

Abstract
Zwitterionic capsular polysaccharides from pathogenic bacteria have peculiar immunological properties. They are capable of eliciting T-cell proliferation and modulating the course of abscess formation. To understand the molecular basis of this characteristic immune response, we are conducting detailed structure–function studies on these polysaccharides. We have identified, purified, and characterized an abscess-modulating polysaccharide, PS A2, from the clinical strain Bacteroides fragilis 638R. Here, we report the elucidation of both the chemical and three-dimensional structures of PS A2 by NMR spectroscopy, chemical methods, gas chromatography–mass spectrometry, and restrained molecular dynamics calculations. PS A2 consists of a pentasaccharide repeating unit containing mannoheptose, N-acetylmannosamine, 3-acetamido-3,6-dideoxyglucose, 2-amino-4-acetamido-2,4,6-trideoxygalactose, fucose, and 3-hydroxybutanoic acid. PS A2 is zwitterionic and carries one cationic free amine and one anionic carboxylate in each repeating unit. It forms an extended right-handed helix with two repeating units per turn and a pitch of 20 Å. Positive and negative charges are exposed on the outer surface of the polymer in a regularly spaced pattern, which renders them easily accessible to other molecules. The helix is characterized by repeated large grooves whose lateral boundaries are occupied by the charges. The three-dimensional structure of PS A2 explicitly suggests mechanisms of interaction between zwitterionic polysaccharides and proteins.
Bacteroides fragilis species are obligate anaerobic Gram-negative bacteria and the most common anaerobes isolated from severe human infections, such as intraabdominal sepsis and abscesses (1–3). Abscesses are a characteristic host response to infection by B. fragilis and cause considerable morbidity and mortality (4). Previous investigations in animal models have demonstrated that capsular polysaccharides (CPs) isolated from B. fragilis are capable of modulating the course of abscess formation via a T-cell-dependent mechanism (5–9). More important, B. fragilis CPs can confer protection against a wide variety of abscess-inducing microorganisms, including B. fragilis itself, Staphylococcus aureus, Streptococcus pneumoniae, and other synergistic microbes (5–9). It is interesting to note that, in contrast to the paradigm that carbohydrate antigens are T-cell independent, B. fragilis CPs activate T cells to proliferate and elicit cytokine responses (9, 10).
Immunomodulating CPs from B. fragilis are structurally distinctive in that they are zwitterionic polysaccharides (ZPSs) carrying a high density of both positive and negative charges. Previous studies have identified two ZPSs, PS A1 and PS B, from the capsule of B. fragilis 9343 (11–13), both of which are potent activators of T cells in vitro and protect animals against abscess formation in vivo (5–8). The charges are critical determinants of immunologic activity, as chemical neutralization of these groups abolishes T-cell stimulation (5–8). More recent studies suggest that T-cell-mediated abscess modulation is a common property of ZPSs (9). The unique immunological properties of these polysaccharides have triggered our interest in characterizing the structures of different ZPSs from various strains of B. fragilis in detail and in addressing the basic structure–activity relationship.
Recently, we have found that the clinical strain B. fragilis 638R expresses a structurally complex capsule that comprises at least three chemically distinct polysaccharides (W.M.K.-M., unpublished work). We have been able to purify the predominant component, PS A2, to homogeneity in quantities sufficient for structural determination and immunological assessment. PS A2 has been named after PS A1, because both share similar immunological properties, and their biosynthesis loci are contained on homologous chromosomal regions of the two strains (9, 14). Here, we report the structure and conformation of PS A2, as determined by a combination of NMR spectroscopy, chemical analysis, gas chromatography–MS (GC-MS), and restrained molecular dynamics calculations.
Materials and Methods
Bacterial Culture and Polysaccharide Purification.
The clinical isolate B. fragilis 638R was stored in peptone–yeast broth at −80°C. The isolation of CPs will be described in detail elsewhere (W.M.K.-M. et al., unpublished work). In brief, after rat-spleen passage, bacteria were grown anaerobically as previously described (11, 12). Polysaccharides were extracted with phenol/water and digested extensively with RNase, DNase, and pronase. Further separation of polysaccharides and purification of PS A2 were achieved by mild acid hydrolysis with subsequent anion exchange and gel filtration chromatography.
Compositional Analysis.
