

Abstract
PPARγ agonists inhibit liver fibrosis, but the mechanisms involved are uncertain. We hypothesized that PPARγ agonists inhibit transforming growth factor (TGF)β1-activation of TGFβ receptor (TGFβR)-1 signaling in quiescent stellate cells, thereby abrogating Smad3-dependent induction of extracellular matrix (ECM) genes, such as PAI-1 and collagen-1αI. To test this, human HSC were cultured to induce a quiescent phenotype, characterized by lipid accumulation and PPARγ expression and transcriptional activity. These adipocytic HSC were then treated with TGFβ1 ± a TGFβR-1 kinase inhibitor (SB431542) or a PPARγ agonist (GW7845). TGFβ1 caused dose- and time-dependent increases in Smad3 phosphorylation, followed by induction of collagen and PAI-1 expression. Like the TGFβR-1 kinase inhibitor, the PPARγ agonist caused dose-dependent inhibition of all of these responses without effecting HSC proliferation or viability. Thus, the anti-fibrotic actions of PPARγ agonists reflect their ability to inhibit TGFβ1-TGFβR1 signaling that initiates ECM gene expression in quiescent HSC.
Introduction
TGFβ modulates expression of extracellular matrix (ECM) genes in hepatic stellate cells (HSC) (1). TGFβ transduces its profibrogenic signals through type 1-TGFβ receptors ((TGFβR1) Activin receptor-like kinase 5 kinase receptor (ALK5)), and increases transcription of genes, such as collagen 1α-I and plasminogen activator inhibitor (PAI)-1, that regulate matrix deposition and degradation (2). TGFβ-induced fibrosis is a Smad3-dependent process following acute liver injury (3). During chronic liver injury, however, Smad3 is constitutively phosphorylated and localizes in nuclei of activated myofibroblastic HSC, irrespective of exogenous TGFβ stimulation (4). These findings suggest that quiescent and activated HSC respond differently to TGFβ. The basis for this remains obscure, but may have clinical relevance because various factors in injured livers alter HSC phenotypes (5).
In healthy livers, most HSC are quiescent and adipocyte-like, i.e. PPARγ expressing and lipid-laden. Liver injury promotes HSC activation, leading to accumulation of myofibroblastic HSC that are relatively depleted of lipid and PPARγ activity, similar to fibroblastic pre-adipocytes (6). Perpetuation of ECM accumulation during chronic liver injury requires recurrent activation of residual, adipocytic HSCs into myofibroblastic cells because activated HSC ultimately undergo apoptosis (5). Hence, inhibitors of TGFβ reduce fibrosis during chronic liver injury (7), although ECM gene expression in myofibroblastic HSC occurs independently of TGFβ stimulation (4). TGFβ-ALK5 signaling plays a major role in this process because blocking ALK5 phosphorylation reduces collagen gene expression in both acute and chronic models of liver fibrosis (8). This suggests that other inhibitors of TGFβ-ALK5 might also prevent fibrosis during chronic liver injury.
The recent demonstration that adenovirus-mediated over-expression of PPARγ inhibited liver fibrosis in bile duct-ligated rats (9) may be pertinent to this point, because PPARγ inhibits TGFβ-ALK5-Smad3-mediated induction of PAI-1 in renal mesangial cells (10). Although PPARγ does not block PAI-1 expression in all cells (11–13), an inhibitory effect on TGFβ1-ALK5 induction of PAI-1 has also been demonstrated in fat cells. Interestingly, PPARγ mediated repression of PAI-1 expression in fat cells promotes adipocytic differentiation (2, 14). Thus, TGFβ-ALK5-Smad3 signaling transduces events that inhibit differentiation of fibroblastic pre-adipocytes, and PPARγ represses this pathway, favoring acquisition of the adipocytic phenotype.
The latter observation is intriguing given similarities in the gene expression profiles of mature adipocytes and quiescent HSC. Both cell types exhibit strong PPARγ activity and express several PPARγ-regulated genes (15). Primary rat HSC maintain their adipocytic phenotype when cultured in medium that promotes pre-adipocyte differentiation into adipocytes (6). We used this culture system to study the effect of PPARγ on TGFβ signaling in human HSC. We hypothesized that TGFβ–ALK5 interactions activate Smad3 and induce ECM gene expression in adipocytic HSC and proposed that PPARγ agonists would abrogate this interaction. If validated, this concept provides a plausible explanation for the benefits of various PPARγ ligands on liver fibrosis.
