

Abstract
We used patch pipettes to record whole-cell currents from single dopamine neurons in slices of rat midbrain. Pharmacological methods were used to isolate EPSCs evoked by focal electrical stimulation.
Baclofen was significantly more potent for inhibiting NMDA receptor-mediated EPSCs (IC50=0.24 μM) compared with inhibition of EPSCs mediated by AMPA receptors (IC50=1.72 μM). The increased potency of baclofen for inhibiting the NMDA component persisted in superfusate that contained zero Mg2+ and when postsynaptic K+ conductances were reduced by Cs+ and QX-314. Effects of baclofen on EPSCs were blocked by the GABAB receptor antagonist CGP-35348.
Adenosine was 20 fold more potent for reducing the NMDA component of transmission (IC50=31 μM) compared with inhibition of AMPA receptor-mediated EPSCs (IC50=654 μM). Effects of adenosine on EPSCs were blocked by the A1 receptor antagonist DPCPX.
Both baclofen and adenosine significantly increased the ratio of EPSCs in paired-pulse studies, suggesting presynaptic sites of action. Although adenosine (1 mM) did not reduce currents evoked by exogenous NMDA (10 μM), baclofen (1 μM) reduced NMDA currents by 29%. Neither baclofen nor adenosine altered currents evoked by exogenous AMPA (1 μM).
We conclude that adenosine acts at presynaptic A1 receptors to cause a preferential reduction in the NMDA component of synaptic transmission. In contrast, baclofen preferentially reduces NMDA EPSCs by acting at both pre- and postsynaptic GABAB receptors. By regulating NMDA receptor function, A1 and GABAB receptors may play important roles in regulating the excitability of dopamine neurons.
Keywords: Adenosine, baclofen, EPSC, GABAB, N-methyl-D-aspartate, presynaptic inhibition, substantia nigra, synaptic transmission, ventral tegmental area, voltage-clamp
Introduction
Stimulation of N-methyl-D-aspartate (NMDA) receptors promotes dopamine neurons of the ventral midbrain to fire bursts of action potentials when recorded either in vitro (Johnson et al., 1992; Wang et al., 1994) or in vivo (Overton & Clark, 1992). Burst firing is thought to be significant because bursts of action potentials have been shown to release more dopamine from nerve terminals than do the same number of evenly spaced spikes (Gonon & Buda, 1985; Suaud-Chagny et al., 1992; Manley et al., 1992). This may have behavioural relevance because burst firing has been implicated in motivation and reward mechanisms (Schultz, 1986; Mirenowicz & Schultz, 1996). Moreover, aberrant regulation of dopamine neuronal firing pattern might contribute to symptoms of cognitive disorders (Grace, 1991). Thus, drugs or transmitters that regulate burst firing of dopamine neurons could have important effects on affect and cognition.
A study by Engberg et al. (1993) showed that baclofen, a GABAB receptor agonist, blocks burst firing in dopamine neurons when injected systemically into rats. Recordings in the rat midbrain slice have also shown that baclofen can convert NMDA-induced burst firing to a single-spike firing pattern in dopamine neurons (Seutin et al., 1994). Membrane hyperpolarization, which causes voltage-dependent block of NMDA receptor-dependent currents by Mg2+, has been proposed as the mechanism by which baclofen blocks burst firing (Seutin et al., 1994; Engberg et al., 1993). Furthermore, this postsynaptic mechanism has also been invoked to explain data from hippocampal slices in which baclofen was found to be more potent for inhibiting the NMDA receptor-mediated component of transmission compared with the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor-mediated component (Morrisett et al., 1991). However, the GABAB receptor can also be located presynaptically where it has a well-established role in causing inhibition of glutamate release (Lanthorn & Cotman, 1981; Dutar & Nicoll, 1993).
The present work was undertaken to investigate the role of GABAB receptors in regulating glutamate-mediated synaptic transmission in dopamine neurons in the rat midbrain slice. We have also extended our studies to include the actions of adenosine, another well-recognized inhibitor of excitatory synaptic transmission. A preliminary report of this work has been published as an abstract (Wu et al., 1996).
Methods
Tissue preparation
Male Sprague-Dawley rats (150–250 g; Bantin & Kingman, Seattle, WA, U.S.A.) were anaesthetized with halothane and killed by severing major thoracic vessels, according to institutional guidelines. The brain was rapidly removed and a block of tissue containing the ventral midbrain was mounted on a plexiglass platform and immersed in artificial cerebrospinal fluid at 4°C. As described previously (Wu et al., 1995), a horizontal midbrain slice (300 μm thick) was cut on a vibratome, placed on a nylon mesh in a recording chamber (500 μl), and immobilized with an electron microscope grid held in place by small platinum weights. The slice was submerged and superfused continuously (2 ml min−1) with artificial cerebrospinal fluid (36°C). The superfusate was saturated with 95% O2 and 5% CO2; it contained (mM): NaCl 126, KCl 2.5, NaH2PO4 1.2, MgCl2 1.2, CaCl2 2.4, glucose 11, and NaHCO3 18 (pH 7.35). Osmolality of the external solution was 295–300 mOsmol l−1 as measured by a Wescor 5500 vapour pressure osmometer (Wescor, Inc., Logan, UT, U.S.A.).
Electrophysiological recordings
Patch pipettes were used to record membrane potentials and currents from dopamine neurons in the whole-cell configuration. Standard internal (pipette) solution contained (mM): K+ gluconate, 125; NaCl, 15; CaCl2, 1; MgCl2, 2; HEPES, 10; EGTA, 11; K2ATP, 1.5; Na3GTP, 0.3. The pH was 7.25, and osmolality was adjusted to 290 mOsmol l−1. Series resistance was 10–40 MΩ after 50–80% electronic compensation. After forming a gigaseal and breaking into the cell, membrane potentials and currents were amplified by an Axopatch 1D amplifier (Axon Instruments, Foster City, CA, U.S.A.). An IBM-type personal computer with a Digidata analog/digital interface and pCLAMP software (Axon Instruments) was used to execute voltage-step commands and record data. Currents were also recorded continuously using a MacLab analog/digital interface, Chart software (AD Instruments, Castle Hill, Australia), and a Macintosh Quadra 630 computer. Voltages were corrected for liquid junction potential (10 mV).