The monosaccharide components of PS A2 were identified by GC-MS analysis of the corresponding alditol acetate derivatives (15). The absolute configurations of the sugar components were established by butanolysis, peracetylation, and GC-MS analysis (16). Commercially available d- and l-fucose (Fuc), d-glycero-d-mannoheptose (Hep), and N-acetyl-d-mannosamine served as standards (Fluka). PS A1 from B. fragilis 9343, which contains 2-acetamido-4-amino-2,4,6-trideoxygalactose, was also used as a standard (11, 12). Because no authentic standard for 3-acetamido-3,6-dideoxyglucose was available, its configuration was derived from molecular modeling (12, 17). The chirality of 3-hydroxybutanoic acid (Bu) was determined by methanolysis and trifluoroacetylation followed by GC-MS analysis on a Lipodex A chiral column (Macherey & Nagel) (18).
GC-MS Analysis.
All analyses were performed on an HP 6890/5973 GC-MS spectrometer (Hewlett–Packard) with a capillary DB17 column (J & W Scientific, Folsom, CA). Mass detection was obtained by electron ionization at 70 eV, and ions were scanned from 45 to 550 m/z.
Mild Base Treatment.
This method was used to identify whether Bu is linked to the polysaccharide through an ether or ester bond, because only the latter is susceptible to mild base cleavage. A 2-mg sample of PS A2 was treated with 0.5 ml of 0.5 M NaOH at room temperature for 2 h, dialyzed extensively against water, and lyophilized. The sample was then examined by 1H NMR spectroscopy.
NMR Spectroscopy.
NMR experiments were performed on Varian Unity 500 and Unity Plus 750 spectrometers. A 10-mg sample of PS A2 dissolved in 0.7 ml of D2O was used for two-dimensional experiments. 1H-1H double-quantum filtered correlation spectroscopy (DQF-COSY) (19), total correlation spectroscopy (TOCSY) (20), and nuclear Overhauser enhancement spectroscopy (NOESY) (21) were carried out with standard pulse sequences provided by Varian. A spectral width of 10 ppm in each dimension was used, and mixing times of 80 and 100 ms were used for NOESY. Spin-lock times were 50, 80, and 100 ms for various TOCSY experiments. 1H-13C heteronuclear multiple quantum coherence (HMQC) (22), distortionless enhancement by polarization transfer-HMQC (23), and heteronuclear multiple bond coherence (HMBC) (24) spectra were recorded with proton and carbon spectral widths of 10 and 200 ppm, respectively. HMQC spectra were obtained with and without carbon decoupling. The latter was used to determine 1JC,H coupling constants for the anomeric carbons. Spectra were generally acquired at 70°C. NOESY spectra were also measured at 37°C. 1H chemical shifts were referenced to the water resonance at 4.36 ppm as calibrated externally. 13C chemical shifts were referenced to an external CH3I standard at 22.5 ppm.
Structure Calculation.
All calculations were performed on an Orion 200 workstation equipped with two R12000 270-MHz and two R10000 180-MHz IP27 central processing units (Silicon Graphics, Mountain View, CA). The insight ii 2000 program package (Molecular Simulations, Waltham, MA) was used for visualization, modeling, force-field calculations, energy minimizations, and molecular dynamics simulations. Surface renderings and qualitative Poisson–Boltzmann electrostatic calculations were carried out with grasp (25) and spock (26). The consistent valence force field with harmonic potential function and including crossterms was used for all calculations (27). The Polak–Ribiere conjugate gradient method was used for minimizations after the initial 50 steps of steepest descent. As described below, each repeating unit of PS A2 contains five monosaccharides (residues a–e) and a dual-charge motif composed of a free amine and a carboxylic acid. Generally, residues a and c were treated uncharged for simulations in vacuo and charged for simulations in water. Individual models of each of the five sugars in one repeating unit were energy minimized in vacuo (ɛ = 4⋅r, no cutoff, final convergence <0.001 kcal⋅mol−1⋅Å−1). ΦH − ΨH total energy maps for each of the five glycosidic bonds were obtained by systematically rotating both angles from 0° to 360° with 10° increments. For every linkage, each of the 362 disaccharide structures was minimized in vacuo (ɛ = 4⋅r, no cutoff, final convergence <0.001 kcal⋅mol−1⋅Å−1), whereas ΦH and ΨH were restrained (cosine restraint with k = 1,000 kcal⋅mol−1). ΦH and ΨH are defined as H1-C1-O1-CX′ and C1-O1-CX′-HX′, respectively (28). Inspection of the energy landscapes allowed the identification of global minima for each pair of ΦH and ΨH as the following (linkages in parentheses): −36°, 35° (ac); −52°, 1° (cd); −27°, −21° (de); 42°, 3° (be); and 57°, −11° (ea). These conformations were confirmed by starting from various glycosidic dihedral angles and demonstrating convergence toward the global minima on minimization. The torsion angle parameters were used to build an initial model of four repeating units. All observed interresidue nuclear Overhauser effect (NOE) distance restraints were added to the model in the form of a flat-bottomed energy term with a proximal target value of 1.8 Å and distal target values of 3.3 Å for strong and 5.0 Å for weak NOE crosspeaks, respectively. The force constants were set to 100 kcal⋅mol−1⋅Å−2 with a scaling factor of 1.
