doi: 10.1016/s1097-2765(04)00026-7. [DOI] [PubMed] [Google Scholar]
- Ferdous A., Gonzalez F., Sun L., Kodadek T., Johnston S.A., Gonzalez F., Sun L., Kodadek T., Johnston S.A., Sun L., Kodadek T., Johnston S.A., Kodadek T., Johnston S.A., Johnston S.A. The 19S regulatory particle of the proteasome is required for efficient transcription elongation by RNA polymerase II. Mol. Cell. 2001;7:981–991. doi: 10.1016/s1097-2765(01)00250-7. [DOI] [PubMed] [Google Scholar]
- Ferdous A., Kodadek T., Johnston S.A., Kodadek T., Johnston S.A., Johnston S.A. A nonprotelytic function of the 19S regulatory subunit of the 26S proteasome is required for efficient activated transcription by human RNA polymerase II. Biochemistry. 2002;41:12798–12805. doi: 10.1021/bi020425t. [DOI] [PubMed] [Google Scholar]
- Fletcher T.M., Xiao N., Mautino G., Baumann C.T., Wolford R., Warren B.S., Hager G.L., Xiao N., Mautino G., Baumann C.T., Wolford R., Warren B.S., Hager G.L., Mautino G., Baumann C.T., Wolford R., Warren B.S., Hager G.L., Baumann C.T., Wolford R., Warren B.S., Hager G.L., Wolford R., Warren B.S., Hager G.L., Warren B.S., Hager G.L., Hager G.L. ATP-dependent mobilization of the glucocorticoid receptor during chromatin remodeling. Mol. Cell. Biol. 2002;22:3255–3263. doi: 10.1128/MCB.22.10.3255-3263.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman B.C., Yamamoto K.R., Yamamoto K.R. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science. 2002;296:2232–2235. doi: 10.1126/science.1073051. [DOI] [PubMed] [Google Scholar]
- Glickman M.H., Rubin D.M., Fried V.A., Finley D., Rubin D.M., Fried V.A., Finley D., Fried V.A., Finley D., Finley D. The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell. Biol. 1998;18:3149–3162. doi: 10.1128/mcb.18.6.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez F., Delahodde A., Kodadek T., Johnston S.A., Delahodde A., Kodadek T., Johnston S.A., Kodadek T., Johnston S.A., Johnston S.A. Recruitment of a 19S proteasome subcomplex to an activated promoter. Science. 2002;296:548–550. doi: 10.1126/science.1069490. [DOI] [PubMed] [Google Scholar]
- Greer S.F., Zika E., Conti B., Zhu X.S., Ting J.P., Zika E., Conti B., Zhu X.S., Ting J.P., Conti B., Zhu X.S., Ting J.P., Zhu X.S., Ting J.P., Ting J.P. Enhancement of CIITA transcriptional function by ubiquitin. Nat. Immunol. 2003;4:1074–1082. doi: 10.1038/ni985. [DOI] [PubMed] [Google Scholar]
- Kim Y.-I., Burton R.E., Burton B.M., Sauer R.T., Baker T.A., Burton R.E., Burton B.M., Sauer R.T., Baker T.A., Burton B.M., Sauer R.T., Baker T.A., Sauer R.T., Baker T.A., Baker T.A. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol. Cell. 2000;5:639–648. doi: 10.1016/s1097-2765(00)80243-9. [DOI] [PubMed] [Google Scholar]
- Kim S.Y., Herbst A., Tworkowski K.A., Slaghetti S.E., Tansey W.P., Herbst A., Tworkowski K.A., Slaghetti S.E., Tansey W.P., Tworkowski K.A., Slaghetti S.E., Tansey W.P., Slaghetti S.E., Tansey W.P., Tansey W.P. Skp2 regulates Myc protein stability and activity. Mol. Cell. 2003;11:1177–1188. doi: 10.1016/s1097-2765(03)00173-4. [DOI] [PubMed] [Google Scholar]
