

Abstract
The effects of atorvastatin (3 mg kg−1) and simvastatin (3 mg kg−1) on hepatic enzyme activities involved in very low density lipoprotein metabolism were studied in coconut oil/cholesterol fed rabbits.
Plasma cholesterol and triglyceride levels increased 19 and 4 fold, respectively, after 7 weeks of feeding.
Treatment with statins during the last 4 weeks of feeding abolished the progression of hypercholesterolaemia and reduced plasma triglyceride levels.
3-Hydroxy-3-methyl-glutaryl Coenzyme A reductase, acyl-coenzyme A:cholesterol acyltransferase, phosphatidate phosphohydrolase and diacylglycerol acyltransferase activities were not affected by drug treatment. Accordingly, hepatic free cholesterol, cholesteryl ester and triglyceride content were not modified.
Simvastatin treatment caused an increase (72%) in lipoprotein lipase activity without affecting hepatic lipase activity.
Atorvastatin caused a reduction in hepatic phospholipid content and a compensatory increase in CTP:phosphocholine cytidylyl transferase activity.
The results presented in this study suggest that, besides the inhibitory effect on 3-hydroxy-3-methyl-glutaryl Coenzyme A reductase, simvastatin and atorvastatin may have additional effects that contribute to their triglyceride-lowering ability.
Keywords: Atorvastatin, simvastatin, high fat/cholesterol fed rabbits, lipoprotein lipase, CTP:phosphocholine cytidylyl transferase
Introduction
Statins interfere with the rate-limiting step in cholesterol biosynthesis and consistently reduce plasma low density lipoprotein (LDL)-cholesterol levels (Davignon et al., 1992). However, their ability to reduce triglyceride concentrations depends on the baseline triglyceride levels and on the potency and efficacy of the statin used (Lea & McTavish, 1997, Jones et al., 1998, Stein et al., 1998).
The mechanisms of the triglyceride-lowering effect of statins are still unclear. It has been proposed that inhibition of 3-hydroxy-3-methyl-glutaryl Coenzyme A (HMG-CoA) reductase leads to a reduction in the concentration of cholesterol in the hepatocytes and up-regulation of LDL-receptors (Ma et al., 1986). Since these receptors bind and internalize not only LDL but also very low density lipoprotein (VLDL) remnants, plasma triglyceride levels are reduced (Grundy et al., 1988). Another possible mechanism may relate to the role of HMG-CoA reductase in VLDL secretion. Some studies have shown that the HMG-CoA reductase-derived cholesterol pool is intricately involved in hepatic VLDL-cholesterol and triglyceride formation and secretion (Khan et al., 1989; Yoshino et al., 1988). Inhibition of HMG-CoA reductase could limit the availability of cholesterol for VLDL production and decrease hepatic VLDL secretion, with the subsequent lowering of plasma triglyceride. Both mechanisms are common to all HMG-CoA reductase inhibitors; however, only a few of these drugs cause marked reductions in plasma triglyceride levels. Although these differences may be related to the potency, duration of action or hepatic clearance of the drugs, some authors have speculated that simvastatin and atorvastatin may alter triglyceride metabolism (Bocan et al., 1994) or phospholipid synthesis (Alegret et al., 1998). Thus, besides the known action on HMG-CoA reductase, some statins might have particular effects that contribute to their triglyceride-lowering ability.
The genetic and metabolic bases of human combined hyperlipidaemia have not yet been defined, although an overproduction of hepatic VLDL is consistently found. Thus, to test the effect of statins on VLDL-triglyceride metabolism, we have chosen an animal model of diet-induced hyperlipidaemia, the coconut oil/cholesterol fed rabbit, in which the diet produces hypercholesterolaemia and moderate hypertriglyceridaemia as a result of an increased hepatic secretion of VLDL (Van Heek & Zilversmit, 1988; 1991).
Since VLDL secretion depends on the availability of lipid constituents, we examined whether atorvastatin and simvastatin alter the key enzyme activities related to cholesterol (HMG-CoA reductase), cholesteryl ester (acyl-coenzyme A:cholesterol acyltransferase), triglyceride (phosphatidate phosphohydrolase and diacylglycerol acyltransferase) and phospholipid (CTP:phosphocholine cytidylyl transferase) synthesis in an animal model of combined dyslipidaemia. In addition, we studied the effect of both statins on the enzyme activities regulating catabolism of triglyceride-rich lipoproteins (lipoprotein lipase and hepatic lipase).
Methods
Chemicals
3-Hydroxy-3-methyl [3-14C]-glutaryl-coenzyme A, RS-[2-14C]-mevalonic acid lactone, [1-14C]-oleoyl-coenzyme A, cholesteryl [1-14C]-oleate, phosphoryl [methyl-14C]-choline ammonium salt, [1-14C]-palmitoyl-coenzyme A and glycerol tri [1-14C]-oleate were purchased from Amersham Iberica (Madrid, Spain). Glucose 6-phosphate, NADP+, dithiothreitol (DTT), glutathione (GSH), 3-hydroxy-3-methyl-glutaryl-coenzyme A, oleoyl-coenzyme A, palmitoyl-coenzyme A, cholesteryl oleate, ADP, CTP, CDP-choline, phosphorylcholine, phosphatidylcholine and fatty acid free bovine serum albumin (BSA) were from Sigma Chemical Co. (Madrid, Spain). Glucose 6-phosphate dehydrogenase and silica gel TLC plates were from Merck (Barcelona, Spain). Scintillation fluids (Co 136 and Co 36) were from Scharlau Co. (Barcelona, Spain). Other general chemicals were obtained from commercial sources and were of analytical grade.
