

Abstract
We show that a portion of the TM2 domain regulates the sensitivity of beta subunit-containing rat neuronal nicotinic AChR to the ganglionic blocker mecamylamine, such that the substitution of 4 amino acids of the muscle beta subunit sequence into the neuronal beta4 sequence decreases the potency of mecamylamine by a factor of 200 and eliminates any long-term effects of this drug on receptor function.
The same exchange of sequence that decreases inhibition by mecamylamine produces a comparable potentiation of long-term inhibition by nicotine.
Inhibition by mecamylamine is voltage-dependent, suggesting a direct interaction of mecamylamine with sequence elements within the membrane field. We have previously shown that sensitivity to TMP (tetramethylpiperidine) inhibitors is controlled by the same sequence elements that determine mecamylamine sensitivity. However, inhibition by bis-TMP compounds is independent of voltage.
Our experiments did not show any influence of voltage on the inhibition of chimeric receptors by nicotine, suggesting that the inhibitory effects of nicotine are mediated by binding to a site outside the membrane's electric field.
An analysis of point mutations indicates that the residues at the 6′ position within the beta subunit TM2 domain may be important for determining the effects of both mecamylamine and nicotine in a reciprocal manner. Single mutations at the 10′ position are not sufficient to produce effects, but 6′ 10′ double mutants show more effect than do the 6′ single mutants.
Keywords: Voltage-clamp, Xenopus oocytes, voltage-dependence, ganglionic blockers, nicotinic AChR, mecamylamine
Introduction
The nicotinic acetylcholine receptor (AChR) of the neuromuscular junction remains the prototype for all synaptic transmitter-gated ion channels. However, the majority of the effects of smoking dosages of nicotine arise from the activation and/or subsequent inhibition (desensitization) of receptors on peripheral and central neurons which are homologous to the muscle-type receptor. Genes cloned from the nervous system, coding for proteins that are related to the subunits of the neuromuscular nicotinic AChR, include eight proteins designated as alpha subunits (α2–α9) and three non-alpha subunits, designated beta subunits (β2–β4) (for review see Papke, 1993). Nicotinic receptors in the nervous system which show high affinity binding of nicotine require the presence of at least one type of neuronal beta-subunit along with at least one type of neuronal alpha subunit. In these beta subunit-containing receptors, the agonist binding sites are believed to be at the interface between the alpha and beta subunits, since both alpha and beta subunits influence the sensitivity to agonists and antagonists (Hussy et al., 1994; Luetje & Patrick, 1991; Luetje et al., 1990; Papke & Heinemann, 1994).
The predominant alpha subunit in the peripheral nervous system is α3. When the α3 subunit is co-expressed in Xenopus oocytes with β4, the major beta subunit of the peripheral nervous system, functional receptors are formed which may be taken to be at least a partial model for the receptors of autonomic ganglia (however, see also Conroy et al., 1992; Corriveau & Berg, 1993; Vernallis et al., 1993).
This study provides insights into the structural basis for inhibitory effects of both nicotine and the ganglionic antagonist mecamylamine on α3-containing AChR. Mecamylamine is generally thought of as a noncompetitive antagonist (Bertrand et al., 1990; Francis & Papke, 1996; Stone et al., 1956; Varanda et al., 1985) and as such would produce inhibition by binding to sites other than the agonist activation sites. Nicotine on the other hand, is an agonist and may produce inhibition via enhanced desensitization, or alternatively, the inhibitory effects of nicotine may also arise from binding to sites similar or identical to those bound by noncompetitive inhibitors.
Noncompetitive inhibition can be classified as either steric in nature, where the ligand directly blocks the conduction path, or allosteric, where the binding of the inhibitory ligand promotes the transition to, or stabilization of, non-conducting states. Steric inhibition is often referred to as open channel block, and such inhibition may be promoted by membrane hyperpolarization (i.e. voltage-dependent) if the steric binding site is within the membrane's electric field. The effects of a noncompetitive inhibitor may be use-dependent for the onset of inhibition (i.e. require open channels) and then inhibition may persist if the ligand remains bound to the receptor.