Material and methods
Reagents
Isobutylmethylxanthine, dexamethazone, insulin, and mouse anti-β actin monoclonal antibody were purchased from SIGMA. Trizol reagent came from Invitrogen; TGFβ1 from R&D Systems; rabbit anti-PPARγ antibody and mouse anti-PAI-1 antibody from Santa Cruz Biotechnology; phospho-Smad3 and Smad2/3 antibody from Cell Signaling Technology; and SB431542 from Tocris Bioscience. GlaxoSmithKline Pharmaceuticals provided the PPARγ agonist GW7845.
Cell culture
Human HSC (LX-2) cells ((16) from Scott L. Friedman, Mount Sinai School of Medicine) were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin-streptomycin at 37 °C in 5% CO2. Cells were seeded at 3×105 in 6cm dishes and grown until 70% confluent, treated with the adipogenic differentiation mixture (MDI, 0.5mM isobutylmethylxanthine, 1μM dexamethazone, and 1μM insulin) for 72h. MDI was replaced with DMEM containing 0.2% FBS with penicillin/streptomycin for 24h before each experimental manipulation.
Staining
LX-2 cells cultured in slide chambers were washed with phosphate-buffered saline and fixed with 4% paraformaldehyde. Oil Red O in propylene-glycol was added, washed away, and lipid droplets were photographed.
RNA extraction, reverse transcription, and real-time PCR analysis
After TGFβ1 treatment, total RNA was extracted and quantified by spectrophotometry, reverse-transcribed to cDNA using the Invitrogen Reverse Transcription System. An iCycler iQ Multicolor Real-Time PCR (RT-PCR) Detection System (Bio-Rad) was used for RT-PCR. cDNA was amplified using iQ-SYBR Green Supermix with specific oligonucleotide primers for target sequences or gylceraldehyde-3-phosphate dehydrogenase (GAPDH, for normalization). H2O was the negative control. Specific oligonucleotide primers were: GAPDH forward-5′TGGGTGTGAACCATGAGAAG-3′, reverse-5′GCTAAGCAGTTGGTGGTGC-3′, PPAR γ forward-5′CGTGGCCGCAGATTTGAA-3′, reverse-5′CTTCCATTACGGAGAGATCCAC-3′, PAI-1 forward 5-CTCTCTCTGCCCTCACCAAC-3′, reverse 5′-GTGGAGAGGCTCTTGGTCTG -3′, Collagen 1α-I forward-5′ACGTCCTGGTGAAGTTGGTC-3′, reverse-5′ACCAGGGAAGCCTCTCTCTC-3′. Threshold cycles (Ct) were automatically calculated by the RT-PCR System. Each Ct value was normalized to the GAPDH Ct value and a control sample. Relative quantization was expressed as fold-induction compared to control conditions.
Assays
LX-2 cells were transiently transfected with a LFABP (DR1)4-TK-GL3 reporter containing four copies of the liver fatty-acid binding PPRE upstream of a luciferase reporter driven by a thymidine kinase promoter using FuGENE 6 (Roche). For each condition, 1μg reporter and 0.1μg PRL-CMV (internal control) were co-transfected. After 24h, cells were incubated in serum-free medium with DMSO (control) or 1μM GW7845 for 18h. For the PAI-1 promoter assay, 1μg of the TGFβ1 responsive reporter 3TK-Luc (Gerry Blobe, Duke University), and 0.1μg PRL-CMV were transfected into LX-2 cells, stimulated with DMSO, 1μM GW7845, or 10μM SB431542, ± TGFβ1 (100pM). Luciferase assays were performed using the Dual-Luciferase Assay System (Promega) after 24h with the relative luciferase activity normalized to PRL-CMV luciferase activity. Western blotting was performed as described (6).
Cytotoxicity, cell number and proliferation
To assess cytotoxicity, LX-2 cells growing in MDI were seeded onto 96-well plates at a density of 5×103 cells/well and incubated for 24h. Cells were serum starved for 24h in DMEM without phenol red, while treated with GW7845, PBS (negative control), or 0.2% Tween-20 (positive control) for 24h. Absorbance (450nm) was assessed. Cell numbers were measured using a Cell Counting Kit-8. (Dojindo) and cell proliferation was assessed using BrdU incorporation ELISA (Roche).
Data Analysis
All experiments were performed at least 3 times. Data were analyzed by Student t test or one-way ANOVA when necessary.
Results
Culture in MDI induces an adipocytic phenotype in LX-2
LX-2 cells were cultured in MDI according to Tsukamoto’s method (6) to determine if an adipocytic phenotype was induced. PPARγ expression was evaluated by real time PCR (Fig 1A) and Western blotting (Fig 1B), and lipid accumulation was assessed by Oil Red O staining (Fig 1C–D). LX2 cells strongly up-regulated expression of the adipogenic transcription factor, PPARγ, and accumulated intracellular lipid, acquiring an adipocytic phenotype when cultured in MDI. To determine if these adipocytic HSC were able to support transcription of PPARγ target genes, the cells were transfected with a PPARγ dependent luciferase reporter, and then treated with either a PPARγ agonist (GW7845) or DMSO vehicle for 24 h. The PPARγ agonist increased PPRE-luciferase activity by 3–4 fold in adipocytic HSC (Fig 1E), confirming that such cells can accomplish PPARγ-dependent transcriptional activation.