Drugs
Atorvastatin calcium was supplied by Parke-Davis (Ann Arbor, MI, U.S.A.) and simvastatin lactone form was a generous gift from Merck (Barcelona, Spain).
Experimental design
Male New Zealand white rabbits (Pi Petit Farm, Barcelona, Spain) were housed in individual cages in a room with constant humidity and temperature (22±2°C) under a 12 h light-dark cycle, and fed a standard diet (Mucedola, Milano, Italy). After 1 week, the animals (average weight 1.86±0.15 kg) were fed 10 g of a chow diet plus 100 g of the same diet supplemented with 0.5% cholesterol and 14% coconut oil (Mucedola, Milano, Italy) for 7 weeks. After 3 weeks on this diet, the animals were randomized based on their 12 h post-meal plasma cholesterol and triglyceride values, into three groups of eight animals with no statistically significant differences between lipid values. For the next 4 weeks, a group of control animals was maintained on the same diet (100 g of coconut oil/cholesterol diet plus 10 g of chow diet) and two groups of animals were fed 100 g of coconut oil/cholesterol diet plus 10 g of chow diet containing simvastatin or atorvastatin in order to provide a dose of 3 mg kg−1 day−1 of each drug. The diets supplemented with the drugs were prepared as described previously (Alegret et al., 1998). No significant differences were found between body weight of control and treated groups. All procedures were conducted in accordance with the principles and guidelines established by the University of Barcelona Bioethics Committee as stated in law 5/1995, 21st July, from the Generalitat de Catalunya.
At the end of the treatment, blood samples for plasma lipid determinations were obtained, after 12 h fasting, by ear puncture and collected in EDTA-tubes, before and 5 min after intravenous heparin injection (100 U kg−1). Plasma was obtained by centrifugation at 3000×g for 10 min at 4°C. The animals were killed by exsanguination under ether anaesthesia and the livers were removed, perfused with ice-cold 0.9% NaCl and homogenized in (mM): KH2 PO4 50, pH 7.4, NaCl 150, DTT 1, EDTA (buffer A) 30, and sucrose 0.25 M, using a Potter Elvehjem homogenizer with a teflon pestle. The post-mitochondrial and microsomal fractions were obtained by differential centrifugation as described elsewhere (Montgomery & Cinti, 1977) and microsomes were resuspended in buffer A devoid of sucrose. The protein content of the subcellular fractions was determined by the method of Bradford (1976) using BSA as a standard.
Lipid analysis
Plasma total cholesterol, triglyceride and phospholipid concentrations were measured with the Boehringer-Mannheim (Barcelona, Spain) colorimetric tests (Monotest Cholesterol CHODPAP No. 290319, Peridochrom Triglyceride GPO-PAP No. 701882 and MPR2 No 691844 phospholipids, respectively). VLDL and LDL from plasma samples were precipitated by using the reagent No. 543004, also from Boehringer Mannheim, and HDL-cholesterol concentration was determined in the supernatant.
Lipid in the liver was extracted according to Bligh & Dyer (1959), using the post-nuclear supernatant. The lipid extract was evaporated under a stream of nitrogen gas and redissolved in absolute ethanol. Total and free cholesterol, triglyceride and phospholipid concentration were determined as described above.
Enzyme assays
3-Hydroxy-3-methyl-glutaryl coenzyme A reductase (EC 1.1.1.34)
This activity was determined in liver microsomes as described previously (Alegret et al., 1998). Briefly, incubations were performed in a total volume of 150 μl which contained buffer A, 0.46 mg BSA ml−1, 3 mM NADP+, 4.25 mM DTT, 30 mM glucose 6-phosphate, 0.3 units of glucose 6-phosphate dehydrogenase and 200 μg of microsomal protein. After a preincubation of 20 min at 37°C, the reaction was carried out with 90 μM HMGCoA (containing 0.45 μCi of [3-14C]-HMGCoA) for 20 min.
Acyl-coenzyme A:cholesterol acyltransferase (EC 2.3.1.26)
This activity was determined using oleoyl-CoA as substrate by the method described by Alegret et al. (1998). The incubation mixture contained (final volume 250 μl) 100 mM KH2PO4 1 mM EDTA buffer pH 7.4, 6 mg BSA ml−1, 0.62 mg GSH−1 ml and 150 μg of liver microsomal protein. After a preincubation of 5 min at 37°C, oleoyl-CoA (containing 0.135 μCi of [1-14C]-oleoyl-CoA) was added to a final concentration of 35 μM and samples were incubated for 4 min.
Phosphatidate phosphohydrolase (EC 3.1.3.4)
This activity was determined in liver post-mitochondrial fraction by using [14C]-phosphatidic acid and phosphatidylcholine emulsion prepared as described by Aridor-Piterman et al. (1992). The reaction was carried out at 37°C for 35 min with 50 μg of post-mitochondrial supernatant in a final volume of 100 μl, according to Martin et al. (1991).