Cell identification
Using a dissection microscope with illumination from below, the substantia nigra compacta (SNC) was identified as a crescent-shaped semi-lucent region rostral and caudal to the medial terminal nucleus of the accessory optic tract, whereas the ventral tegmental area (VTA) was identified as the region lateral to fasciculus retroflexus and medial to the medial terminal nucleus of the accessory optic tract (Paxinos & Watson, 1986). Dopamine-containing neurons in the SNC were identified by well-established electrophysiological and pharmacological characteristics (Yung et al., 1991; Lacey et al., 1989). Neurons were considered dopaminergic if they exhibited large (>200 pA) time-dependent and hyperpolarization-activated current (Ih) during a voltage step from −60 to −120 mV, if dopamine (30 μM) evoked an outward current of 50–150 pA, and if neurons fired spontaneously at a rate of 1–5 Hz with broad (1–2 ms) action potentials. Data from dopamine neurons in the SNC and VTA were similar; therefore data from these regions were pooled.
Synaptic potentials and currents
Bipolar stimulation electrodes (tip separation 300–500 μm) were made from electrolytically sharpened tungsten wire, and their tips were placed in the slice within 500 μm of the recording pipette. Single square-wave electrical pulses (100 μs) of constant current were used to evoke synaptic potentials and currents. As reported previously (Johnson & North, 1992), a single focal electrical stimulus to the slice evokes an EPSP (or EPSC) mediated by NMDA and AMPA receptors followed by an IPSP (or IPSC) mediated by GABAA receptors. Excitatory synaptic events were isolated pharmacologically. When studying synaptic events mediated by NMDA receptors, the slice was superfused with 6-cyano-7-nitroquinoxalone (CNQX; 10 μM) to block AMPA receptors. When studying synaptic events mediated by AMPA receptors, the slice was perfused with ±5-aminophosphovalerate (AP5; 50 μM) in order to block NMDA receptors. All experiments on EPSPs or EPSCs were performed in the presence of the GABAA antagonist bicuculline methiodide (30 μM).
Drugs
All drugs were added to the superfusate which entered the recording chamber within 30 s of turning a tap, the delay being necessary for passage of the solution through a heat exchanger. Complete exchange of the bath solution occurred within 2 min. The following drugs were obtained from Research Biochemicals Int. (Natick, MA, U.S.A.): adenosine, AMPA AP5, baclofen hydrochloride, CNQX, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), and QX-314 (lidocaine N-ethyl bromide). Bicuculline methiodide NMDA and tetrodotoxin (TTX) were obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.). CGP-35348 was a gift from CIBA-GEIGY (Basel, Switzerland).
Data analysis
All data in figures and text are expressed as mean±s.e.mean. Statistical tests were performed on an IBM-type personal computer using SigmaStat software (Jandel Scientific, San Rafael, CA, U.S.A.). Two-way analysis of variance (ANOVA) with repeated measures was used to test for statistically significant differences between concentration-response curves. In some experiments, data were generated in control and treatment groups that contained the same population of neurons. In these situations, we used either one-way ANOVA with repeated measures or the paired t-test to test for statistical significance. We also calculated IC50 values (concentrations producing 50% maximal inhibition) and tested these for significant differences using Student's t-test (two-tails). An IC50 was calculated for each cell (with at least 3 data points per cell) using the KaleidaGraph curve-fitting program (Synergy Software, Reading, PA, U.S.A.) on a Macintosh computer. Concentration-response curves were fitted to the equation y=a*x/(x+b) where y is the magnitude of drug effect, a is the maximum drug effect, x is concentration of drug, and b is the IC50. Data from all cells were then averaged to yield a mean IC50±s.e.mean. Because concentration-response curves contain all relevant data, whereas IC50s are values extrapolated from data, we considered the ANOVA analysis of concentration-response curves to be more sensitive and reliable for statistical purposes than were comparisons of IC50 values. Differences were considered statistically significant when P⩽0.05.
Results
Effects of baclofen under current-clamp
When recording in current-clamp mode, baclofen produced a concentration-dependent reduction in the amplitudes of EPSPs mediated by either NMDA or AMPA receptors (Figure 1a). However, as seen in Figure 1b, the concentration-response curve for baclofen-induced inhibition of NMDA EPSPs was significantly to the left of that for inhibition of AMPA receptor-mediated EPSPs. The estimated IC50 value of baclofen for inhibition of NMDA EPSPs (0.24±0.06 μM, n=22) was also significantly smaller than that for inhibition of AMPA EPSPs (1.28±0.33 μM, n=17). Baclofen also produced a concentration-dependent hyperpolarization (EC50=0.84±0.20 μM, n=24) (Figure 1c) which is consistent with the ability of baclofen to increase a K+ conductance (Lacey et al., 1988). The highest concentration of baclofen tested (10 μM) produced a hyperpolarization of 20.1±1.3 mV (n=15). Effects of baclofen began within 2 min of perfusion, and recovery was complete 5–10 min after washout.
Figure 1.
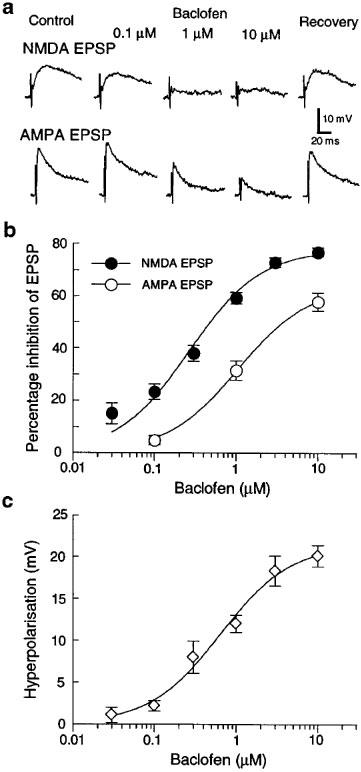
Effects of baclofen on EPSPs and membrane potential recorded in dopamine neurons under current-clamp. (a) Baclofen inhibits NMDA (upper traces) and AMPA (lower traces) EPSPs in concentration-dependent manner. EPSPs were isolated pharmacologically: NMDA EPSPs were recorded in CNQX (10 μM), whereas AMPA EPSPs were recorded in AP5 (50 μM). Bicuculline (30 μM) was present throughout. (b) Concentration-response curves show that baclofen is significantly more potent for reducing the amplitude of EPSPs mediated by NMDA receptors (•) compared with those mediated by AMPA receptors (○). Each data point represents 10–18 neurons. (c) Baclofen produces a concentration-dependent hyperpolarization under current-clamp. Each data point represents 9–19 neurons.