Fig.1.
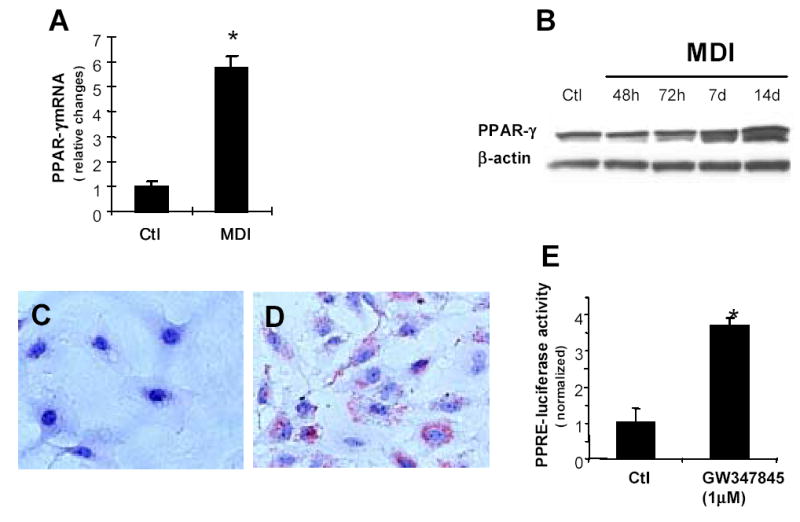
Culture in MDI promotes acquisition of an adipocytic phenotype in human stellate cells. LX-2 cells were treated in MDI for 48h, 72h, 7d or 14d. Total RNA was prepared from triplicate wells after 72h, and PPARγ mRNA expression was assessed by Real time quantitative PCR (A), the PPARγ mRNA level was normalized to GAPDH. * p<0.05. Total protein was extracted from triplicate wells and separated by SDS-PAGE, transferred to membranes and evaluated by immunoblotting for PPARγ Membranes were re-probed for β-actin to confirm equal protein loading (B). LX-2 cells were fixed with 4% paraformaldehyde and stained with Oil Red O after incubation in regular medium(C) or MDI for 72h (D). (E) LX-2 cells were cultured in MDI for 72h, co-transfected with a PPAR response element–luciferase reporter and PRL-CMV-renila control plasmid, and then treated with GW7845X (1μM), or DMSO vehicle for 24h. Luciferase activity was normalized to PRL-CMV activity, and the luciferase activity of mock-transfected cells was subtracted from all values. The results shown are the means ± SEM of triplicate wells. *p<0.05 vs mock-transfected controls.
TGFβ1 induces ECM gene expression and activates Smad3
To characterize the response of adipocytic HSC to TGFβ1, cells were cultured in MDI with recombinant TGFβ1 (0.5, 10, 50, 100, and 500pM) for up to 72h and expression of PAI-1, a TGFβ1-target gene, was assessed. Treatment with 100pM TGFβ1 induced PAI-1 maximally (data not shown). Therefore, subsequent studies were conducted with this dose. TGFβ1 evoked gradual accumulation of PAI-1 protein in LX2 cells, with peak PAI-1 levels observed from 6–24h after stimulation (Fig 2A-B). Increases in PAI-1 protein content were preceded by increased expression of PAI-1 mRNA, and levels of PAI-1 mRNA remained about 3-fold increased for 24h (Fig 2C). TGFβ1 caused a comparable increase in collagen 1α-I gene expression in LX2 cells (Fig 2D). Hence, adipocytic human HSC responded to TGFβ1 by coordinately up-regulating expression of type 1 collagen, a major ECM component, and PAI-1, an inhibitor of ECM degradation.
Fig.2.