Two solvent simulation approaches were taken for structure calculation and further refinement. (i) The tetramer was coated on all sides with a 15-Å-thick equilibrated water layer surrounded by a 2.5-Å-thick outer water shell that was kept fixed in space to prevent solvent evaporation (3,207 mobile water molecules). The ensemble was minimized to a final convergence of <0.01 kcal⋅mol−1⋅Å−1 with group-based summation (9.5 Å cutoff, 1.0 Å spline, 0.5 Å buffer) and ɛ = 1. (ii) The tetramer was immersed in an equilibrated water box of dimensions 60 × 30 × 30 Å3 (1,571 water molecules), and the system was set up as a periodic boundary condition simulation with group-based summation (15.0 Å cutoff, 2.0 Å spline, 1.0 Å buffer) and ɛ = 1. To equilibrate the water box further, the tetramer was constrained in space, and only water molecules were allowed to move. Minimization was carried out for 1,000 steps. The constraint was then released, and further minimization was performed for 500 steps. At this stage, both 10-ps and 300-ps NOE-restrained molecular dynamics simulations of the entire equilibrated ensemble were started. The system was kept at constant volume and 298 ± 10 K by the Andersen method as implemented in discover 98.0 (Molecular Simulations). Time steps of 1.0 fs and the Verlet velocity integration method were used. The ensembles were minimized to <0.01 kcal⋅mol−1⋅Å−1.
Results and Discussion
Composition of PS A2.
The components of PS A2, as identified by chemical derivatization and GC-MS analysis, are l-Fuc, d-N-acetylmannosamine, d-Hep, 3-acetamido-3,6-dideoxy-d-glucose, 2-amino-4-acetamido-2,4,6-trideoxy-d-galactose, and (S)-Bu. The exact structures and substituents of these components were further established and confirmed by NMR spectroscopy. The 1H NMR spectrum (Fig. 1) of PS A2 reveals five anomeric proton signals at 5.26, 4.97, 4.84, 4.79, and 4.70 ppm. They are correlated with five carbon resonances at 98.41, 100.18, 97.43, 100.48, and 103.81 ppm, respectively, as shown in the 1H-13C HMQC spectrum of PS A2 (Fig. 2). These chemical shifts are characteristic of anomeric protons and carbons of pyranoses (29). The five monosaccharides of PS A2 are designated as residues a, b, c, d, and e, according to their proton chemical shifts (Fig. 1). The complete 1H and 13C chemical shifts of all components were determined by two-dimensional NMR spectroscopy, as described below.
Figure 1.

1H NMR spectrum of PS A2 recorded at 70°C.
Figure 2.

1H-13C HMQC spectrum of PS A2. Each crosspeak corresponds to a C-H pair. Note that a1 refers to position 1 of residue a. Unlabeled peaks arise from contaminants.
Assignment of Residue a (2-amino-4-acetamido-2,4,6-trideoxy-α-dgalactose).
The chemical shifts of all six protons of this residue were readily obtained from the 1H-1H DQF-COSY spectrum of the polysaccharide (not shown). Because only protons attached to adjacent carbons show correlation in a COSY spectrum, a proton chemical shift can be determined once the adjacent proton in a sequence has been assigned. Starting from the known anomeric resonance, this strategy was used to trace all six protons in the sugar ring. Because H1 (δ 5.26 ppm) displays a crosspeak with a resonance at 3.99 ppm, the latter was unambiguously assigned to H2. The H2 signal also shows a crosspeak with a signal at 4.32 ppm, which is obviously because of H3. Similarly, H3 correlates with a signal at 4.46 ppm, which was assigned to H4. Furthermore, H4 correlates with a resonance at 4.59 ppm (H5), and H5 has a crosspeak with a signal at 1.10 ppm (H6). The complete proton chemical shift assignment was thus obtained (Table 1).