Concentration-dependent effects of baclofen on EPSCs
To minimize baclofen-induced changes on postsynaptic membrane potential, subsequent experiments were done under voltage-clamp (at −60 mV). However, Figure 2a shows that baclofen was significantly more potent for reducing NMDA EPSCs than it was for reducing AMPA receptor-mediated EPSCs. The baclofen IC50 for reducing NMDA EPSCs (0.24±0.05 μM, n=6) was also significantly smaller than that for reducing AMPA EPSCs (1.72±0.52 μM, n=9). Moreover, these baclofen IC50s were not significantly different from those for inhibition of EPSPs recorded under current-clamp (compare Figures 2a to 1b). Thus, the increased potency of baclofen for reducing the NMDA component of transmission–as opposed to reducing the AMPA component–persisted under voltage-clamp.
Figure 2.
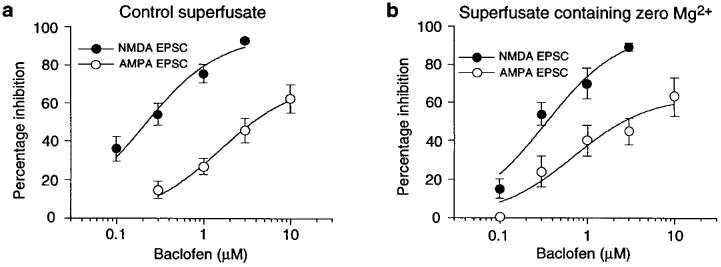
Effects of baclofen on EPSCs recorded under voltage-clamp. (a) Baclofen is significantly more potent for inhibition of NMDA EPSCs (•; n=6) compared with AMPA EPSCs (○; n=9). (b) In the presence of perfusate containing zero Mg2+, baclofen remains significantly more potent for inhibition of NMDA EPSCs (•; n=7) compared with AMPA EPSCs (○; n=6).
Despite the above results, inadequate space clamp could still permit baclofen to hyperpolarize distal dendrites and thereby impose voltage-dependent block of NMDA currents by Mg2+ (Seutin et al., 1994; Wu & Johnson, 1996). But this hypothesis is discouraged by data in Figure 2b which show that baclofen remained significantly more potent for reducing the NMDA component of transmission when the superfusate contained zero Mg2+. Moreover, zero Mg2+ superfusate did not significantly alter the IC50 of baclofen for inhibition of NMDA EPSCs (0.43±0.08 μM, n=7) compared with the baclofen IC50 obtained in control superfusate (compare Figure 2a to b). Similarly, the IC50 of baclofen for inhibition of the AMPA component of transmission in zero Mg2+ (0.85±0.26 μM, n=6) was not significantly different from the IC50 obtained in control superfusate. These data suggest that voltage-dependent block by Mg2+ does not explain the greater potency of baclofen for inhibition of NMDA EPSCs.
Recordings with Cs+ and QX-314 in pipettes
Although the use of voltage-clamp minimized the tendency of baclofen to hyperpolarize the postsynaptic membrane, an increase in postsynaptic conductance could still mediate inhibitory actions of baclofen on synaptic transmission. Therefore, recordings were also performed under voltage-clamp with pipette solutions that contained the K+ channel blocker QX-314 (5 mM) (Nathan et al., 1990) and Cs+ gluconate in place of the K+ salt. Under these conditions, baclofen (1 μM) evoked only 16±9 pA of outward current (n=10); this represents an 80% reduction in the amount of current that is usually evoked by this concentration of baclofen under control conditions (86±15 pA, n=12). Despite the presence of intracellular Cs+ and QX-314, baclofen reduced EPSCs mediated by both NMDA and AMPA receptors in a concentration-dependent manner (Figure 3a). Furthermore, Figure 3b shows that baclofen remained significantly more potent for inhibition of the NMDA receptor-mediated component of transmission compared with reducing the AMPA component. However, the baclofen IC50 for inhibition of NMDA EPSCs (0.45±0.06 μM, n=7) was not significantly different from that for inhibition of AMPA EPSCs (0.70± 0.13 μM, n=6).
Figure 3.
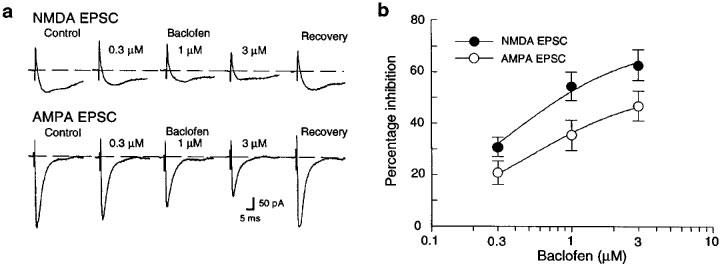
Effects of baclofen on EPSCs recorded under voltage-clamp with pipettes containing the K+ channel blockers Cs+ gluconate and QX-314 (5 mM). (a) Baclofen inhibits both NMDA and AMPA EPSCs in a concentration-dependent manner. The broken line indicates zero holding current. All data are from the same dopamine neuron. (b) Concentration-response curves showing that baclofen is significantly more potent for reducing NMDA EPSCs (•; n=7) compared with reducing AMPA EPSCs (○; n=6).
Although baclofen remained more potent for inhibition of the NMDA component of transmission when K+ conductances were reduced, it also appears that the use of Cs+ and QX-314 narrowed the difference between the concentration-response curves for baclofen-induced inhibition of NMDA and AMPA EPSCs (compare Figures 3a to 2a). These results support the hypothesis that postsynaptic changes in conductance contribute to the inhibitory effect of baclofen on NMDA currents.