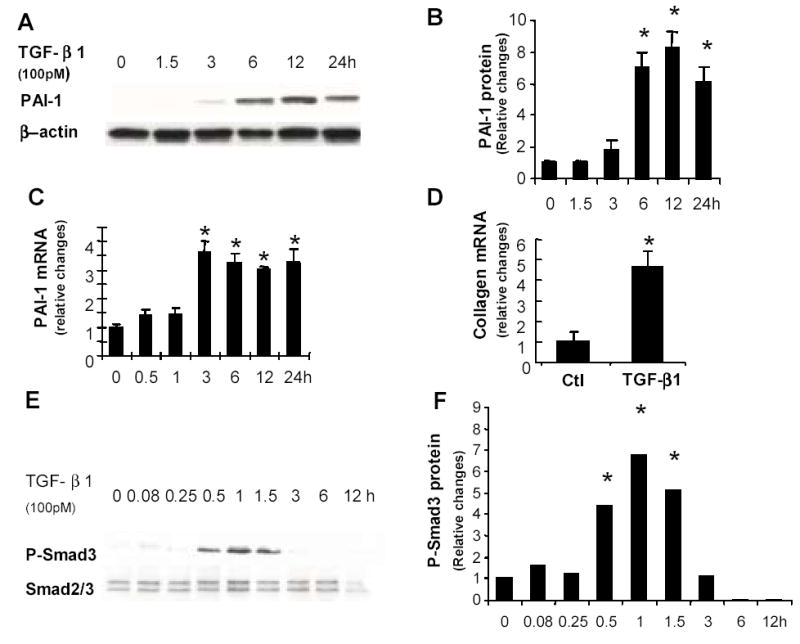
TGFβ1 induces ECM gene expression and phosphorylation of Smad3 in adipocytic human HSC. LX-2 cells were cultured in MDI for 72h and TGFβ1 (100 pM) or vehicle were added, then cells were harvested from triplicate wells after 1.5, 3, 6, 12, 24 or 48h. Total protein was extracted and subjected to immuno-blotting for PAI-1 (A, B) and P-Smad3 (E, F). Membranes were stripped and reprobed for β-actin (A) and Smad2/3 (E) to confirm equal protein loading. Total RNA was extracted from parallel triplicate cultures and expression of PAI-1 mRNA (C) and α (1) collagen mRNA (D) were assessed by real time PCR. Results are normalized to the expression of β-actin (for protein studies) or GAPDH (for RNA studies) in the same sample. Reported data are the mean ± SEM of three independent experiments. *p<0.05 versus control.
Induction of ECM gene expression by TGFβ1 may involve various signaling mechanisms, including activation of the mitogen activated protein kinases (MAPK), Erk1/2 and p38, as well as Smad3 (1, 2, 17, 18). Changes in the phosphorylation status of these factors were examined following TGFβ1 exposure. Although no changes in MAPK phosphorylation were detected at any of the evaluated points (data not shown), significant, albeit transient, increases in phospho-Smad3 were observed. The content of phospho-Smad3 in HSC increased 4–6 fold within the initial 1.5h following TGFβ1 stimulation, and then quickly fell to baseline by 3h (Fig 2E–F). Thus, TGFβ1 rapidly and selectively activated Smad3 in adipocytic human HSC, leading to a transient accumulation of phospho-Smad3 that preceded the subsequent induction of PAI-1 and collagen gene expression. Because Smad3 activation occurs downstream of TGFβ1-ALK5 interactions in many cells, TGFβ1 may be mediating its effects on HSC ECM gene expression via an ALK5-dependent process.
TGFβR1 kinase inhibitor SB431542 inhibits TGFβ-mediated activation of Smad3
SB431542 is a specific inhibitor of TGFβ superfamily type I receptors. To determine if blockade of TGFβR1 kinase activity abrogated the effects of TGFβ1 in LX2 cells, they were cultured with MDI for 72h and then treated with different doses of SB431542 for 18h. Vehicle or TGFβ1 was added and cultures were harvested at 1h (to assess Smad3 phosphorylation) or 12h (to assess PAI-1 and collagen mRNA). SB431542 caused a dose-dependent inhibition of TGFβ1 induction of both ECM genes (Fig 3A–C) and Smad3 phosphorylation (Fig 3D). Maximal inhibition of TGFβ1 action was observed following exposure to 10μM SB431542. TGFβ1-induced phosphorylation of Smad3 was particularly sensitive to inhibition of the TGFβR1 kinase, decreasing significantly after treatment with only 0.1μM SB431542. The maximally effective dose of the TGFβR1 kinase inhibitor (10μM) had no effect on any of these parameters in the absence of exogenous TGFβ, demonstrating that TGFβR1 must be stimulated by TGFβ1 in order to activate Smad3-dependent signaling in adipocytic human HSC.
Fig.3.