Desensitization can be defined as a decrease in response that occurs in the continued presence of an agent that initially activates the receptor. It may be argued that true desensitization is promoted only by the binding of agonist at the same sites which promote activation (Katz & Thesleff, 1957). Such desensitization occurs as a first-order process(s) with the rate constants for the conversion to desensitized states being faster for channels that have bound agonist and/or have opened. This gives the desensitization process an apparent concentration dependence. However, the interpretation of data obtained in the continued presence of agonist is complicated by the fact that agonists are known to bind to multiple sites on the receptor and decrease response via channel block or allosteric effects (Hussy et al., 1994; Rozental et al., 1989; Sine & Steinbach, 1984). Moreover, desensitization and noncompetitive inhibition are tied together, at least in the sense that noncompetitive inhibitors have been documented to promote the same sort of high affinity for agonists that is associated with desensitization (Changeux et al., 1987). We have previously described how such noncompetitive inhibition by agonist can limit the effectiveness of nicotine and other agonists for the activation of α4β2 receptors, the predominant beta-subunit-containing receptor of the brain (Papke et al., 1997).
In the present study we examine inhibition produced by the agonist nicotine and the ganglionic blocker mecamylamine. We determine the relative importance of sequence elements within the ion permeation pathway, and whether inhibition is affected by transmembrane voltage. Our results suggest that the pore-forming domain of the receptor is essential for determining sensitivity of nicotinic AChR to multiple and diverse forms of receptor inhibition.
Methods
Design of chimeric subunits
Hydrophobicity analysis and sequence comparison of the nicotinic subunits suggests that they all have similar basic structures, with a large amino-terminal extracellular domain containing the primary ligand binding sites followed by three hydrophobic domains, the primary intracellular domain, and a fourth transmembrane domain located downstream of the large intracellular segment, so that the carboxy-terminus of the protein is extracellular. Many lines of evidence have converged to indicate that the second transmembrane domain forms the ion channel (Changeux et al., 1987; Charnet et al., 1990; Cohen et al., 1992; Dennis et al., 1986; Giraudat et al., 1986; Labarca et al., 1995; Leonard et al., 1988; Revah et al., 1991). This sequence of 20 amino acids (see below) contains essential elements to regulate the conductance, selectivity and desensitization of the channel. The TM2 (second transmembrane) domain of the muscle receptor subunits has been shown to also contain the binding site for local anaesthetics and other non-competitive inhibitors. While much of the TM2 domain is likely to be within the membrane's electric field, we have recently shown (Francis et al., 1998) that sequence elements deep within the TM2 domain also control the accessibility of sites outside the membrane's electric field to the slowly dissociating ganglionic blocker bis-tetramethyl-piperidinyl sebacate (BTMPS).
The sequences of the TM2 domains of the relevant subunits are shown below. Adopting the terminology proposed by Leonard et al. (1988), the 20 residues in the putative transmembrane sequence are identified as 1′–20′. Initial experiments were conducted with wild-type subunits and two chimeric subunits designated β4 (β1TM2) and β1 (β4TM2). These subunits have been previously characterized (Francis et al., 1998) and contain reciprocal exchanges of 8 amino acids from positions 4′–11′ (inclusive) in the TM2, shown in underline below. This amounts to total differences of only 4 amino acids (at sites 4′, 6′, 7′ and 10′) between the wild-type and chimeric proteins.
 |
For our experiments we used the rat cDNA clones for the neuronal receptors and the mouse muscle cDNA clones (Heinemann et al., 1986). It is interesting to note that not only is the TM2 amino acid sequence 100% identical in the rat neuronal β2 and β4 subunits, but the TM2 amino acid sequences are 100% conserved between the rodent beta subunits and their human homologues.
Production of TM2 chimeras and sequencing
Chimeric genes were constructed by the method of overlap extension PCR (polymerase chain reaction) (Horton et al., 1989). Clones were evaluated by both restriction analysis and sequencing through the PCR-generated region by the dideoxy chain termination method (Sanger et al., 1977) using the Sequenase 2.0 kit from United States Biochemical Corporation (Cleveland, OH, U.S.A.).