Baclofen receptor pharmacology
In order to ascertain that baclofen inhibits synaptic transmission via the GABAB receptor, we tested the ability of CGP-35348, a GABAB antagonist (Olpe et al., 1990), to block effects of baclofen on evoked synaptic currents. Although baclofen (1 μM) reduced NMDA receptor-mediated EPSCs by 59±8%, this concentration of baclofen reduced NMDA EPSCs by only 1±10% in the presence of CGP-35348 (300 μM; n=5). Furthermore, baclofen (3 μM) reduced AMPA EPSCs by only 1±11% in the presence of CGP-35348 (300 μM), whereas this concentration reduced AMPA EPSCs by 44±12% under control conditions (n=5). Thus, it is clear that baclofen acts at GABAB receptors to reduce NMDA and AMPA receptor-mediated EPSCs.
Adenosine reduces EPSCs
In order to test whether or not the preferential inhibition of NMDA EPSCs was unique to baclofen, we next tested effects of adenosine. Adenosine was chosen because we have shown previously that it causes presynaptic inhibition of synaptic transmission while having little or no effect on postsynaptic membrane properties of dopamine neurons (Wu et al., 1995).
As seen in Figure 4, adenosine produced concentration-dependent reductions in the amplitudes of both NMDA and AMPA EPSCs. However, adenosine was approximately 20 fold more potent for reducing NMDA EPSCs (IC50=31±5 μM, n=5) compared with inhibition of AMPA EPSCs (IC50= 654±111 μM, n=8). These results show that adenosine, similar to baclofen, exerts a strong concentration-preference for reducing the NMDA component of excitatory transmission in dopamine cells.
Figure 4.
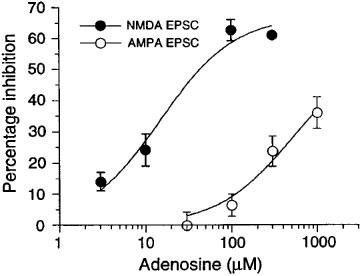
Adenosine is significantly more potent for reducing the amplitude of EPSCs mediated by NMDA receptors (•) compared with inhibition of AMPA receptor-mediated EPSCs (○). Each data point represents 3–8 neurons.
Adenosine receptor pharmacology
In four neurons, adenosine (1 mM) reduced EPSCs by 55±13% (from a control amplitude of 36±3 pA). In the presence of the adenosine A1 receptor antagonist DPCPX (300 nM) (Lee & Reddington, 1986), this concentration of adenosine had no effect on EPSC amplitude (EPSCs were reduced by 4±7%). Thus, adenosine reduces glutamate EPSCs by acting at the adenosine A1 receptor.
On the site of action of baclofen and adenosine
Two methods were used to establish site of action: (1) paired-pulse depression, and (2) perfusion with NMDA and AMPA. In paired-pulse depression, pairs of EPSCs were evoked using an inter-stimulus interval (30–50 ms) that caused the second EPSC to be smaller than the first. As seen in Figure 5a, baclofen (1 μM) reduced the amplitudes of both EPSCs in each pair. However, the paired-pulse ratio (ratio of the amplitude of the second EPSC divided by the first) was significantly increased by baclofen (1 μM), as seen in the averaged data displayed in Figure 5b. Baclofen increased the paired-pulse ratio of EPSCs from the control value of 0.64±0.04 to 1.08±0.07 (n=6). Adenosine (1 mM) also reduced the first EPSC more than the second, as seen in Figure 5c. The averaged data in Figure 5d show that adenosine (1 mM) increased the paired-pulse ratio from the control value of 0.50±0.07 to 1.03±0.05 (n=5). By increasing paired-pulse ratios, these results suggest that both baclofen and adenosine have presynaptic sites of action that reduce glutamate release (Harris & Cotman, 1985).
Figure 5.
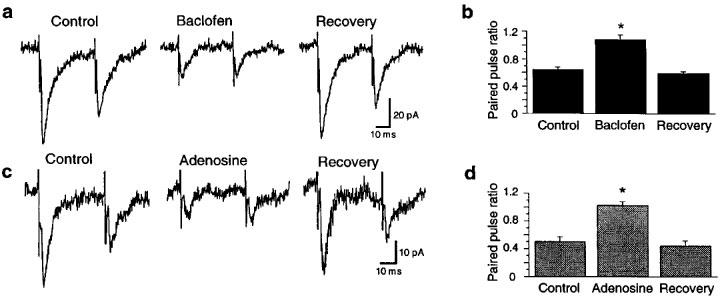
Evidence that baclofen and adenosine act presynaptically to reduce EPSCs. (a) Baclofen (1 μM) reduces each EPSC, but the second is reduced less than the first. The baclofen-evoked outward current is not shown. (b) Averaged data showing that baclofen (1 μM) reversibly increases the EPSC paired-pulse ratio (the ratio of the second EPSC divided by the first). Data in each bar are from the same six cells. (c) Adenosine (1 mM) reduces each EPSC, but the second is reduced less than the first. (d) Averaged data showing that adenosine (1 mM) reversibly increases the EPSC paired-pulse ratio. Data in each bar are from the same five cells. All recordings were made in bicuculline (30 μM) under voltage-clamp (−60 mV). Asterisks indicate a significant difference from control value.
Effects of adenosine and baclofen on inward currents evoked by NMDA (10 μM) are shown in Figure 6. As seen in the continuous current trace (Figure 6a), adenosine (1 mM) actually produced a small increase in the amplitude of current evoked by NMDA. Adenosine (1 mM) also evoked a small but statistically significant net outward current of 9±2 pA (n=13). In contrast, baclofen (1 μM) evoked a more prominent outward current (91±11 pA, n=5) during which the net amplitude of the NMDA-induced inward current was slightly reduced (Figure 6a). Figure 6b and c summarize paired data obtained from five dopamine neurons, including data shown in Figure 6a. Baclofen (1 μM) reduced NMDA currents by 28% from a control value of 85±8 pA to 61±8 pA (Figure 6b). In contrast, adenosine (1 mM) significantly increased net NMDA (10 μM) currents by 24% from a control value of 85±8 pA to 105±9 pA. These data clearly show that baclofen–but not adenosine–has a postsynaptic site of action that inhibits the amplitude of NMDA currents.