Table 1.
Complete 1H and 13C chemical shift assignments of PS A2 residues
| Residue | 1 | 2 | 3 | 4 | 5 | 6 | 7 | NAc* (C = O) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| a | →3)-α-d-AATp-(1→ | 1H | 5.26 | 3.99 | 4.32 | 4.46 | 4.59 | 1.10 | ||
| 13C | 98.41 | 50.92 | 74.25 | 55.27 | 66.56 | 17.08 | 175.51 | |||
| b | α-l-Fucp-(1→ | 1H | 4.97 | 3.69 | 3.59 | 3.65 | 4.14 | 1.17 | ||
| 13C | 100.18 | 69.06 | 71.02 | 72.97 | 67.80 | 16.87 | ||||
| c | →2)-α-d-Hepp-[6→Bu]-(1→ | 1H | 4.84 | 3.85 | 3.75 | 3.92 | 3.41 | 4.08 | 3.77 | |
| 13C | 97.43 | 76.21 | 75.17 | 67.69 | 76.33 | 70.03 | 64.03 | |||
| d | →3)-β-d-ManNAcp-(1→ | 1H | 4.79 | 4.60 | 3.95 | 3.56 | 3.44 | 3.84, 3.94 | ||
| 13C | 100.48 | 50.85 | 78.24 | 66.65 | 77.77 | 61.97 | 175.67 | |||
| e | →4)-β-d-ADGp-[2→](1→ | 1H | 4.70 | 3.48 | 4.01 | 3.43 | 3.54 | 1.29 | ||
| 13C | 103.81 | 76.83 | 56.60 | 82.84 | 73.11 | 18.14 | 175.68 | |||
| Bu-(3→ | 1H | 2.56 | 4.23 | 1.26 | ||||||
| 13C | 175.42 | 45.99 | 66.34 | 23.02 |
Chemical shifts are reported in ppm. AAT, 2-amino-4-acetamido-2,4,6-trideoxygalactose; ManNAc, N-acetyl mannosamine; ADG, 3-acetamido-3,6-dideoxyglucose.
The methyls in all three NAc groups have the same chemical shifts: δH 2.05 ppm and δC 23.41 ppm.
Once the protons had been identified, the chemical shifts of their corresponding carbons were readily determined from heteronuclear correlations between carbon and proton in C-H pairs. As revealed by the 1H-13C HMQC spectrum (Fig. 2), C1-C6 of residue a appear at 98.41, 50.92, 74.25, 55.27, 66.56, and 17.08 ppm, respectively. The chemical shifts of both H6 (δ 1.10 ppm) and C6 (δ 17.08 ppm) indicate that the 6-position bears a methyl group. The chemical shift of C2 (δ 50.92 ppm) is characteristic of a carbon substituted with an amine, whereas that of C4 (δ 55.27 ppm) suggests an acetamido group at C4. The latter was confirmed by the three-bond correlation between H4 and the acetamide carbonyl carbon (δ 175.51 ppm) in the 1H-13C HMBC spectrum of PS A2 (not shown).
On the basis of the compositional analysis by GC-MS, residue a is expected to assume a galacto configuration. This conclusion is supported by intraresidue NOEs obtained from the 1H-1H NOESY spectrum of PS A2 (Fig. 3). H4 displays strong NOEs to both H3 and H5, which indicates that H4 is in close proximity to and thus on the same side of the sugar ring as H3 and H5. Furthermore, residue a adopts an α-configuration at its anomeric center, which is evident from the down-field chemical shift of H1 (δ 5.26 ppm, singlet) as well as the characteristic 3JH1,H2 (<3 Hz) and 1JC1,H1 (194 Hz) coupling constants (29). Combination of these data identified residue a as 2-amino-4-acetamido-2,4,6-trideoxy-α-d-galactopyranose.
Figure 3.

1H-1H NOESY spectrum of PS A2 recorded at 37°C and 750 MHz field strength. The methyl proton region is not shown. Seven interresidue NOE crosspeaks are labeled. Note that a1-c2 refers to the correlation between H1 of residue a and H2 of residue c.
Assignment of Residue b (α-l-Fuc).