Construction of site-directed mutants
Site-directed mutagenesis was conducted with QuickChange™ kits (Stratagene, LaJolla, CA, U.S.A.). In brief, two complimentary oligonucleotides were synthesized which contain the desired mutation flanked by 10–15 bases of unmodified nucleotide sequence. Using a thermal cycler, Pfu DNA polymerase extended the sequence around the whole vector, generating a plasmid with staggered nicks. Each cycle built only off the parent strands, therefore there was no amplification of misincorporations. After 12–16 cycles, the product was treated with DpnI, which digested the methylated parent DNA into numerous small pieces. The product was then transformed into E. coli cells, which repaired the nicks.
Chemicals
Fresh acetylcholine (Sigma; St. Louis, MO, U.S.A.) stock solutions were made daily in Ringer's solution and diluted. Mecamylamine (N-2,3,3-tetramethylbicyclo[2.2.1]heptan-2-amine), (−)-Nicotine ([−]-1-methyl-2-[3-pyridyl]-pyrrolidine), and all other chemicals for electrophysiology were obtained from Sigma Chemical Co. (St. Louis MO, U.S.A.).
Preparation of RNA and expression in Xenopus oocytes
Mature (>9 cm) female Xenopus laevis African toads (Nasco, Ft. Atkinson, WI, U.S.A.) were used as a source of oocytes. Prior to surgery, frogs were anaesthetized by placing the animal in a 2 g l−1 solution of MS222 (3-aminobenzoic acid ethyl ester). Eggs were removed from an incision made in the abdomen. Prior to the ligation of the ovarian tissue, the ovaries were sutured with 4–0 silk. The incisions were disinfected with gentamicin, sutured with 4–0 gut, and the animals were allowed to recover from the anaesthesia in a humid environment. Postoperative, animals were kept in isolation tanks and checked daily before being returned to the colony. No more than four survival surgeries were performed on any animal. After the final surgery, animals were again anaesthetized in MS222 and euthanasia was accomplished by decapitation. Animal protocols are annually reviewed and approved by the university's Animal Use Committee.
After linearization and purification of cloned cDNAs, RNA transcripts were prepared in vitro using the appropriate mMessage mMachine kit from Ambion Inc. (Austin, TX, U.S.A.). Harvested oocytes were treated with collagenase from Worthington Biochemical Corporation (Freehold, NJ, U.S.A.) for 2 h at room temperature in calcium-free Barth's solution (in mM NaCl 88, HEPES pH 7.6 10, MgS04 0.33, 0.1 mg−1 ml gentamicin sulphate). Subsequently, stage five oocytes were isolated and injected with 50 nl each of a mixture of the appropriate subunit cRNAs following harvest. Recordings were made 1–7 days after injection depending on the cRNAs being tested.
Electrophysiology
Oocyte recordings were made with a Warner Instruments (Hamden, CT, U.S.A.) OC-725C oocyte amplifier and RC-8 recording chamber interfaced to a Macintosh personal computer. Data were acquired using Labview software (National Instruments) and filtered at a rate of 6 Hz. Oocytes were placed in a Warner recording chamber with a total volume of about 0.6 ml and perfused at room temperature by frog Ringer's solution (mM) NaCl 115, KCl 2.5, HEPES pH 7.3 10, CaCl2 1.8) containing 1 μM atropine to inhibit potential muscarinic responses. A Mariotte flask filled with Ringer's solution was used to maintain a constant hydrostatic pressure for drug deliveries and washes. Drugs were diluted in perfusion solution and loaded into a 2 ml loop at the terminus of the perfusion line. A bypass of the drug-loading loop allowed bath solution to flow continuously while the drug loop was loaded, and then drug application was synchronized with data acquisition by using a 2-way electronic valve. The rate of bath solution exchange and all drug applications was 6 ml min−1. Current electrodes were filled with a solution containing (mM) CsCl 250, CsF 250 and EGTA 100 and had resistances of 0.5–2 MΩ. Voltage electrodes were filled with 3 M KCl and had resistances of 1–3 MΩ. Oocytes with resting membrane potentials more positive than −30 mV were not used.