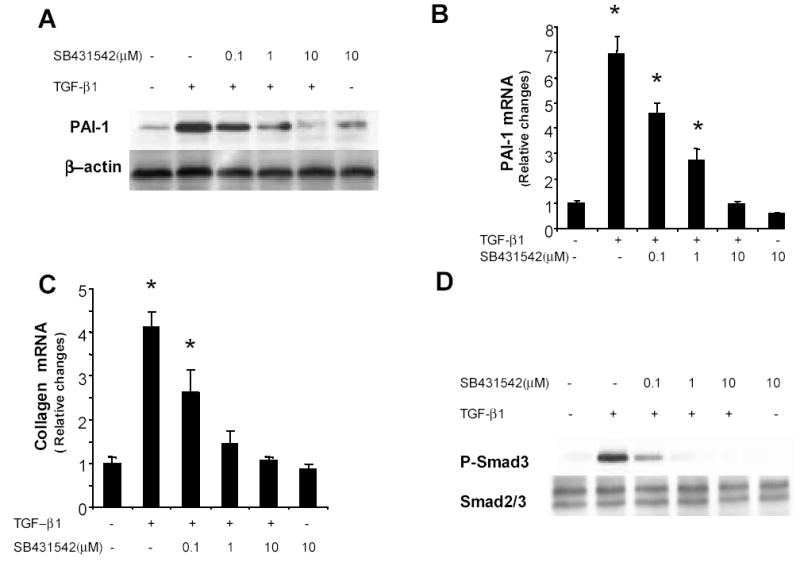
SB 431542 inhibits TGFβ1 induction of ECM expression and blocks TGFβ1-mediated activation Smad3. LX-2 cells were cultured in MDI for 72h, incubated with different doses of SB 431542, an inhibitor of TGFβR1 kinase, or vehicle overnight, and then treated with TGFβ1 (100pM). Triplicate wells were harvested after 1h (to evaluate Smad3) or 12h (to evaluate PAI-1 and collagen). Whole–cell lysates were immunoblotted with specific antibodies against PAI-1 or P-Smad3. Membranes were reprobed for β-actin, Smad2/3 to confirm equal protein loading (A, D). Total RNA was extracted from three parallel cultures, and the expression of PAI-1 and α (1) | collagen mRNA were assessed by real time PCR (B, C). mRNA results were normalized to GAPDH expression in each sample. The graph depicts the mean ± SEM of three separate experiments, each analyzed samples from triplicate wells.*p<0.05 versus control.
PPARγ agonist GW7845 interferes with Smad3 phosphorylation and causes a dose–dependent inhibition of TGFβ1’s fibrogenic effects
To determine if increasing PPARγ activity also inhibited TGFβ effects, adipocytic LX-2 cells were pretreated for 24h with different doses of GW7845, and then exposed to various doses of TGFβ1. Following TGFβ1 exposure, cultures were harvested at 1h (to assess Smad3 phosphorylation) or 12h (to evaluate PAI-1 and collagen). Results were compared to parallel cultures treated with 10μM of TGFβR1 inhibitor, SB431542, before exposure to TGFβ1. When adipocytic LX2 cells were cultured in MDI without supplemental TGFβ1, levels of phospho-Smad3 were virtually undetectable (data not shown) and the PPARγ agonist GW7845 had no appreciable effect on PAI-1 or collagen expression (data not shown). However, treatment with GW7845 inhibited TGFβ-dependent induction of PAI-1 (Fig 4A-B), collagen (Fig 4C), and phospho-Smad3 (Fig 4D) in a dose-dependent fashion. Maximal inhibition occurred following pre-treatment with 1μM GW7845 and this dose was nearly as effective as the maximally inhibitory dose (10μM) of SB431542. Like SB431542, GW7845 had no effect on basal levels of phospho-Smad3, PAI-1 or collagen in adipocytic HSC. Also, as was observed with the TGFβR1 kinase inhibitor, TGFβ-mediated activation of Smad3 was particularly sensitive to PPARγ effects.
Fig.4.