The assignment of proton resonances was not as straightforward as in the case of residue a. Starting from the H1 resonance at 4.97 ppm, H2 (δ 3.69 ppm) and H3 (δ 3.59 ppm) were assigned according to the H1-H2 and H2-H3 DQF-COSY crosspeaks. However, as no H3-H4 correlation is observable in the DQF-COSY, the assignment of H4 relies on the total correlation originating from H1. In the TOCSY spectrum (not shown), H1 correlates with a total of three signals at 3.69, 3.59, and 3.65 ppm, respectively. Because the first two originate from H2 and H3, the third resonance has to stem from H4. In the NOESY spectrum of PS A2 (Fig. 3), both H3 and H4 correlate with a signal at 4.14 ppm, and H2 shows an NOE to a signal at 1.17 ppm. These two protons (δ 4.14 and 1.17 ppm) are in close proximity to H2, H3, and H4 of residue b. Thus, they are also located on this residue. Furthermore, because the signals at 4.14 and 1.17 ppm correlate with each other in both the DQF-COSY and TOCSY spectra, they were assigned to H5 and H6, respectively. On the basis of the proton assignments, the chemical shifts of C1-C6 were readily obtained from the 1H-13C HMQC spectrum (Fig. 2 and Table 1).
Although H5 and H6 were initially assigned with certain ambiguity, they were confirmed by multiple-bond H-C correlations from the HMBC spectrum. The three-bond correlations between H1 and C5, H5 and C4, and H6 and C5 unambiguously show that H5 and H6 are located on residue b. Both carbon and proton chemical shifts (Table 1) are typical of 6-deoxyhexopyranose. Because l-Fuc was the only such sugar identified by GC-MS analysis, residue b had to be the fucose. Moreover, H1 appears as a singlet (3JH1,H2 < 3 Hz) in the 1H-NMR spectrum (Fig. 1), and the 1JC1,H1 is 185 Hz. Both values indicate an α-configuration at the anomeric center. Thus, residue b was identified as α-l-fucopyranoside.
Assignments of Remaining Residues.
Using similar approaches, we obtained the complete proton and carbon chemical shifts of the remaining residues (Table 1). The chemical shifts identify residues c, d, and e as Hep, N-acetylmannosamine, and 3-acetamido-3,6-dideoxyglucose, respectively. The 1JC1,H1 coupling constants for residues c and d are 168 and 171 Hz, respectively, which indicates an α-configuration at the anomeric centers (29). H1 (4.70 ppm) of residue e appears as a doublet with a 3JH1,H2 of 8 Hz (Fig. 1), which strongly indicates a β-configuration at the anomeric center.
Sequence Determination.
The next task was to determine the linkage positions and sequence of the polymer. Such information can be obtained directly from long-range 1H-13C correlations across the glycosidic bond between two residues (24). The HMBC spectrum of PS A2 reveals a crosspeak with δH 3.85 ppm and δC 98.41 ppm, which arises from H2 of residue c (H2c) and C1 of residue a (C1a) and corresponds to a three-bond coupling across the glysidic linkage H2c-C2c-O1a-C1a. Therefore, residues c and a are connected, and the linkage involves position 1 of residue a and position 2 of residue c, thus establishing a fragment with the sequence of a-(1→2)-c. Similarly, because H1c (δH 4.84 ppm) and C3d (δC 78.24 ppm) display a crosspeak in the HMBC spectrum, residues c and d are connected in a c-(1→3)-d sequence. A d-(1→4)-e fragment is indicated by the correlation between C1d and H4e. Furthermore, residue e is linked to residue a in the form of e-(1→3)-a, as established from the long-range correlation between H1e and C3a.
Connecting the above fragments yields a linear sequence of a-(1→2)-c-(1→3)-d-(1→4)-e-(1→3)-a. Therefore, the fundamental building unit c-d-e-a repeats itself to produce the backbone of the polymer. In addition, residue b is connected to e via a 1→2 linkage, as indicated by the correlation between C1b and H2e. Residue e is connected with residues a, b, and d and forms a branching point in the structure. Thus, the repeating unit of PS A2 is a branched pentasaccharide with the sequence of →2)-c-(1→3)-d-(1→4)[b-(1→2)]-e-(1→3)-a-(1→. Furthermore, Hep (residue c) is substituted with Bu through an ether linkage between the C3 of Bu and C6 of c. The linkage position is confirmed by the long-range correlation between C3 of Bu and H6c in the HMBC spectrum. The observation that Bu is not susceptible to mild base treatment strongly supports ether instead of ester linkage. The chemical structure of PS A2 was thus completely determined and is shown in Fig. 4.