Experimental protocols and data analysis
Current responses to drug application were studied under two-electrode voltage clamp at a holding potential of −50 mV unless otherwise noted. Holding currents immediately prior to agonist application were subtracted from measurements of the peak response to agonist. All drug applications were separated by a wash period of 5 min unless otherwise noted. At the start of recording, all oocytes received two initial control applications of ACh. Subsequent drug applications were normalized to the second ACh application in order to control for the level of channel expression in each oocyte. The second application of control ACh was used to minimize the affects of rundown that occasionally occurred after the initial ACh-evoked response. In order to measure residual inhibitory effects, an experimental application of ACh with inhibitor or of inhibitor alone was followed by a second application of ACh alone and compared to the pre-application control ACh response. Means and standard errors (s.e.mean) were calculated from the normalized responses of at least four oocytes for each experimental concentration.
For each of the receptor subtypes tested, a control ACh concentration was selected that was sufficient to stimulate the receptors to a level representing a reasonably high value of popen at the peak of the response while minimizing rundown with successive ACh applications. For potent use-dependent inhibitors, we have found that such conditions are adequate to achieve maximal inhibition (Francis & Papke, 1996; Papke et al., 1994). The control ACh concentration used for the α3-containing receptors was 100 μM. We noted that the ACh EC50 for the α3β4(β1TM2) receptors (≈75 μM) was somewhat reduced compared to α3β4 receptors (EC50≈50 μM). This was presumably because at higher ACh concentrations, the inhibitory effects of ACh itself (see results) limited the maximum response obtainable, shifting the concentration response curve to the left. Specific concentrations for other receptor subtypes are as noted in the figures.
For concentration-response relations, data were plotted using Kaleidagraph 3.0.2 (Abelbeck Software; Reading, PA, U.S.A.), and curves were generated from the Hill equation (Luetje & Patrick, 1991):
where Imax denotes the maximal response for a particular agonist/subunit combination, and n represents the Hill coefficient. Imax, n, and the EC50 were all unconstrained for the fitting procedures. Negative Hill slopes were applied for the calculation of IC50 values.
For experiments assessing voltage-dependence of inhibition, oocytes were initially voltage-clamped at a holding potential of −50 mV, and a control application of ACh alone was delivered. The holding potential was stepped to the designated voltage for 30–60 s prior to either co-application of ACh with mecamylamine or the application of nicotine alone. Thirty to sixty seconds after the peak of the co-application response, voltage was stepped back to −50 mV, and residual inhibition was evaluated with a subsequent application of ACh alone after a 5 min wash period. The e-folding voltages were calculated by the method of Woodhull (1973), from the exponential fits to the value Z-1 against voltage, where Z=control response divided by the response after the application of agonist and inhibitor.
Results
The sensitivity of neuronal nicotinic AChR to long term inhibition by mecamylamine can be regulated by sequence in the beta subunit transmembrane domain
The co-application of mecamylamine with 100 μM ACh produced a potent inhibition of α3β4 responses (Figure 1A), with an IC50 of approximately 150 nM. In contrast, receptors containing the β4(β1TM2) chimeric subunit were 200 fold less sensitive to inhibition by mecamylamine (IC50≈30 μM, Figure 1A). In addition to lowering the potency of mecamylamine, the substitution of the muscle beta subunit sequence into β4 also produced a qualitative change in the nature of the inhibition. Specifically, a residual inhibition of α3β4 receptors was observed after the application of mecamylamine, while α3β4(β1TM2) receptors recovered fully following a 5 min wash period. The after-effects of mecamylamine are shown in Figure 1B, which shows the amplitude of control responses to ACh alone following the co-application of ACh and mecamylamine. The IC50 for this form of inhibition of α3β4 receptors was approximately 1 μM. This persistent inhibition was use dependent, such that when 1 μM mecamylamine was applied in the absence of agonist, post-application ACh controls were 98±5% (n=4) of the pre-application controls.