Figure 6.
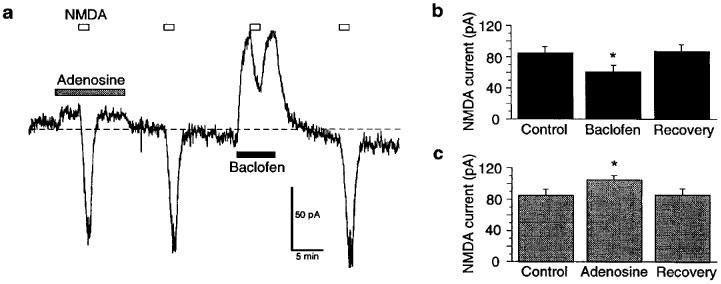
Effects of adenosine and baclofen on inward currents evoked by NMDA (10 μM) added to the superfusate. (a) Continuous current trace (at −60 mV) showing effects of adenosine (1 mM) and baclofen (1 μM) on NMDA currents. Horizontal bars indicate durations of drug application. Note that current evoked by NMDA was slightly larger in the presence of adenosine, whereas current was smaller in the presence of baclofen. Baclofen also causes a relatively large outward shift in holding current compared to adenosine. Broken line indicates zero holding current. (b) Baclofen (1 μM) reduces currents evoked by NMDA (10 μM). (c) Adenosine (1 mM) increases currents evoked by NMDA. Data in (b) and (c) were from the same five neurons; asterisks indicate a significant difference from control value. TTX (0.3 μM) was present throughout.
Effects of adenosine and baclofen on inward currents evoked by AMPA (1 μM) are shown in Figure 7. This concentration of AMPA evoked 99±14 pA of inward current (n=17). Neither adenosine (1 mM) nor baclofen (1 μM) altered current evoked by AMPA (Figure 7a). Currents evoked by AMPA were 100±5% of control in baclofen (Figure 7b; n=12), and they were 96±5% of control in adenosine (Figure 7c; n=5). These data do not support the hypothesis that adenosine and baclofen act postsynaptically to reduce AMPA EPSCs.
Figure 7.
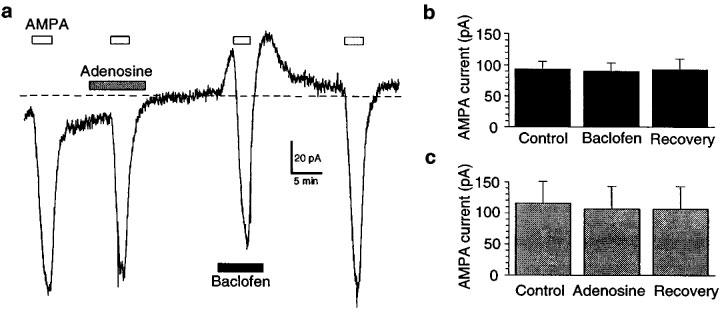
Adenosine (1 mM) and baclofen (1 μM) fail to alter inward currents evoked by AMPA (1 μM). (a) Continuous current trace (at −60 mV) showing lack of effect of adenosine and baclofen on AMPA currents. Horizontal bars indicate durations of drug application. Broken line indicates zero holding current. Averaged data show that (b) baclofen (n=12) and (c) adenosine (n=5) do not alter currents evoked by AMPA. TTX (0.3 μM) was present throughout.
Discussion
Our results show that baclofen and adenosine are more potent for reducing the NMDA receptor-mediated component of synaptic transmission compared with inhibition of the AMPA receptor-mediated component in midbrain dopamine neurons. Our data also strongly suggest that baclofen and adenosine reduce glutamate release by acting at presynaptic GABAB and A1 receptors, respectively. However, baclofen, but not adenosine, also has a postsynaptic site of action which reduces the amplitude of NMDA-gated currents. By preferentially reducing the NMDA component of transmission, we suggest that adenosine A1 and GABAB receptors may play important roles in modulating the firing pattern of dopamine neurons.
Evidence that inhibition of the NMDA component of transmission occurs by a presynaptic mechanism is strongest for adenosine. This conclusion is based upon our findings that adenosine markedly reduced paired-pulse depression of EPSCs, and it failed to reduce the amplitudes of currents evoked by exogenous NMDA and AMPA. The finding that adenosine produced a small but significant increase in currents evoked by exogenous NMDA was surprising, especially because adenosine has been reported to reduce NMDA currents in hippocampus (de Mendonça & Ribeiro, 1993) and in striatum (Nörenberg et al., 1997). It is possible that the adenosine-induced potentiation of NMDA currents in dopamine neurons is mediated by an increase in cyclic AMP-dependent kinase activity, as has been shown in spinal cord neurons (Cerne et al., 1993). Future experiments are planned to test this hypothesis, as well as the hypothesis that the same mechanism is responsible for the small outward current produced by adenosine. Nevertheless, the fact that adenosine did not reduce currents evoked by exogenous NMDA shows conclusively that adenosine does not act postsynaptically to reduce NMDA EPSCs. Furthermore, our results from paired-pulse experiments strongly suggest that adenosine acts presynaptically to cause a preferential reduction in neurotransmission mediated by NMDA receptors.
Our data also suggest that baclofen has a presynaptic site of action that reduces glutamate release. In support of this conclusion, we found that baclofen (1) significantly reduced paired-pulse depression of EPSCs, (2) failed to reduce currents evoked by exogenous AMPA, and (3) exerted a significant concentration-preference for reducing NMDA EPSCs even when postsynaptic K+ conductances were reduced by intracellular Cs+ and QX-314. However, our finding that baclofen reduced currents evoked by exogenous NMDA confirms results of an earlier study (Seutin et al., 1994) and suggests that baclofen also has a postsynaptic site of action for reducing the NMDA component of synaptic transmission. Most likely, it is the baclofen-induced hyperpolarization of distal dendrites that inhibits NMDA currents by imposing a voltage-dependent block of conductance by Mg2+ (Wu & Johnson, 1996). However, we have not ruled out the possibility that baclofen reduces NMDA currents by triggering a second messenger system that is not coupled to postsynaptic ion channels.