Figure 4.

Chemical structure of one repeating unit of PS A2.
NOE Distance Restraints.
A total of 41 intraresidue and 10 interresidue NOE crosspeaks per repeating unit are observed (Fig. 3). Intraresidue NOEs were used to determine the absolute configurations of the individual residues a–e. Interresidue NOEs were incorporated into the global structure calculation as distance restraints. NOE crosspeak intensities were classified as either strong (s) or weak (w) and interpreted as 1.8–3.3-Å and 1.8–5.0-Å distance intervals, respectively. The detected interresidue NOEs are the following: c1-a5 (s), c2-a1 (s), c1-d3 (s), c5-d2 (s), d1-e6 (s), d2-e6 (w), e1-a3 (s), e2-b1 (s), a2-b5 (w), and a2-b6 (w). The last two NOEs occur between nonadjacent residues. It is noteworthy that by itself the initial model of four repeating units based on ΦH-ΨH grid searches already satisfies eight interresidue distances perfectly, with the remaining two being close to target values.
Three-Dimensional Structure of PS A2.
Preferred solution conformations of one tetramer (four repeating units) of PS A2 were computed by energy minimization from NOE-restrained molecular mechanics and dynamics calculations. The final models are in excellent agreement and are consistent with all intra- and interresidue NOE crosspeaks. The tetramer describes a right-handed helix with two repeating units per turn and a pitch of 20 Å (Fig. 5). The molecule is covered with a high density of charges, as illustrated by the electrostatic surface representation (Fig. 5B). All charges are exposed on the outmost surface of the molecule and are in favorable positions for binding interactions. Positive and negative charges alternate along the sides of the helical chain and follow a zigzag pattern with approximately equal distances of 10 Å. The helix is characterized by a regular series of grooves that are oriented roughly perpendicular to its long axis. These grooves are about 10 Å wide, 10 Å long, and 5 Å deep. All four edges of a specific groove are occupied by charges from residues ax-1 (amine), cx (carboxylate), ax (amine), and cx+1 (carboxylate), respectively, where x denotes a given repeating unit. The first charge projects toward the outside of the groove, whereas the other three face toward the inside. The tetramer contains two complete grooves formed between repeating units 1–3 and 2–4, respectively, as counted from left to right (arrows in Fig. 5B).
Figure 5.

(A) Stick model of the helical structure of one tetramer (four repeating units) of PS A2. Carbons are colored green, oxygens red, nitrogens blue, and hydrogens white. (B) Electrostatic surface representation of the tetramer. Positive charges are colored blue and negative charges, red. Two grooves are indicated by arrows. Both models are oriented identically, with the left sides tilted slightly toward the observer to make all eight charges visible simultaneously.
Models for the Structure–Activity Relationship in ZPSs.
The conformational model of PS A2 suggests plausible mechanisms for the interaction of ZPSs with other molecules. In one scenario, PS A2 binds to other molecules primarily “along its sides” where they display a high density of alternating opposite charges. High binding affinities would be achieved via abundant electrostatic interactions supplemented by the potential for numerous hydrogen bonds to hydrophilic hydroxyls and, to a lesser extent, van der Waals interactions. In another scenario, the grooves of PS A2 serve as the primary binding domains. The geometry of each groove would be able to accommodate the insertion of an α helix from a protein. To test this idea, we docked hypothetical α helices (10–14 mers) with charged side chains at their termini into PS A2 grooves in silico and found that they indeed fit very well (Fig. 6). The charges at the edges of a groove would help anchor the peptide. In addition to multiple salt bridges, the complex would be stabilized by primarily hydrophobic interactions along the inner surface of the groove. In either of these scenarios, charged groups are located at critical positions and contribute significantly to binding, which would explain why charges are essential determinants of the biological activity of ZPSs.
Figure 6.
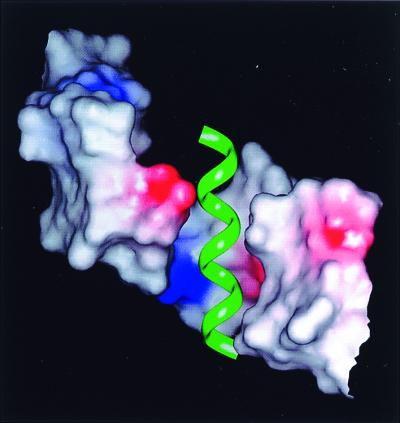
Model of a protein α helix bound to one of the PS A2 grooves. The electrostatic surface of PS A2 is shown within 15 Å of the helix. The helix is represented by a green ribbon, and the coloring scheme for the surface is identical to that in Fig. 5.