Diacylglycerol acyltransferase (EC 2.3.1.20)
Acylation of sn-1,2-diacylglycerol with [1-14C]-palmitoyl-CoA was determined according to Coleman (1992). The reaction was carried out exactly as described, using liver microsomes (15 μg of protein in a total volume of 200 μl of incubation mixture), for 15 min at 25°C.
Lipoprotein lipase (EC 3.1.1.34) and hepatic lipase (EC 3.1.1.3)
These activities were determined in post-heparin plasma samples by using a glycerol tri[1-14C]-oleate and phosphatidylcholine emulsion according to the method described by Alegret et al. (1998). Lipoprotein lipase (LPL) activity was calculated by subtracting hepatic lipase from total lipase activity. The reactions were carried out at 37°C for 30 min with 10 μl of post-heparin plasma and 50 μl of each emulsion (143.3 nmol, 200,000 c.p.m.).
CTP:phosphocholine cytidylyl transferase (EC 2.7.7.15)
This activity was determined in liver post-mitochondrial supernatant (30 μg of protein in 100 μl final volume of incubation mixture) by measuring the formation of radioactive CDP-choline from phosphoryl [methyl-14C]-choline by the charcoal adsorption method, exactly as described by Weinhold & Feldman (1992).
Statistics
The results are the mean±s.e.mean of n experiments assayed in duplicate. Significant differences were established by an ANOVA or Student's t-test, depending on the number of groups compared, using the computer programme GraphPad InStat. When the number of animals was small, a non-parametric test was performed (Kruskal-Wallis test). Logarithms of HMG-CoA reductase activity, CTP:phosphocholine cytidylyl transferase activity and plasma triglyceride concentration were used to calculate statistics, as the variance was not homogeneous. When significant variations were found, the Dunnett multiple comparisons test was performed. Linear correlation between variables was calculated by using the above mentioned programme.
Results
Plasma and liver lipid content
Plasma total cholesterol, HDL-cholesterol and triglyceride levels rose 16, 1.6 and 2.1 fold, respectively during the first 3 weeks of coconut oil/cholesterol feeding (Table 1). Following randomization of the animals in their respective treatment groups, plasma total cholesterol and triglyceride levels in the control group continued to increase over the following 4 weeks, from day 21 to 49 (Table 2). When plasma cholesterol levels were compared in each group to their pretreatment values (day 49 vs day 21), HMG-CoA reductase inhibitors had no lowering effect on plasma total cholesterol or apoB-cholesterol levels, but prevented the rise in these parameters induced by the diet. In contrast, plasma HDL-cholesterol levels were significantly increased after atorvastatin treatment.
Table 1.
Effect of coconut oil/cholesterol feeding on plasma lipid levels

Table 2.
Plasma lipid levels at the beginning (day 21) and the end (day 49) of drug treatment
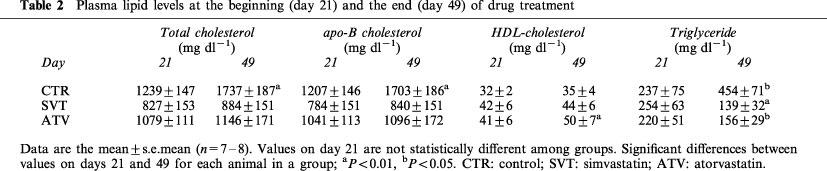
Plasma triglyceride levels were reduced relative to control by atorvastatin and simvastatin at the end of the treatment (day 49). Both statins also significantly lowered plasma triglyceride levels over the 4 weeks of treatment, as compared to their pretreatment values (day 49 vs day 21) (Table 2). No significant changes in plasma phospholipids were observed at the end of the treatment (404±73 and 635±177 mg dl−1, for simvastatin and atorvastatin, respectively; vs 822±169 mg dl−1, for control on day 49).
Figure 1 shows the effect of drug treatment on liver lipid content. Whereas liver cholesterol and triglyceride content were not significantly modified, phospholipid concentration was reduced 28%, but only in atorvastatin-treated animals.
Figure 1.
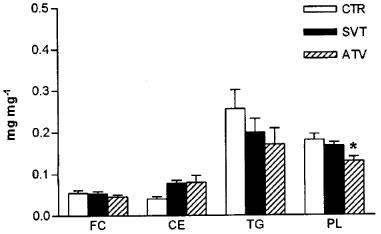
Effect on simvastatin (SVT) and atorvastatin (ATV) on liver lipid content. FC: free cholesterol; CE: cholesteryl esters; TG: triglyceride; PL: phospholipid. The results are expressed as mg per mg of protein of post-nuclear fraction and are the mean±s.e.mean (n=4). *Values significantly different from control (P<0.05).
When we compared data from all animals, we obtained a positive correlation between liver phospholipid content and plasma triglyceride levels (n=11; r2=0.477; P=0.0186).
Enzyme activities involved in cholesterol-metabolism
As is shown in Table 3, neither atorvastatin nor simvastatin affected HMG-CoA reductase activity, the rate limiting enzyme in hepatic cholesterol synthesis. Acyl-coenzyme A:cholesterol acyltransferase (ACAT), the enzyme that catalyzes cholesteryl ester formation, was also unaffected by treatment (Table 3). In addition, the dietary cholesterol entering the liver is almost immediately esterified by the enzyme ACAT, increasing its activity by about 30 fold (Table 4).