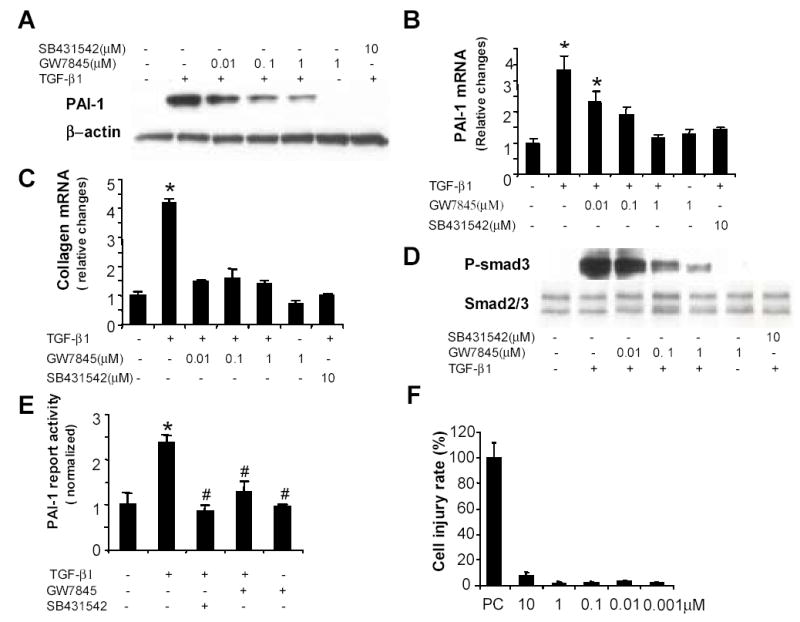
GW7845 inhibits TGFβ1-dependent induction of ECM genes and activation of Smad3 without inducing toxicity in human HSC. LX-2 cells were cultured in MDI for 72h, pretreated overnight with vehicle or different doses of GW7845 or SB 431542 (10μM) and then stimulated with TGFβ1 (100 pM). Cultures were harvested after 1h (for Smad3 analysis) or 12h (to evaluate PAI-1, collagen). Cell lysates were immunoblotted with antibodies against PAI-1 and P-Smad3, membranes were reprobed for β-actin, Smad2/3 to confirm equal protein loading (A, D). Total RNA was extracted, subjected to reverse transcription, and then assessed for expression of PAI-1 and α (1) | collagen mRNA by real time PCR. In each sample, mRNA levels of PAI-1 or collagen were normalized to GAPDH (B, C). The results are expressed as relative fold induction compared to control. Data shown were as mean ± SEM of three independent experiments, each of which analyzed samples from triplicate wells. *p<0.05 versus control. (D) LX-2 cells were cotransfected with a PAI-1–luciferase reporter and PRL-CMV-renila control plasmid and treated with SB 431542 (10μM), GW7845 (1μM), or DMSO ± TGFβ1 for 24h. Luciferase activity was normalized to PRL-CMV activity, and the activity of mock-transfected cells was subtracted from all values. The results shown are the means ± SEM of triplicate wells. * p<0.05 versus control, # p<0.05 versus group treated with TGFβ1. (E). To assess PPARγ agonist cytotoxicity, LX-2 cells growing in MDI were re-plated onto 96-well plates (0.5×104 cells/well) for 24h. The cells were serum-starved for 24h in DMEM while treated with various doses of GW7845, or 0.2% Tween-20. LDH activity in the medium was measured. After subtracting LDH values in the negative (untreated) control cultures from the other values, a cell injury rate was calculated ({LDH value in treated cultures minus LDH value in untreated cultures} divided by 24h). Results are expressed as percentage of the cell injury rate in the positive control (PC) cultures that were totally lysed following Tween-20 treatment (F).
Because phosphorylated Smad3 mediates TGFβ1-ALK5 transcriptional activation of ECM target genes, we next examined how GW7845 influenced TGFβ1-dependent induction of PAI-1 promoter activity. Adipocytic LX2 cells were transiently transfected with 3-TP-Luc, a plasmid that reports activation of TGFβ-responsive elements in the PAI-1 gene. As expected, TGFβ1 increased the activity of 3-TP–Luc by approximately 2.5 fold and pre-incubation with the TGFβR1 kinase inhibitor SB431542 abolished this activity (Fig 4E). Pre-incubation with the PPARγ agonist GW7845 similarly inhibited TGFβ from activating the PAI-1 reporter. This finding supports our other evidence that PPARγ agonists prevent TGFβ1 from inducing ECM genes by blocking TGFβ1-TGFβR1–initiated signals that promote phosphorylation of Smad3 and resultant transcription of Smad3 target genes.
Increasing PPARγ activity does not induce toxicity in adipocytic LX-2
One explanation for the reduced collagen 1α-I and PAI-1 expression that followed treatment with GW7845 is that this PPARγ agonist is toxic to adipocytic HSC. To evaluate this, LX-2 cells were cultured in MDI with different doses of GW7845 for 24h and LDH release was assessed as a measure of cell death. Results were compared to LDH release elicited by exposure to 0.2% Tween-20, a cytolytic detergent. The latter served as a positive control because 24h exposure to Tween-20 was lethal to HSC. Results in the cultures treated with GW7845 are displayed as a percentage of the cell injury that occurred in the positive control cultures (Fig 4F).
There was no evidence of cell toxicity in LX-2 cells exposed to GW7845, even when cells were incubated with doses that elicited maximal inhibition of TGFβ1 signaling. To validate this impression, we repeated these studies and assessed cell viability and proliferation. No effect on HSC number (0.63 ± 0.02AU for no treatment vs. 0.59 ± 0.06AU for 10μM GW7845) or BrdU incorporation (452,000 ± 32,500AU for no treatment vs. 420,000 ± 4, 100AU for 10μM GW7845) was noted, confirming that PPARγ agonist GW7845 is non-toxic to LX2 cells.