The “groove-binding model” offers an attractive explanation for the T-cell-stimulating activity of ZPSs. In general, T-cell activation is initiated by the specific recognition of antigens bound to major histocompatibility (MHC) molecules on antigen-presenting cells. The physical binding of T-cell receptors to antigen–MHC complexes triggers specific T-cell responses to infectious microorganisms (30). Crystallographic studies have revealed that α helices form the lateral boundaries of antigen-binding clefts in MHC molecules (31, 32). In this regard, it is possible that PS A2 forms a complex with MHC molecules by capturing α helices in its grooves. It is noteworthy that PS A2 is polymeric, with hundreds of repeating units, and thus provides a large number of binding sites. Furthermore, PS A2 may crosslink T-cell receptors and MHC molecules by clamping α helices on both proteins. We are currently investigating these proposed binding mechanisms.
Previous studies have demonstrated that molecular charges of ZPSs are the critical determinants of their immunological effects (5–9, 13). PS A2 further supports this charge–activity relationship. Thus far, several immunoactive ZPSs have been identified, including PS A2, PS A1, and PS B from B. fragilis and type 1 capsular polysaccharide from S. pneumoniae (7, 9). Realizing that the primary structures of these compounds differ significantly, we hypothesize that the structural basis for the activity of ZPSs lies in their three-dimensional conformation and overall spatial charge organization. Different ZPSs may assume similar three-dimensional structures, e.g., helices, which would define a common scaffold for the presentation of charges. Preliminary conformational studies on PS A1 support this assumption (data not shown).
In summary, our study provides a picture of the structure and conformation of an important class of abscess-modulating zwitterionic polysaccharides. The structural information suggests interesting binding and interaction mechanisms. It affords a basis on which to rationalize and investigate the intriguing immunological properties of these biopolymers.
Acknowledgments
We thank Dr. Gregory Heffron for expert NMR technical assistance and Julie McCoy and Jaylyn Olivo for editorial service. This study is supported by National Institutes of Health grant AI 39576 (D.L.K.).
Abbreviations
- Bu
3-hydroxybutanoic acid
- CP
capsular polysaccharide
- DQF-COSY
double-quantum filtered correlation spectroscopy
- Fuc
fucose
- GC-MS
gas chromatography–MS
- Hep
d-glycero-d-mannoheptose
- HMBC
heteronuclear multiple bond coherence
- HMQC
heteronuclear multiple quantum coherence
- MHC
major histocompatibility complex
- NOE
nuclear Overhauser effect
- NOESY
nuclear Overhauser enhancement spectroscopy
- TOCSY
total correlation spectroscopy
- ZPS
zwitterionic polysaccharide
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Gorbach S L, Thadepalli H, Norsen J. Anaerobic Microorganisms in Intraabdominal Infections. Springfield, IL: Thomas; 1974. pp. 399–407. [Google Scholar]
- 2.Aldridge K E. Am J Surg. 1995;169:2S–7S. [PubMed] [Google Scholar]
- 3.Polk B J, Kasper D L. Ann Intern Med. 1977;86:567–571. doi: 10.7326/0003-4819-86-5-569. [DOI] [PubMed] [Google Scholar]
- 4.Cross A S. Lancet. 1994;343:248–249. doi: 10.1016/s0140-6736(94)91106-1. [DOI] [PubMed] [Google Scholar]
- 5.Brubaker J O, Li O, Tzianabos A O, Kasper D L, Finberg R W. J Immunol. 1999;162:2235–2242. [PubMed] [Google Scholar]