Table 3.
Effect of drug treatment on enzyme activities involved in cholesterol and triglyceride synthesis

Table 4.
Enzyme activities involved in lipid metabolism in normolipaemic (NLP) and hyperlipaemic (HLP) rabbits

Enzyme activities involved in triglyceride metabolism
Phosphatidate phosphohydrolase (PAP) and diacylglycerol acyltransferase (DGAT), the two enzyme activities regulating the synthesis of triglyceride were not modified after atorvastatin or simvastatin treatment (Table 3). As regards the enzyme activities related to catabolism of triglyceride-rich lipoproteins, neither statin modified hepatic lipase (HL) activity (Figure 2), but simvastatin caused a significant increase in lipoprotein lipase (LPL) activity (172% of control value) (Figure 2). When data from animals of the three groups were included, a negative correlation between LPL activity and plasma triglyceride levels at day 49 was obtained (n=22; r2=0.199; P=0.0375). However, in the control coconut oil/cholesterol fed animals plasma triglyceride levels were not highly correlated to LPL activity. In contrast, we obtained a high inverse correlation for simvastatin-treated animals: about 65% of the triglyceride lowering effect of simvastatin in coconut oil/cholesterol fed rabbits may be explained by the increase in LPL activity (Figure 3). However, hypotriglyceridaemic effect of atorvastatin does not appear to be related to triglyceride catabolism.
Figure 2.
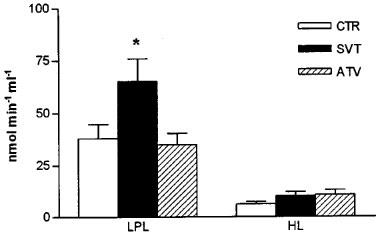
Effect of simvastatin (SVT) and atorvastatin (ATV) on lipoprotein lipase (LPL) and hepatic lipase (HL) activities. The results are the mean±s.e. mean from 7–8 experiments performed in duplicate. *Values significantly different from control (P<0.05).
Figure 3.
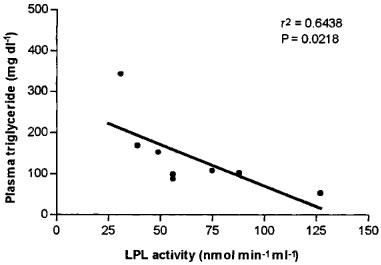
Correlation between plasma triglyceride levels and lipoprotein lipase (LPL) activity for simvastatin-treated animals (n=8).
Enzyme activities related to phospholipid synthesis
Figure 4 shows the effect of drug treatment on CTP:phos-phocholine cytidylyl transferase (CT), the rate limiting enzyme in phospholipid synthesis. This enzyme activity was unaffected by simvastatin treatment, but significantly increased by atorvastatin (147% of control value).
Figure 4.
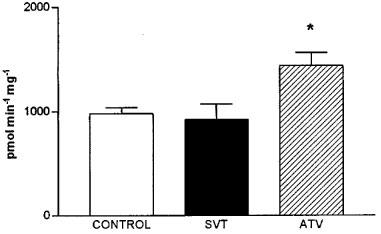
Effect of simvastatin (SVT) and atorvastatin (ATV) on CTP: phosphocholine cytidylyl transferase activity. The results are the mean±s.e.mean from 7–8 experiments performed in duplicate. *Values significantly different from control (P<0.05).
Discussion
Rabbits fed a coconut oil/cholesterol diet develop hypercholesterolaemia with increases in apoB-cholesterol and HDL cholesterol as shown in this and previous studies (Van Heek & Zilversmit, 1988; 1991). In cholesterol fed rabbits the increase in apoB-cholesterol is associated with diminished LDL receptor function and the addition of oils containing saturated fatty acids causes a larger down-regulation of LDL receptor which led to higher cholesterol levels. Further, as a larger percentage of LDL-cholesterol is degraded in the periphery, the total amount of cholesterol returning to the liver is increased, and thus plasma HDL-cholesterol rises (Dietschy et al., 1993). These effects are the consequence of a series of changes in hepatic sterol metabolism. Initially, cholesterol synthesis is suppressed by downregulation of HMG-CoA reductase activity (Xu et al., 1995).
Previous studies have shown that plasma triglyceride and apoB increase with high fat/high cholesterol diets and that increase results from an overproduction of VLDL (Bocan et al., 1994; Van Heek & Zilversmit, 1988; 1991). Accordingly, in our study, plasma triglyceride levels and enzyme activities regulating triglyceride and phospholipid synthesis, such as DGAT and CT were increased. Given the increase in DGAT activity and the lack of significant change in LPL activity, it seems likely that overproduction of hepatic triglyceride is the major determinant of the hypertriglyceridaemia developed in this animal model.