Although our results clearly suggest that adenosine and baclofen cause presynaptic inhibition of glutamate release, this raises an apparent paradox. How can presynaptic inhibition contribute to a preferential reduction in the NMDA component of transmission? We can conceive of three possible situations, illustrated in Figure 8, that would be consistent with our data. In the first model (Figure 8a), the afferent nerve terminal would release glutamate onto a postsynaptic membrane in which most of the NMDA receptors are located peripherally in the synapse. Therefore, presynaptic inhibition of transmitter release would preferentially reduce the glutamate concentration at the NMDA receptors located in the margins of the synapse. Although an analogous non-homogenous distribution of metabotropic glutamate receptors has been reported in synapses in the cerebellar cortex (Nusser et al., 1994), we are not aware of any report describing a similar distribution of NMDA receptors.
Figure 8.
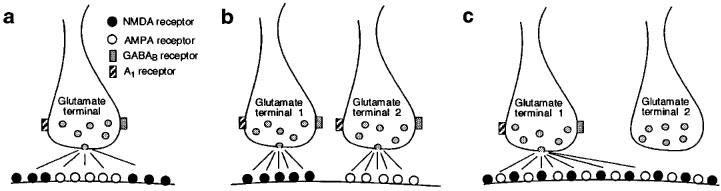
Three different models of glutamate-containing synapses. Postsynaptic membranes contain NMDA (•) and AMPA (○) receptors. (a) Postsynaptic membrane expresses a non-homogenous population of NMDA and AMPA receptors. AMPA receptors are located in the center of the synapse whereas NMDA receptors are located distally. A reduction in glutamate release would be expected to preferentially decrease the stimulation of the peripherally located NMDA receptors. (b) Two different glutamate inputs innervate separate populations of NMDA and AMPA receptors on the same cell. The concentration-preference of adenosine and baclofen for inhibition of NMDA receptor-mediated transmission would be due to either differences in receptor subtype or to differences in the efficiency by which these presynaptic receptors couple to intracellular effector mechanisms. (c) Spillover of glutamate activates distant NMDA receptors on an adjacent synapse on the same cell. Due to the high affinity of NMDA receptors for glutamate, dilute concentrations of glutamate in the adjacent synapse are sufficient for NMDA receptor stimulation, but this concentration is too low to stimulate AMPA receptors. We hypothesize that when adenosine or baclofen reduces glutamate release, it is the NMDA receptors located far from the synapse that will most likely fail to be stimulated by transmitter.
A second possibility is shown in Figure 8b. In this case, the same postsynaptic neuron receives two separate inputs in which one releases glutamate onto NMDA receptors and the other releases glutamate onto AMPA receptors. Although this model would predict that both glutamate inputs express GABAB and A1 receptors, the concentration-preference for inhibition of NMDA receptor-mediated transmission could be due to differences in receptor subtype or to differences in the efficiency by which these presynaptic receptors couple to intracellular effector mechanisms. Although NMDA and AMPA receptors are clearly co-localized in many synapses (Bekkers & Stevens, 1989; O'Brien et al., 1997), there is evidence that NMDA and AMPA receptors receive separate innervation in frontal cortex (Kang, 1995). Furthermore, the presence of synaptic transmission mediated solely by NMDA receptors in the hippocampus has been attributed to functionally ‘silent' AMPA receptors (Isaac et al., 1995; Liao et al., 1995). Although these reports provide some precedent for postulating separate inputs to NMDA and AMPA receptors, there is as yet no direct evidence for separate glutamate inputs to NMDA and AMPA receptors in the ventral midbrain.
We favour a third possibility which is illustrated in Figure 8c. This model proposes that a substantial amount of NMDA receptor-mediated current is due to ‘spillover' of glutamate onto adjacent excitatory synapses. Because the affinity of glutamate for the NMDA receptor is 100 fold greater than its affinity for the AMPA receptor (Jonas & Sakmann, 1992; Patneau & Mayer, 1990), the dilute concentration of glutamate in adjacent synapses might activate NMDA receptors but not AMPA receptors. Consequently, presynaptic inhibition of glutamate release would be expected to produce a disproportionate reduction in the NMDA component of transmission. It is important to note that this model would not be valid if synaptic concentrations of glutamate are high enough to saturate glutamate receptors (Clements et al., 1992; Jonas & Sakmann, 1992). Nevertheless, evidence for selective stimulation of NMDA receptors during spillover of glutamate has been presented by Kullmann et al. (1996) in the hippocampal slice. However, this phenomenon was reportedly observed only when temperatures were significantly lower than that (36°C) used in our studies (Asztély et al., 1997).
Our results are markedly different from those of several others who recorded from hippocampus. Perkel & Nicoll (1993) showed that adenosine and baclofen reduced both NMDA and AMPA components of transmission equally well. They used their results to argue for an ‘all-or-nothing' type of transmitter release, as opposed to a ‘graded' release that would provide a range of concentrations of transmitter in the synapse. Similarly, Tong & Jahr (1994) also found that adenosine and baclofen reduced both components of transmission equally well, and they suggested that postsynaptic receptors may be saturated with glutamate during synaptic transmission. In contrast, our results suggest that glutamate receptors are not normally saturated, and that presynaptic inhibitors of release can vary the concentration of glutamate in a graded fashion. Although it is unclear why our results are so strikingly different from those of others, this could be due to differences in the microanatomy of excitatory synapses in the ventral midbrain compared to the hippocampus.
At first glance, our findings might appear similar to those of Morrisett et al. (1991) who demonstrated that baclofen selectively reduces the NMDA as opposed to the AMPA component of transmission in the hippocampal slice. However, the study by Morrisett et al. (1991) presented evidence that baclofen reduced NMDA EPSPs solely by acting at a postsynaptic site of action. In contrast, our study clearly shows that baclofen has both pre- and postsynaptic sites of action that reduce NMDA receptor-mediated transmission in ventral midbrain dopamine neurons.