Discussion
Here we demonstrate that the PPARγ agonist, GW7845, is as effective as a pharmacologic inhibitor of TGFβR1 kinase in blocking TGFβ1-mediated activation of Smad3 and induction of ECM gene expression in adipocytic human HSC. PPARγ ligands also inhibit expression of TGFβ1-regulated genes, such as connective tissue growth factor (CTGF) in activated, myofibroblastic HSC (19). However, the mechanisms involved may differ. For example, in aortic vascular smooth muscle cells, PPARγ prevents TGFβ1 from inducing CTGF transcription by physically interacting with nuclear-localized Smad3. In adipocytic HSC, however, PPARγ agonists inhibit Smad3 phosphorylation, a cytoplasmic process that precedes Smad3 nuclear localization and Smad-regulated transcriptional activity. Also, PPARγ agonists markedly reduce the viability and proliferation of myofibroblastic HSC (20), but they down-regulate ECM gene expression without effecting the viability or proliferative activity of adipocytic HSC. Hence, intrinsic differences in the phenotypes of adipocytic and myofibroblastic HSC may modulate their responses to PPARγ agonists.
One of the genes that is differentially expressed in quiescent and activated HSC is PAI-1 (21). PAI-1 blocks the generation of plasmin, thereby silencing a major mechanism for ECM degradation and promoting ECM accumulation (22). PAI-1 expression is a characteristic of various immature fibrocytic cells, including bone marrow progenitors and pre-adipocytes (2, 14). In such cells, PAI-1 maintains the fibroblastic phenotype by inhibiting cellular differentiation. Treatment of 3T3-L1 pre-adipocytes with neutralizing antibodies to PAI-1 is sufficient to increase PPARγ activity and induce adipocytic differentiation (14). Adipocytic differentiation is similarly enhanced in mice by PAI-1 gene disruption (23). Interestingly, such PAI-1-deficient mice are also protected from liver fibrosis when subjected to bile duct ligation (22). Therefore, by blocking TGFβ-mediated induction of PAI-1, PPARγ agonists may prevent adipocytic HSC from acquiring a myofibroblastic phenotype during chronic liver injury. Further research is necessary to evaluate this concept. Nevertheless, by demonstrating molecular mechanisms for PPARγ inhibition of TGFβ1 signaling in quiescent human HSC, our results complement and extend knowledge that is emerging from rodent models of fibrosis. In aggregate, the data provide compelling support for evaluation of PPARγ agonists as potential anti-fibrotic therapies in human liver disease.
Footnotes
Support:
This work was supported by NIH grants RO1-AA010154 and DK53792 (AMD) and a research agreement between Duke University and GlaxoSmithKline, Inc.
References
- 1.Liu X, Wang W, Hu H, Tang N, Zhang C, Liang W, Wang W. Smad 3 specific inhibitor, naringenin, decreases the expression of extracellular matrix induced by TGF-beta1 in cultured rat hepatic stellate cells. Pharm Res. 2006;23:82–89. doi: 10.1007/s11095-005-9043-5. [DOI] [PubMed] [Google Scholar]
- 2.Zhou S, Lechpammer S, Breenberger JS, Glowacki J. Hypoxia inhibition of adipocytogenesis in human bone marrow stromal cells requires transforming growth factor-beta/Smad3 signaling. J Biol Chem. 2005;280:22688–22696. doi: 10.1074/jbc.M412953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tahashi Y, Matsuzaki K, Date M, Yoshida K, Furukawa F, Sugano Y, Matsushita M, et al. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 2002. 2002;35:49–61. doi: 10.1053/jhep.2002.30083. [DOI] [PubMed] [Google Scholar]
- 4.Inagaki Y, Mamura M, Kanamaru Y, Greenwel P, Nemoto T, Takehara K, Ten Dijki P, et al. Constitutive phosphorylation and nuclear localization of Smad3 are correlated with increased collagen gene transcription in activated hepatic stellate cells. J Cell Physiol. 2001;187:117–123. doi: 10.1002/1097-4652(2001)9999:9999<00::AID-JCP1059>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 5.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;2:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.She H, Xiong S, Hhazra S, Tsukamoto H. Adipogenic transcritpional regulation of hepatic stellate cells. J Biol Chem. 2005;280:4959–4967. doi: 10.1074/jbc.M410078200. [DOI] [PubMed] [Google Scholar]