Treatment of coconut oil/cholesterol fed rabbits with HMG-CoA reductase inhibitors markedly decreased plasma cholesterol levels due to a reduction in apoB cholesterol concentration, as plasma HDL cholesterol levels were not modified relative to control animals. However, atorvastatin treatment caused an increase in plasma HDL-cholesterol concentration compared to pretreatment values. From our data, it seems that HMG-CoA reductase inhibitors prevent the progression of hypercholesterolaemia during treatment, though plasma cholesterol levels remain much higher than in normolipidaemic animals. Since hepatic HMG-CoA reductase activity and LDL receptor function is markedly decreased in rabbits chronically fed cholesterol (Kovanen et al., 1981; Xu et al., 1995), the mechanism for the observed cholesterol lowering in this animal model is unclear: (i) some authors have postulated that plasma cholesterol changes may be caused by the inhibition of intestinal HMG-CoA reductase, which would limit cholesterol absorption (Bocan et al., 1994) and (ii) inhibition of HMG-CoA reductase could lower hepatic VLDL secretion and, subsequently, plasma cholesterol and triglyceride (Khan et al., 1990; Auerbach et al., 1995). VLDL secretion may be controlled by cholesteryl ester availability (Thompson et al., 1996); any intervention that reduces the ACAT free cholesterol substrate pool, such as HMG-CoA reductase inhibition, may modulate ACAT activity and hence VLDL secretion (Huff & Burnett, 1997). Nevertheless, in our coconut oil/cholesterol fed rabbits ACAT activity and the concentration of liver free or esterified cholesterol were not affected by either simvastatin or atorvastatin. The effect of HMG-CoA reductase inhibitors on ACAT activity depends on the tissue and the species studied (Conde et al., 1996; Owens et al., 1991). Thus, simvastatin inhibited rabbit intestinal (Ishida et al., 1989), but not liver ACAT activity (Alegret et al., 1998).
Bocan et al. (1994) showed that different HMG-CoA inhibitors reduced VLDL-cholesterol to a similar extent, but only simvastatin and atorvastatin decreased triglyceride levels, and they speculate that these two drugs may alter also triglyceride synthesis. Moreover, most authors accept that apoB secretion can be modulated by triglyceride synthesis and availability (Benoist & Grand-Perret, 1996). In the present study we have examined the effect of simvastatin and atorvastatin on key enzymes involved in triglyceride synthesis, PAP and DGAT, but neither of them affected these enzyme activities. Therefore, the triglyceride lowering effect does not seem to be caused by inhibition of the biosynthesis of triglyceride, consequently, liver triglyceride levels remained unchanged in our studies after drug treatment. In order to test whether statins accelerate triglyceride catabolism, we assayed the effect of simvastatin and atorvastatin on LPL activity. As was shown, there was no significant difference between atorvastatin-treated rabbits and control animals. In contrast, simvastatin treatment significantly increased LPL activity without affecting HL activity. These data are consistent with studies performed in other species (Sato et al., 1991; Castro-Cabezas et al., 1993; Cabezas et al., 1993). To elucidate whether the triglyceride-lowering effect of simvastatin was attributable to LPL induction, we studied the correlation between LPL activity and plasma triglyceride levels on day 49, and found a significant negative correlation overall but this was mainly due to a high inverse correlation in the simvastatin-treated animals.
Some authors have suggested that apoB secretion can also be regulated by phosphatidylcholine synthesis (Dixon & Ginsberg, 1993). Most phosphatidylcholine is synthesized through the CDP-choline pathway (Vance, 1990). As CT is the rate-limiting enzyme of this pathway (Boggs et al., 1995), it becomes crucial for the regulation of phosphatidylcholine synthesis. Treatment of coconut oil/cholesterol fed rabbits with simvastatin had no effect on CT activity, while in atorvastatin-treated animals there was a significant increase in the enzyme activity. When liver phospholipids were analysed, atorvastatin, but not simvastatin caused a decrease in phospholipid concentration. In high fat fed guinea-pigs atorvastatin treatment also produced a slight reduction in microsomal phospholipid content (Conde et al., 1996). The decrease in liver phospholipid content suggests that phospholipid synthesis may be inhibited in the hepatocytes of high fat/cholesterol fed rabbits treated with atorvastatin, despite CT activity being increased.
We have previously shown that atorvastatin administration to normolipidaemic rabbits reduces CT activity about 60%. Nevertheless, the remaining activity seemed to be enough to maintain VLDL secretion, as plasma triglyceride levels were not affected by drug treatment (Alegret et al., 1998). Therefore, the effect of atorvastatin on CT activity and phospholipid synthesis seems to depend on the experimental model used. We suggest that atorvastatin treatment may inhibit CT activity, reducing phospholipid content inside the hepatocyte of coconut oil/cholesterol fed rabbits. This decrease in liver phospholipid concentration could cause a compensatory increase in CT activity in order to maintain VLDL synthesis, but not high enough to overcome the inhibitory effect of atorvastatin or its metabolites on CT activity.
Some enzyme inhibitors such as HMG-CoA reductase inhibitors also induced an increase in the enzyme activity due to a compensatory response of the organism (Qin et al., 1992). Although atorvastatin was not able to inhibit CT activity when the drug was added to the assay medium at 50 μM (Alegret et al., 1998), we cannot discard that one of its metabolites could inhibit this enzyme activity.