Our results have important implications for the regulation of burst firing in dopamine neurons. We (Johnson et al., 1992) and others (Overton & Clark, 1992; Wang et al., 1994) have demonstrated that the firing of action potentials in bursts is dependent upon stimulation of NMDA receptors. The report that baclofen blocks burst firing without interfering with single-spike firing of dopamine neurons in vivo (Engberg et al., 1993) is consistent with our hypothesis that activation of pre- and postsynaptic GABAB receptors selectively reduces NMDA receptor-dependent transmission. Moreover, our data predict that stimulation of presynaptic adenosine A1 receptors should also selectively block burst firing in these neurons.
In conclusion, we have presented evidence that presynaptic inhibition of glutamate release preferentially reduces the NMDA receptor-mediated component of transmission. Our working hypothesis for this phenomenon is that presynaptic inhibition reduces the synaptic concentration of glutamate in a graded fashion, which causes a reduction in postsynaptic spillover of glutamate onto NMDA receptors. We suggest that by regulating the degree of NMDA receptor stimulation, GABAB and adenosine A1 receptors may play important roles in regulating the firing pattern of midbrain dopamine neurons.
Acknowledgments
This work was funded by USPHS grants MH40416 and NS35437. We thank Dr Johan Grenhoff for his critical review of the manuscript.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AP5
±5-aminophosphovalerate
- CNQX
6-cyano-7-nitroquinoxalone
- DPCPX
8-cyclopentyl-1,3-dipropylxanthine
- EPSC
excitatory postsynaptic current
- EPSP
excitatory postsynaptic potential
- GABA
γ-aminobutyric acid
- NMDA
N-methyl-D-aspartate
- QX-314
lidocaine N-ethyl bromide
- SNC
substantia nigra compacta
- VTA
ventral tegmental area
References
- ASZTÉLY F., ERDEMIL G., KULLMANN D.M. Extrasynaptic glutamate spillover in the hippocampus: Dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- BEKKERS J.M., STEVENS C.F. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- CERNE R., RUSIN K.I., RANDI M. Enhancement of the N-methyl-D-aspartate response in spinal dorsal horn neurons by cAMP-dependent protein kinase. Neurosci. Lett. 1993;161:124–128. doi: 10.1016/0304-3940(93)90275-p. [DOI] [PubMed] [Google Scholar]
- CLEMENTS J.D., LESTER R.A.J., TONG G., JAHR C.E., WESTBROOK G.L. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- DE MENDONÇA A., RIBEIRO J.A. Adenosine inhibits the NMDA receptor-mediated excitatory postsynaptic potential in the hippocampus. Brain Res. 1993;606:351–356. doi: 10.1016/0006-8993(93)91007-f. [DOI] [PubMed] [Google Scholar]
- DUTAR P., NICOLL R.A.GABAB receptor-mediated inhibition of synaptic transmission in the hippocampus: Pharmacology and intracellular mechanisms Presynaptic receptors in the mammalian brain 1993Boston: Birkhäuser; 14–26.(eds. Dunwiddie, T.V. & Lovinger, D.M.) pp [Google Scholar]
- ENGBERG G., KLING-PETERSEN T., NISSBRANDT H. GABAB-receptor activation alters the firing pattern of dopamine neurons in the rat substantia nigra. Synapse. 1993;15:229–238. doi: 10.1002/syn.890150308. [DOI] [PubMed] [Google Scholar]
- GONON F.G., BUDA M.J. Regulation of dopamine release by impulse flow and by autoreceptors as studied by in vivo voltammetry in the rat striatum. Neurosci. 1985;14:765–774. doi: 10.1016/0306-4522(85)90141-1. [DOI] [PubMed] [Google Scholar]
- GRACE A.A. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neurosci. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- HARRIS E.W., COTMAN C.W. Effects of synaptic antagonists on perforant path paired-pulse plasticity: Differentiation of pre- and postsynaptic antagonism. Brain Res. 1985;334:348–353. doi: 10.1016/0006-8993(85)90230-6. [DOI] [PubMed] [Google Scholar]
- ISAAC J.T., NICOLL R.A., MALENKA R.C. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- JOHNSON S.W., NORTH R.A. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J. Physiol. (Lond.) 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON S.W., SEUTIN V., NORTH R.A. Burst firing in dopamine neurons induced by N-methyl-D-aspartate: Role of electrogenic sodium pump. Science. 1992;258:665–667. doi: 10.1126/science.1329209. [DOI] [PubMed] [Google Scholar]
- JONAS P., SAKMANN B. Glutamate receptor channels in isolated patches from CA1 and CA3 pyramidal cells of rat hippocampal slices. J. Physiol. (Lond.) 1992;455:143–171. doi: 10.1113/jphysiol.1992.sp019294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG Y. Differential paired pulse depression of non-NMDA and NMDA currents in pyramidal cells of the rat frontal cortex. J. Neurosci. 1995;15:8268–8280. doi: 10.1523/JNEUROSCI.15-12-08268.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KULLMANN D.M., ERDEMLI G., ASZTÉLY F. LTP of AMPA and NMDA receptor-mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron. 1996;17:461–474. doi: 10.1016/s0896-6273(00)80178-6. [DOI] [PubMed] [Google Scholar]
- LACEY M.G., MERCURI N.B., NORTH R.A. On the potassium conductance increase activated by GABAB and dopamine D2 receptors in rat substantia nigra neurones. J. Physiol. (Lond.) 1988;401:437–453. doi: 10.1113/jphysiol.1988.sp017171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LACEY M.G., MERCURI N.B., NORTH R.A. Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the actions of dopamine and opioids. J. Neurosci. 1989;9:1233–1241. doi: 10.1523/JNEUROSCI.09-04-01233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANTHORN T.H., COTMAN C.W. Baclofen selectively inhibits excitatory transmission in the hippocampus. Brain Res. 1981;255:171–178. doi: 10.1016/0006-8993(81)90326-7. [DOI] [PubMed] [Google Scholar]