- Kodadek T., Sikder D., Nalley K., Sikder D., Nalley K., Nalley K. Maintaining control: Proteolytic and non-proteolytic mechanisms for limiting the activity of transcriptional activators. Cell. 2006;127:261–264. doi: 10.1016/j.cell.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Kurosu T., Peterlin B.M., Peterlin B.M. VP16 and ubiquitin: Binding of P-TEFb via its activation domain and ubiquitin facilitates elongation of transcription of target genes. Curr. Biol. 2004;14:1112–1116. doi: 10.1016/j.cub.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Lee J.W., Ryan F., Swaffield J.C., Johnston S.A., Moore D.D., Ryan F., Swaffield J.C., Johnston S.A., Moore D.D., Swaffield J.C., Johnston S.A., Moore D.D., Johnston S.A., Moore D.D., Moore D.D. Interaction of thyroid-hormone receptor with a conserved transcriptional mediator. Nature. 1995;374:91–94. doi: 10.1038/374091a0. [DOI] [PubMed] [Google Scholar]
- Lee D.H., Ezhkova E., Li B., Pattenden S.G., Tansey W.P., Workman J.L., Ezhkova E., Li B., Pattenden S.G., Tansey W.P., Workman J.L., Li B., Pattenden S.G., Tansey W.P., Workman J.L., Pattenden S.G., Tansey W.P., Workman J.L., Tansey W.P., Workman J.L., Workman J.L. The proteasome regulatory particle alters the SAGA coactivator to enhance its interactions with transcriptional activators. Cell. 2005;123:423–436. doi: 10.1016/j.cell.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Leuther K.K., Johnston S.A., Johnston S.A. Nondissociation of GAL4 and GAL80 in invo after galactose induction. Science. 1992;256:1333–1335. doi: 10.1126/science.1598579. [DOI] [PubMed] [Google Scholar]
- Levchenko I., Luo L., Baker T.A., Luo L., Baker T.A., Baker T.A. Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes & Dev. 1995;9:2399–2408. doi: 10.1101/gad.9.19.2399. [DOI] [PubMed] [Google Scholar]
- Lipford J.R., Smith G.T., Chi Y., Deshaies R.J., Smith G.T., Chi Y., Deshaies R.J., Chi Y., Deshaies R.J., Deshaies R.J. A putative timulatory role for activator turnover in gene expression. Nature. 2005;438:113–116. doi: 10.1038/nature04098. [DOI] [PubMed] [Google Scholar]
- Lonard D.M., Nawaz Z., Smith C.L., O’Malley B.W., Nawaz Z., Smith C.L., O’Malley B.W., Smith C.L., O’Malley B.W., O’Malley B.W. The 26S proteasome is required for estrogen receptor-α and coactivator turnover and for efficient estrogen receptor-α transactivation. Mol. Cell. 2000;5:939–948. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- Métivier R., Penot G., Hübner M.R., Reid G., Brand H., Ko M., Gannon F., Penot G., Hübner M.R., Reid G., Brand H., Ko M., Gannon F., Hübner M.R., Reid G., Brand H., Ko M., Gannon F., Reid G., Brand H., Ko M., Gannon F., Brand H., Ko M., Gannon F., Ko M., Gannon F., Gannon F. Estrogen Receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- Morris M.C., Kaiser P., Rudyak S., Baskerville C., Watson M.H., Reed S.I., Kaiser P., Rudyak S., Baskerville C., Watson M.H., Reed S.I., Rudyak S., Baskerville C., Watson M.H., Reed S.I., Baskerville C., Watson M.H., Reed S.I., Watson M.H., Reed S.I., Reed S.I. Cks1-dependent proteasome recruitment and activation of CDC20 transcription in budding yeast. Nature. 2003;423:1009–1013. doi: 10.1038/nature01720. [DOI] [PubMed] [Google Scholar]
- Muratani M., Tansey W.P., Tansey W.P. How the ubiquitin–proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 2003;4:192–201. doi: 10.1038/nrm1049. [DOI] [PubMed] [Google Scholar]