On the other hand, we cannot rule out that the effect on CT activity was dependent on HMG-CoA reductase inhibition. Although in the present study atorvastatin, but not simvastatin, affected CT activity and liver phospholipid concentration, this difference could be a matter of potency or liver selectivity, as atorvastatin is concentrated in the liver to a greater extent (Bocan et al., 1992). Further, simvastatin decreased CT activity in cell culture (Yanagita et al., 1994) and lovastatin treatment caused an increase in CT activity in rats (Linscheer et al., 1995).
In order to assess whether the decrease in liver phospholipid content contributes to the triglyceride-lowering effect of atorvastatin, we studied the correlation between liver phospholipid content and plasma triglyceride levels and found a positive correlation. Thereby, the decrease in liver phospholipid content caused by atorvastatin treatment could reduce VLDL secretion and contributes partly to the hypotriglyceridaemic effect of the drug. However, as we only have studied the effect of statins on enzyme activities involved in hepatic lipid synthesis, we cannot discard that these drugs may affect other cellular targets as MTP (microsomal triglyceride transfer protein), that catalyzes the transference of triglyceride and cholesteryl esters to apoB, or ASP (acylation stimulating protein), essential for triglyceride storage in adipose tissue.
In conclusion we have studied the effect of simvastatin and atorvastatin on the key enzyme activities involved in the synthesis and catabolism of VLDL in an animal model of combined dyslipidaemia. The most significant finding was that, besides their action on cholesterol biosynthesis, both statins have specific effects on lipid metabolism: simvastatin increases the degradation of VLDL by induction of LPL activity and atorvastatin decreases hepatic phospholipid synthesis. These effects may contribute to the triglyceride-lowering effect of these agents, at least in the coconut oil/cholesterol fed rabbit.
Acknowledgments
Financial support was partly provided by FPCNL, Parke-Davis (981-SPA-01), CICYT (SAF97-0215 and SAF98-0105) and grant SGR 96-84 from Generalitat de Catalunya. We also thank Mr R. Rycroft (Language Advice Service of the University of Barcelona) for his helpful assistance.
Abbreviations
- ACAT
acyl-coenzyme A:cholesterol acyltransferase
- CT
CTP:phosphocholine cytidylyl transferase
- DGAT
diacylglycerol acyltransferase
- HL
hepatic lipase
- HMG-CoA
3-Hydroxy-3-methyl-glutaryl Coenzyme A
- LDL
low density lipoproteins
- LPL
lipoprotein lipase
- PAP
phosphatidate phosphohydrolase
- VLDL
very low density lipoproteins
References
- ALEGRET M., VERD J.C., DIAZ C., HERNANDEZ G., ADZET T., SANCHEZ R.M., LAGUNA J.C. Effect of hypolipidemic drugs on key enzyme activities related to lipid metabolism in normolipidemic rabbits. Eur. J. Pharmacol. 1998;347:283–291. doi: 10.1016/s0014-2999(98)00096-x. [DOI] [PubMed] [Google Scholar]
- ARIDOR-PITERMAN O., LAVIE Y., LISCOVITCH M. Bimodal distribution of phosphatidic acid phosphohydrolase in NG108-15 cells. Eur. J. Biochem. 1992;204:561–568. doi: 10.1111/j.1432-1033.1992.tb16668.x. [DOI] [PubMed] [Google Scholar]
- AUERBACH B.J., KRAUSE B.R., BISGAIER C.L., NEWTON R.S. Comparative effects of HMG-CoA reductase inhibitors on apoB production in the casein-fed rabbit: Atorvastatin versus lovastatin. Atherosclerosis. 1995;115:173–180. doi: 10.1016/0021-9150(94)05508-g. [DOI] [PubMed] [Google Scholar]
- BENOIST F., GRAND-PERRET T. Apo B secretion by HepG2 cells is regulated by the rate of triglyceride biosynthesis but not by intracellular lipid pools. Arterioscler. Thromb. Vasc. Biol. 1996;16:1229–1235. doi: 10.1161/01.atv.16.10.1229. [DOI] [PubMed] [Google Scholar]
- BLIGH E.G., DYER W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- BOCAN T.M.A., FERGUSON E., MCNALLY W., UHLENDORF P.D., MUELLER S.B., DEHART P., SLISKOVIC D.R., ROTH B.D., KRAUSE B.R., NEWTON R.S. Hepatic and nonhepatic sterol synthesis and tissue distribution following administration of a liver selective HMG-CoA reductase inhibitor, CI-981: comparison with selected HMG-CoA reductase inhibitors. Biochem. Biophys. Acta. 1992;1123:133–144. doi: 10.1016/0005-2760(92)90103-3. [DOI] [PubMed] [Google Scholar]