- LEE K.S., REDDINGTON M. 1,3-Dipropyl-8-cyclopentylxanthine (DPCPX) inhibition of [3H]N-ethylcarboxamidoadenosine (NECA) binding allows the visualization of putative non-A1 adenosine receptors. Brain Res. 1986;368:394–398. doi: 10.1016/0006-8993(86)90589-5. [DOI] [PubMed] [Google Scholar]
- LIAO D., HESSLER N.A., MALINOW R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- MANLEY L.D., KUCZENSKI R., SEGAL D.S., YOUNG S.J., GROVES P.M. Effects of frequency and pattern of medial forebrain bundle stimulation of caudate dialysate dopamine and serotonin. J. Neurochem. 1992;58:1491–1498. doi: 10.1111/j.1471-4159.1992.tb11369.x. [DOI] [PubMed] [Google Scholar]
- MIRENOWICZ J., SCHULTZ W. Preferential activation of midbrain dopamine neurons by appetitive rather than aversive stimuli. Nature. 1996;379:449–451. doi: 10.1038/379449a0. [DOI] [PubMed] [Google Scholar]
- MORRISETT R.A., MOTT D.D., LEWIS D.V., SWARTZWELDER H.S., WILSON W.A. GABAB receptor-mediated inhibition of the N-methyl-D-aspartate component of synaptic transmission in the rat hippocampus. J. Neurosci. 1991;11:203–209. doi: 10.1523/JNEUROSCI.11-01-00203.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NATHAN T., JENSEN M.S., LAMBERT J.D.C. The slow inhibitory postsynaptic potential in rat hippocampal CA1 neurones is blocked by intracellular injection of QX-314. Neurosci. Lett. 1990;110:309–313. doi: 10.1016/0304-3940(90)90865-7. [DOI] [PubMed] [Google Scholar]
- NÖRENBERG W., WIRKNER K., ILLES P. Effect of adenosine and some of its structural analogues on the conductance of NMDA receptor channels in a subset of rat neostriatal neurones. Br. J. Pharmacol. 1997;122:71–80. doi: 10.1038/sj.bjp.0701347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NUSSER Z., MULVIHILL E., STREIT P., SOMOGYI P. Subsynaptic segregation of metabotropic and ionotropic glutamate receptors as revealed by immunogold localization. Neurosci. 1994;61:421–427. doi: 10.1016/0306-4522(94)90421-9. [DOI] [PubMed] [Google Scholar]
- O'BRIEN J.A., ISAACSON J.S., BERGER A.J. NMDA and non-NMDA receptors are co-localized at excitatory synapses of rat hypoglossal motoneurons. Neurosci. Lett. 1997;227:5–8. doi: 10.1016/s0304-3940(97)00293-0. [DOI] [PubMed] [Google Scholar]
- OLPE H.R., KARLSSON G., POZZA M.F., BRUGGER F., STEINMANN M., VAN RIEZEN H., FAGG G., HALL R.G., FROESTL W., BITTIGER H. CGP 35348: a centrally active blocker of GABAB receptors. Eur. J. Pharmacol. 1990;187:27–38. doi: 10.1016/0014-2999(90)90337-6. [DOI] [PubMed] [Google Scholar]
- OVERTON P., CLARK D. Iontophoretically administered drugs acting at the N-methyl-D-aspartate receptor modulate burst firing in A9 dopamine neurons in the rat. Synapse. 1992;10:131–140. doi: 10.1002/syn.890100208. [DOI] [PubMed] [Google Scholar]
- PATNEAU D.K., MAYER M.L. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. J. Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1986. [Google Scholar]
- PERKEL D.J., NICOLL R.A. Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation. J. Physiol. (Lond.) 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHULTZ W. Responses of midbrain dopamine neurons to behavioral trigger stimuli in the monkey. J. Neurophysiol. 1986;56:1439–1461. doi: 10.1152/jn.1986.56.5.1439. [DOI] [PubMed] [Google Scholar]
- SEUTIN V., JOHNSON S.W., NORTH R.A. Effect of dopamine and baclofen on N-methyl-D-aspartate-induced burst firing in rat ventral tegmental neurons. Neurosci. 1994;58:201–206. doi: 10.1016/0306-4522(94)90167-8. [DOI] [PubMed] [Google Scholar]
- SUAUD-CHAGNY M.F., CHERGUI K., CHOUVET G., GONON F. Relationship between dopamine release in the rat nucleus accumbens and the discharge activity of dopaminergic neurons during local in vivo application of amino acids in the ventral tegmental area. Neurosci. 1992;49:63–72. doi: 10.1016/0306-4522(92)90076-e. [DOI] [PubMed] [Google Scholar]
- TONG G., JAHR C.E. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- WANG T., O'CONNOR W.T., UNGERSTEDT U., FRENCH E.D. N-methyl-D-aspartic acid biphasically regulates the biochemical and electrophysiological response of A10 dopamine neurons in the ventral tegmental area: in vivo microdialysis and in vitro electrophysiological studies. Brain Res. 1994;666:255–262. doi: 10.1016/0006-8993(94)90780-3. [DOI] [PubMed] [Google Scholar]
- WU Y.-N., CAMERON W.E., JOHNSON S.W. Effect of baclofen on NMDA mediated synaptic responses in rat midbrain dopamine neuron in vitro. Soc. Neurosci. (Abstr.) 1996;22:1294. [Google Scholar]
- WU Y.-N., JOHNSON S.W. Pharmacological characterization of inward current evoked by N-methyl-D-aspartate in dopamine neurons in the rat brain slice. J. Pharmacol. Exp. Therap. 1996;279:457–463. [PubMed] [Google Scholar]
- WU Y.-N., MERCURI N.B., JOHNSON S.W. Presynaptic inhibition of γ-aminobutyric acidB-mediated synaptic current by adenosine recorded in vitro in midbrain dopamine neurons. J. Pharmacol. Exp. Therap. 1995;273:576–581. [PubMed] [Google Scholar]
- YUNG W.H., HÄUSSER M.A., JACK J.J.B. Electrophysiology of dopaminergic and non-dopaminergic neurones of the guinea-pig substantia nigra pars compacta in vitro. J. Physiol. (Lond.) 1991;436:643–667. doi: 10.1113/jphysiol.1991.sp018571. [DOI] [PMC free article] [PubMed] [Google Scholar]