- Muratani M., Kung C., Shokat K.M., Tansey W.P., Kung C., Shokat K.M., Tansey W.P., Shokat K.M., Tansey W.P., Tansey W.P. The F box Dsg1/Mdm30 is a transcriptional coactivator that stimulates Gal4 turnover and cotranscriptional mRNA processing. Cell. 2005;120:887–899. doi: 10.1016/j.cell.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Nalley K., Johnston S.A., Kodadek T., Johnston S.A., Kodadek T., Kodadek T. Proteolytic turnover of the Gal4 transcription factor is not required for function in vivo. Nature. 2006;442:1054–1057. doi: 10.1038/nature05067. [DOI] [PubMed] [Google Scholar]
- Peng G., Hopper J.E., Hopper J.E. Evidence for Gal3p’s cytoplasmic location and Gal80p’s dual cytoplasmic–nuclear location implicates new mechanisms for controlling Gal4p activity in Saccharomyces cerevisiae. Mol. Cell. Biol. 2000;20:5140–5148. doi: 10.1128/mcb.20.14.5140-5148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G., Hopper J.E., Hopper J.E. Gene activation by interaction of an inhibitor with a cytoplasmic signaling protein. Proc. Natl. Acad. Sci. 2002;99:8548–8553. doi: 10.1073/pnas.142100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V., Aggarwal A., Glass C.K., Rose D.W., Rosenfeld M.G., Aggarwal A., Glass C.K., Rose D.W., Rosenfeld M.G., Glass C.K., Rose D.W., Rosenfeld M.G., Rose D.W., Rosenfeld M.G., Rosenfeld M.G. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- Reid G., Hübner M.R., Metivier R., Brand H., Denger S., Manu D., Beaudouin J., Ellenberg J., Gannon F., Hübner M.R., Metivier R., Brand H., Denger S., Manu D., Beaudouin J., Ellenberg J., Gannon F., Metivier R., Brand H., Denger S., Manu D., Beaudouin J., Ellenberg J., Gannon F., Brand H., Denger S., Manu D., Beaudouin J., Ellenberg J., Gannon F., Denger S., Manu D., Beaudouin J., Ellenberg J., Gannon F., Manu D., Beaudouin J., Ellenberg J., Gannon F., Beaudouin J., Ellenberg J., Gannon F., Ellenberg J., Gannon F., Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an intergral feature of estrogen signaling. Mol. Cell. 2003;11:695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- Russell S.J., Johnston S.A., Johnston S.A. Proteolysis of Gal4 can not explain the transcriptional effects of proteasome ATPase mutations. J. Biol. Chem. 2001;276:9825–9831. doi: 10.1074/jbc.M010889200. [DOI] [PubMed] [Google Scholar]
- Russell S.J., Reed S.H., Huang W., Friedberg E.C., Johnston S.A., Reed S.H., Huang W., Friedberg E.C., Johnston S.A., Huang W., Friedberg E.C., Johnston S.A., Friedberg E.C., Johnston S.A., Johnston S.A. The 19S regulatory complex of the proteasome functions independently of proteolysis in nucleotide excision repair. Mol. Cell. 1999a;5:687–696. doi: 10.1016/s1097-2765(01)80001-0. [DOI] [PubMed] [Google Scholar]
- Russell S.J., Steger K.A., Johnston S.A., Steger K.A., Johnston S.A., Johnston S.A. Subcellular localization, stoichiometry, and protein levels of 26S proteasome subunits in yeast. J. Biol. Chem. 1999b;274:21943–21952. doi: 10.1074/jbc.274.31.21943. [DOI] [PubMed] [Google Scholar]
- Salghetti S.E., Caudy A.A., Chenoweth J.G., Tansey W.P., Caudy A.A., Chenoweth J.G., Tansey W.P., Chenoweth J.G., Tansey W.P., Tansey W.P. Regulation of transcriptional activation domain function by ubiquitin. Science. 2001;293:1651–1653. doi: 10.1126/science.1062079. [DOI] [PubMed] [Google Scholar]
- Sikder D., Johnston S.A., Kodadek T., Johnston S.A., Kodadek T., Kodadek T. Widespread, but non-identical, association of proteasomal 19s and 20s proteins with yeast chromatin. J. Biol. Chem. 2006;281:27346–27355. doi: 10.1074/jbc.M604706200. [DOI] [PubMed] [Google Scholar]