- 7.Yata Y, Gotwals P, Koteliansky V, Rockey DC. Dose-dependent inhibition of hepatic fibrosis in mice by a TGF-beta soluble receptor: implications for antifibrotic therapy. Hepatology. 2002;35:1022–1030. doi: 10.1053/jhep.2002.32673. [DOI] [PubMed] [Google Scholar]
- 8.de Gouville AC, Boullay V, Krysa G, Pilot J, Brusq JM, Loriollo F, Gauthier JM, et al. Inhibition of TGF-beta signaling by an ALK5 inhibitor protects rats from dimethylnitoramine-induced fibrosis. Br J Pharmacol. 2005;145:166–177. doi: 10.1038/sj.bjp.0706172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Chen CC, Kwon OS, Liu S, McGhee J, Stimpton S, Chen L, et al. Regulation of peroxisome proliferator-activated receptor {gamma} (PPAR{gamma}) in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2006 doi: 10.1152/ajpgi.00124.2006. Epub. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Wen X, Spataro BC, Hu K, Dai C, Liu Y. Hepatocyte growth factor is a downstream effector that mediates the antifibrotic action of peroxisome proliferator-activated receptor-gamma agonists. J AM Soc Nephrol. 2006;17:54–65. doi: 10.1681/ASN.2005030257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sawai H, Liu J, Reber HA, Hines OJ, Eibl G. Activation of peroxisome proliferator-activated receptor-gamma decreases pancreatic cancer cell invasion throught modulation of plasminogen activator system. Mol Cancer Res. 2006;4:159–167. doi: 10.1158/1541-7786.MCR-05-0257. [DOI] [PubMed] [Google Scholar]
- 12.Ye P, Hu X, Zhao Y. The increase in plasminogen activator inhibitor type-1 expression by stimulation of activators for peroxisome proliferator-activated receptors in human endothelial cells. Chin Med Sci J. 2002;17:112–116. [PubMed] [Google Scholar]
- 13.Marx N, Bourcier T, Sukhova GK, Libby P, Plutzky J. PPARgamma activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression: PPARgamma as a potential mediator in vascular disease. Arterioscler Thromb Vasc Biol. 1999;19:546–551. doi: 10.1161/01.atv.19.3.546. [DOI] [PubMed] [Google Scholar]
- 14.Liang X, Kanjanabuch T, Mao SL, Hao CM, Tang YW, Declerck PJ, Hasty AH, et al. Plasminogen activator inhibitor-1 modulates adipocyte differentiation. Am J Physiol Endocrinol Metab. 2006;290:E103–E113. doi: 10.1152/ajpendo.00605.2004. [DOI] [PubMed] [Google Scholar]
- 15.Tsukamoto H. Fat paradox in liver disease. Keio J Med. 2005;54:190–192. doi: 10.2302/kjm.54.190. [DOI] [PubMed] [Google Scholar]
- 16.Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM, Blaner WS, Mukherjee P, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, Yamagata H, et al. p38 MAPK mediates fibroenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38:879–889. doi: 10.1053/jhep.2003.50384. [DOI] [PubMed] [Google Scholar]
- 18.Fu M, Zhang J, Lin Y, Zhu X, Zhao L, Ahmad M, Ehrengruber MU, et al. Early stimulation and late inhibition of peroxisome proliferator-activated receptor gamma gene expression by transforming frowth factor beta in human aortic smoot muscle cells: role of early growth -reponse factor-1 (Egf-1), activator protein 1 (AP1) and Smads. Biochem J. 2003;370:1019–1025. doi: 10.1042/BJ20021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun K, Wang Q, Huang XH. PPAR gamma inhibits growth of rat hepatic stellate cells and TGF beta-induced connective growth factor expression. Acta Pharmacol Sin. 2006;27:715–723. doi: 10.1111/j.1745-7254.2006.00299.x. [DOI] [PubMed] [Google Scholar]
- 20.Fu M, Zhang J, Zhu X, Myles DE, Willson TM, Liu X, Chen YE. Peroxisome proliferator-activated receptor gamma inhibits transforming growth factor beta-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with Smad3. J Biol Chem. 2001;276:45888–45994. doi: 10.1074/jbc.M105490200. [DOI] [PubMed] [Google Scholar]
- 21.Leyland H, Gentry J, Arthur MJ, Benyon RC. The plasminogen-activating system in hepatic stellate cells. Hepatology. 1996;24:1172–1178. doi: 10.1002/hep.510240532. [DOI] [PubMed] [Google Scholar]
- 22.Bergheim I, Guo L, Davis MA, Duveau I, Arteel GE. Critical role of plasminogen activator inhibitor-1 in cholestatic liver injury and fibrosis. J Pharmacol Exp Ther. 2006;31:592–600. doi: 10.1124/jpet.105.095042. [DOI] [PubMed] [Google Scholar]
- 23.Ma LJ, Mao SL, Taylor KL, Kanjanabuch T, Guan Y, Zhang Y, Brown NJ, et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes. 2004;53:336–346. doi: 10.2337/diabetes.53.2.336. [DOI] [PubMed] [Google Scholar]