- BOCAN T.M.A., MAZUR M.J., MUELLER S.B., BROWN E.Q., SLISKOVIC D.R., O'BRIEN P.M., CRESWELL M.W., LEE H., UHLENDORF P.D., ROTH B.D., NEWTON R.S. Antiatherosclerotic activity of inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase in cholesterol-fed rabbits: a biochemical and morphological evaluation. Atherosclerosis. 1994;111:127–142. doi: 10.1016/0021-9150(94)90198-8. [DOI] [PubMed] [Google Scholar]
- BOGGS K.P., ROCK C.O., JACKOWSKI S. Lysophos-phatidylcholine and 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine inhibit the CDP-choline pathway of phosphatidylcholine synthesis at the CTP: phosphocholine cytidylyltransferase step. J. Biol. Chem. 1995;270:7757–7764. doi: 10.1074/jbc.270.13.7757. [DOI] [PubMed] [Google Scholar]
- BRADFORD M. A rapid sensitive method for the quantitation of microgram quantities of protein utilizing the principles of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CABEZAS M.C., DE-BRUIN T.W., KOCK L.A., KORTLANDT W., VAN-LINDE-SIBENIUS-TRIP M., JANSEN H., ERKELENS D.W. Simvastatin improves chylomicron remnant removal in familial combined hyperlipidemia. Metabolism. 1993;42:497–503. doi: 10.1016/0026-0495(93)90109-2. [DOI] [PubMed] [Google Scholar]
- CASTRO-CABEZAS M., DE-BRUIN T.W., VAN-LINDEN-SIBENIUS-TRIP M., KOCK L.A., JANSEN H., ERKELENS D.W. Lipoprotein (a) plasma concentrations associated with lipolytic activities in eight kindreds with familial combined hyperlipidemia and normolipidemic subjects. Metabolism. 1993;42:756–761. doi: 10.1016/0026-0495(93)90245-j. [DOI] [PubMed] [Google Scholar]
- COLEMAN R.A. Diacylglycerol acyltransferase and monoacylglycerol acyltransferase from liver and intestine. Method. Enzymol. 1992;209:98–101. doi: 10.1016/0076-6879(92)09013-s. [DOI] [PubMed] [Google Scholar]
- CONDE K., VERGARA-JIMENEZ M., KRAUSE B.R., NEWTON R.S., FERNANDEZ M.L. Hypocholesterolemic actions of atorvastatin are associated with alterations on hepatic cholesterol metabolism and lipoprotein composition in the guinea pig. J. Lipid Res. 1996;37:2372–2382. [PubMed] [Google Scholar]
- DAVIGNON J., MONTIGNY M., DUFOUR R. HMG-CoA reductase inhibitors: a look back and a look ahead. Can. J. Pharmacol. 1992;8:843–864. [PubMed] [Google Scholar]
- DIETSCHY J.M., TURLEY S.D., SPADY D.K. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 1993;34:1637–1659. [PubMed] [Google Scholar]
- DIXON J.L., GINSBERG H.N. Regulation of hepatic secretion of apolipoprotein B-containing lipoproteins: information obtained from cultured liver cells. J. Lipid Res. 1993;34:167–179. [PubMed] [Google Scholar]
- GRUNDY S.M. HMG-CoA reductase inhibitors for treatment of hypercholesterolaemia. N. Engl. J. Med. 1988;319:24–32. doi: 10.1056/NEJM198807073190105. [DOI] [PubMed] [Google Scholar]
- HUFF M.W., BURNETT J.R. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors and hepatic apolipoprotein B secretion. Curr. Opin. Lipidol. 1997;8:138–145. doi: 10.1097/00041433-199706000-00003. [DOI] [PubMed] [Google Scholar]
- ISHIDA F., SATO A., LIZUKA Y., KITANI K., SAWASAKI Y., KAMEI T. Effects of MK-733 (simvastatin), an inhibitor of 3-hydroxy-3-methyl-glutaryl coenzyme A reductase on intestinal acylcoenzyme A: cholesterol acyltransferase activity in rabbits. Biochim. Biopys. Acta. 1989;1004:117–123. doi: 10.1016/0005-2760(89)90221-x. [DOI] [PubMed] [Google Scholar]
- JONES P., KAFONEK S., LAURORA I., HUNNINGHAKE D. and , researchers of CURVES study Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin and fluvastatin in patients with hyper cholesterolemia (The Curves Study) Am. J. Cardiol. 1998;81:582–587. doi: 10.1016/s0002-9149(97)00965-x. [DOI] [PubMed] [Google Scholar]
- KHAN B., WILCOX H.G., HEIMBERG M. Cholesterol is required for secretion of very-low density lipoprotein by rat liver. Biochem. J. 1989;259:807–816. doi: 10.1042/bj2580807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHAN B.V., FUNGWE T.V., WILCOX H.G., HEIMBERG M. Cholesterol is required for the secretion of very low density lipoprotein: in vivo studies. Biochim. Biophys. Acta. 1990;1044:297–304. doi: 10.1016/0005-2760(90)90073-7. [DOI] [PubMed] [Google Scholar]