- Strickland E., Hakala K., Thomas P.J., DeMartino G.N., Hakala K., Thomas P.J., DeMartino G.N., Thomas P.J., DeMartino G.N., DeMartino G.N. Recognition of misfolding proteins by PA700, the regulatory subcomplex of the 26S proteasome. J. Biol. Chem. 2000;275:5565–5572. doi: 10.1074/jbc.275.8.5565. [DOI] [PubMed] [Google Scholar]
- Sulahian R., Sikder D., Johnston S.A., Kodadek T., Sikder D., Johnston S.A., Kodadek T., Johnston S.A., Kodadek T., Kodadek T. The proteasomal ATPase complex is required for stress-induced transcription in yeast. Nucleic Acids Res. 2006;34:1351–1357. doi: 10.1093/nar/gkl012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Johnston S.A., Kodadek T., Johnston S.A., Kodadek T., Kodadek T. Physical association of the APIS complex and general transcription factors. Biochem. Biophys. Res. Commun. 2002;296:991–999. doi: 10.1016/s0006-291x(02)02026-0. [DOI] [PubMed] [Google Scholar]
- Swaffield J.C., Bromberg J., Johnston S.A., Bromberg J., Johnston S.A., Johnston S.A. Alterations in a yeast protein resembling HIV Tat-binding protein relieve the requirement for an acidic activation domain of GAL4. Nature. 1992;357:698–700. doi: 10.1038/357698a0. [DOI] [PubMed] [Google Scholar]
- Swaffield J.C., Melcher K., Johnston S.A., Melcher K., Johnston S.A., Johnston S.A. A highly conserved ATPase protein as a mediator between acidic activation domains and the TATA-binding protein. Nature. 1995;374:88–91. doi: 10.1038/374088a0. [DOI] [PubMed] [Google Scholar]
- Tansey W. Transcriptional activation: Risky business. Genes & Dev. 2001;15:1045–1050. doi: 10.1101/gad.896501. [DOI] [PubMed] [Google Scholar]
- Thrower J.S., Hoffman L., Rechsteiner M., Pickart C.M., Hoffman L., Rechsteiner M., Pickart C.M., Rechsteiner M., Pickart C.M., Pickart C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R., Chen S., Feldman R., Schieltz D., Yates J., Dohmen J., Deshaies R.J., Chen S., Feldman R., Schieltz D., Yates J., Dohmen J., Deshaies R.J., Feldman R., Schieltz D., Yates J., Dohmen J., Deshaies R.J., Schieltz D., Yates J., Dohmen J., Deshaies R.J., Yates J., Dohmen J., Deshaies R.J., Dohmen J., Deshaies R.J., Deshaies R.J. Proteasomal proteomics: Identification of nucleotide-sensitive proteasome-interacting proteins by mass spectrometric analysis of affinity-purified proteasomes. Mol. Biol. Cell. 2000;11:3425–3439. doi: 10.1091/mbc.11.10.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Lehr N., Johansson S., Wu S., Bahram F., Castell A., Cetinkaya C., Hydbring P., Weidung I., Nakayama K., Nakayama K.I., Johansson S., Wu S., Bahram F., Castell A., Cetinkaya C., Hydbring P., Weidung I., Nakayama K., Nakayama K.I., Wu S., Bahram F., Castell A., Cetinkaya C., Hydbring P., Weidung I., Nakayama K., Nakayama K.I., Bahram F., Castell A., Cetinkaya C., Hydbring P., Weidung I., Nakayama K., Nakayama K.I., Castell A., Cetinkaya C., Hydbring P., Weidung I., Nakayama K., Nakayama K.I., Cetinkaya C., Hydbring P., Weidung I., Nakayama K., Nakayama K.I., Hydbring P., Weidung I., Nakayama K., Nakayama K.I., Weidung I., Nakayama K., Nakayama K.I., Nakayama K., Nakayama K.I., Nakayama K.I., et al. THe F-box protein Skp2 participates in c-Myc proteasomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol. Cell. 2003;11:1189–1200. doi: 10.1016/s1097-2765(03)00193-x. [DOI] [PubMed] [Google Scholar]