- 6.Tzianabos A O, Onderdonk A B, Zaleznik D F, Smith R S, Kasper D L. Infect Immun. 1995;62:4881–4886. doi: 10.1128/iai.62.11.4881-4886.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tzianabos A O, Kasper D L, Cisneros R L, Smith R S, Onderdonk A B. J Clin Invest. 1995;96:2727–2731. doi: 10.1172/JCI118340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalka-Moll W M, Tzianabos A O, Wang Y, Carey V J, Finberg R W, Onderdonk A B, Kasper D L. J Immunol. 2000;164:719–724. doi: 10.4049/jimmunol.164.2.719. [DOI] [PubMed] [Google Scholar]
- 9.Tzianabos A O, Finberg R W, Wang Y, Chan M, Onderdonk A B, Jennings H J, Kasper D L. J Biol Chem. 2000;275:6733–6740. doi: 10.1074/jbc.275.10.6733. [DOI] [PubMed] [Google Scholar]
- 10.Stein K E. J Infect Dis. 1992;165:S49. doi: 10.1093/infdis/165-supplement_1-s49. [DOI] [PubMed] [Google Scholar]
- 11.Pantosti A, Tzianabos A O, Onderdonk A B, Kasper D L. Infect Immun. 1991;59:2075–2082. doi: 10.1128/iai.59.6.2075-2082.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baumann H, Tzianabos A O, Brisson J R, Kasper D L, Jennings H J. Biochemistry. 1992;31:4081–4089. doi: 10.1021/bi00131a026. [DOI] [PubMed] [Google Scholar]
- 13.Tzianabos A O, Onderdonk A B, Rosner B, Cisneros R L, Kasper D L. Science. 1993;262:416–419. doi: 10.1126/science.8211161. [DOI] [PubMed] [Google Scholar]
- 14.Coyne M J, Kalka-Moll W, Tzianabos A O, Kasper D L, Comstock L E. Infect Immun. 2000;68:6176–6181. doi: 10.1128/iai.68.11.6176-6181.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Hollingsworth R I. Carbohydr Res. 1994;260:305–317. doi: 10.1016/0008-6215(94)84048-2. [DOI] [PubMed] [Google Scholar]
- 16.Gerwig G J, Kamerling J P, Vliegenthart J F. Carbohydr Res. 1978;62:349–357. doi: 10.1016/s0008-6215(00)83788-x. [DOI] [PubMed] [Google Scholar]
- 17.Bock K. Pure Appl Chem. 1983;55:605–622. [Google Scholar]
- 18.Hermansson K, Perry M B, Altman E, Brisson J-R, Garcia M M. Eur J Biochem. 1993;212:801–809. doi: 10.1111/j.1432-1033.1993.tb17721.x. [DOI] [PubMed] [Google Scholar]
- 19.Derome A E, Williamson M P. J Magn Reson. 1990;88:177–185. [Google Scholar]
- 20.Bax A, Davis D G. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 21.Bodenhausen G, Kofler H, Ernst R R. J Magn Reson. 1984;58:370–388. doi: 10.1016/j.jmr.2011.08.033. [DOI] [PubMed] [Google Scholar]
- 22.Bax A, Subramanian S. J Magn Reson. 1986;67:565–569. [Google Scholar]
- 23.Bendall M R, Doddrell D M, Pegg D T. J Am Chem Soc. 1981;103:4603–4605. [Google Scholar]
- 24.Lerner L, Bax A. Carbohydr Res. 1987;166:35–46. doi: 10.1016/0008-6215(87)80042-3. [DOI] [PubMed] [Google Scholar]
- 25.Nicholls A, Sharp K A, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 26.Christopher J A. spock: The Structural Properties Observation and Calculation Kit. Texas A&M University, College Station, TX: Center for Macromolecular Design; 1998. [Google Scholar]
- 27.Martin-Pastor M, Bush C A. Biochemistry. 2000;39:4674–4683. doi: 10.1021/bi992050q. [DOI] [PubMed] [Google Scholar]
- 28.Homans S W. Biochemistry. 1990;29:9110–9118. doi: 10.1021/bi00491a003. [DOI] [PubMed] [Google Scholar]
- 29.Gorin P A. Adv Carbohydr Chem Biochem. 1981;38:13–104. doi: 10.1016/s0096-5332(08)60172-1. [DOI] [PubMed] [Google Scholar]
- 30.Janeway C A, Jr, Travers P. Immunobiology. New York: Garland; 1994. pp. 4:1–4:35. [Google Scholar]
- 31.Stern L J, Brown J H, Jardetzky T S, Gorga J C, Urban R G, Strominger J L, Wiley D C. Nature (London) 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 32.Reinherz E L, Tan K, Tang L, Kern P, Liu J H, Xiong Y, Hussey R E, Smolyar A, Hare B, Zhang R, et al. Science. 1999;286:1913–1921. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]