- KOVANEN P.T., BROWN M.S., BASU S.K., BILHEIMER D.W., GOLDSTEIN J.L. Saturation and suppression of hepatic lipoprotein receptor: a mechanism for the hypercholesterolemia of cholesterol-fed rabbits. Proc. Natl. Acad. Sci. U.S.A. 1981;78:1396–1400. doi: 10.1073/pnas.78.3.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEA A.P., MCTAVISH D. Atorvastatin. A review of its pharmacology and therapeutic potential in the management of hyperlipidemias. Drugs. 1997;53:828–847. doi: 10.2165/00003495-199753050-00011. [DOI] [PubMed] [Google Scholar]
- LINSCHEER W.G., ATREYEE B., MURTHY UMA K., JOHN W., SANDOR N., JYOTIRMOY N. Lovastatin induces synthesis of cholesterol, which acts as a secretagogue of biliary phospholipids in rats. Am. J. Physiol. 1995;268:G242–G250. doi: 10.1152/ajpgi.1995.268.2.G242. [DOI] [PubMed] [Google Scholar]
- MA P.T.S., GIL G., SUDHOF T.C., BILHEIMER D.W., GOLDSTEIN J.L., BROWN M.S. Mevinolin, an inhibitor of cholesterol synthesis, induces mRNA for low density lipoprotein receptor in livers of hamsters and rabbits. Proc. Natl. Acad. Sci. USA. 1986;83:8370–8374. doi: 10.1073/pnas.83.21.8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN A., GOMEZ-MUÑOZ A., JAMAL Z., BRINDLEY D.N. Characterization and assay of phosphatidate phosphatase. Methods Enzymol. 1991;197:554–564. doi: 10.1016/0076-6879(91)97183-y. [DOI] [PubMed] [Google Scholar]
- MONTGOMERY M.R., CINTI D.L. Pyridine nucleotide-dependent electron transport in kidney cortex microsomes: interaction between desaturase and other mixed function oxidases. Mol. Pharmacol. 1977;13:60–69. [PubMed] [Google Scholar]
- OWENS D., COLLINS P., JOHNSON A., TIGHE O., ROBINSON K., TOMKIN G.H. Hypercholesterolemia: simvastatin and pravastatin alter cholesterol metabolism by different mechanisms. Biochem. Biophys. Acta. 1991;1127:57–66. doi: 10.1016/0005-2760(91)90206-w. [DOI] [PubMed] [Google Scholar]
- QIN W., INFANTE J., WANG S.-R., INFANTE R. Regulation of HMG-CoA reductase, apoprotein-B and LDL receptor gene expression by the hypocholesterolemic drugs simvastatin and ciprofibrate in Hep G2, human and rat hepatocytes. Biochem. Biophys. Acta. 1992;1127:57–66. doi: 10.1016/0005-2760(92)90201-6. [DOI] [PubMed] [Google Scholar]
- SATO A., WATANABE K., FUKUZUMI H., HASE K., ISHIDA F., KAMEI T. Effect of simvastatin (MK-733) on plasma triacylglycerol levels in rats. Biochem. Pharmacol. 1991;41:1163–1172. doi: 10.1016/0006-2952(91)90654-n. [DOI] [PubMed] [Google Scholar]
- STEIN E.A., LANE M.L., LASKARZEWSKI P. Comparison of statins in hypertriglyceridemia. Am. J. Cardiol. 1998;81:66B–69B. doi: 10.1016/s0002-9149(98)00041-1. [DOI] [PubMed] [Google Scholar]
- THOMPSON G.R., NAUMOVA R.P., WATTS G.F. Role of cholesterol in regulating apolipoprotein B secretion by the liver. J. Lipid Res. 1996;37:439–447. [PubMed] [Google Scholar]
- VAN HEEK M., ZILVERSMIT D.B. Evidence for an inverse relation between plasma triglyceride and aortic cholesterol in the coconut oil/cholesterol fed rabbit. Atherosclerosis. 1988;71:185–192. doi: 10.1016/0021-9150(88)90142-6. [DOI] [PubMed] [Google Scholar]
- VAN HEEK M., ZILVERSMIT D.B. Mechanisms of hypertriglyceridemia in the coconut oil/cholesterol-fed rabbit. Increased secretion and decreased catabolism of very low density lipoprotein. Arterioscl. Thromb. Vasc. Biol. 1991;11:918–927. doi: 10.1161/01.atv.11.4.918. [DOI] [PubMed] [Google Scholar]
- VANCE D. Phosphatidylcholine metabolism: masochistic enzymology, metabolic regulation and lipoprotein assembly. Biochem. Cell Biol. 1990;68:1151–1165. doi: 10.1139/o90-172. [DOI] [PubMed] [Google Scholar]
- WEINHOLD P.A., FELDMAN D.A. Choline-phosphate cytidylyltransferase. Method. Enzymol. 1992;209:248–258. doi: 10.1016/0076-6879(92)09031-w. [DOI] [PubMed] [Google Scholar]
- XU G., SALEN G., SHEFER S., NESS G.C., NGUYEN L.B., PARKER T.S., CHEN T.S., ZHAO Z., DONNELLY T.M., TINT G.S. Unexpected inhibition of cholesterol 7 α-hydroxylase by cholesterol in New Zealand white and Watanabe hyperlipidemic rabbits. J. Clin. Invest. 1995;95:1497–1504. doi: 10.1172/JCI117821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANAGITA T., YAMAMOTO K., ISHIDA S., SONDA K., MORITO F., SAKU K., SAKAI T. Effects of simvastatin, a cholesterol synthesis inhibitor, on phosphatidylcholine synthesis in HepG2 cells. Clin. Ther. 1994;16:200–208. [PubMed] [Google Scholar]
- YOSHINO G., KAZUMI T., KASAMA T., IWAI M., IWATANI I., MATSUBA K., MATSUSHITA M., BABA S. Effect of CS-514 (pravastatin) on VLDL kinetics in rats. Atherosclerosis. 1988;73:191–195. doi: 10.1016/0021-9150(88)90041-x. [DOI] [PubMed] [Google Scholar]