Figure 1.
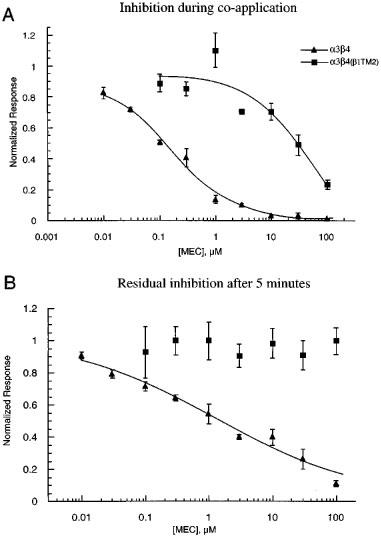
Concentration-response curves for the effect of mecamylamine on α3β4 and α3β4(β1TM2) receptors. (A) The concentration response relationship for mecamylamine's inhibition of the peak currents of α3β4 and α3β4(β1TM2) receptors when 100 μM ACh was co-applied with mecamylamine at the indicated concentrations. Data from each oocyte was normalized to that cell's response to 100 μM ACh alone. (B) Mecamylamine's residual effects on ACh responses of α3β4 and α3β4(β1TM2) receptors. The data plotted represent the responses to 100 μM ACh when it was applied 5 min after 100 μM ACh had been co-applied with mecamylamine at the indicated concentrations. Data were normalized to each cell's prior control response to 100 μM ACh alone. Each point represents the average normalized response of 4–7 cells (±s.e.mean).
The representative traces in Figure 2 illustrate the greater potency of mecamylamine for the inhibition of α3β4 receptors compared to α3β4(β1TM2) receptors. While 300 nM mecamylamine produced a greater than 50% inhibition of the α3β4 peak response, a 100 fold higher concentration of mecamylamine was required to produce similar inhibition of α3β4(β1TM2) receptors. The inserts in Figure 2 show the control and inhibited responses scaled to the same amplitude. Mecamylamine produced an accelerated time to peak in the α3β4 receptor responses (Table 1), consistent with use-dependent inhibition. Note that the ACh control responses of α3β4(β1TM2) receptors were accelerated compared to those obtained in α3β4 receptors (Figure 2 and Table 1). The responses of α3β4(β1TM2) receptors to mecamylamine co-application were not significantly faster than their respective controls. However, with the α3β4(β1TM2) receptors, but not the α3β4 receptors, a small relaxation out of an inhibited state could be seen as the drug concentrations fell in the chamber (arrow in Figure 2).
Figure 2.
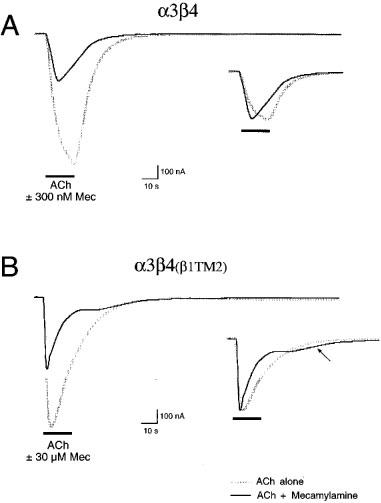
Representative traces illustrating the effect of mecamylamine on α3β4 and α3β4(β1TM2) receptors. (A) The main figure shows an initial control response of an α3β4 -expressing oocyte to the application of 100 μM ACh and the diminished response when 100 μM ACh was co-applied with 300 nM mecamylamine. The insert shows the control and experimental responses scaled to the same amplitude to illustrate the accelerated peak of the mecamylamine-inhibited current and the lack of any late-phase or relaxation current in the presence of mecamylamine. (B) The main figure shows an initial control response of an α3β4(β1TM2) expressing oocyte to the application of 100 μM ACh and the diminished response when 100 μM ACh was co-applied with 30 μM mecamylamine. The insert shows the responses scaled to the same amplitude to illustrate the accelerated peak of the mecamylamine-inhibited current and the presence of a small late-phase or relaxation current as the mecamylamine is washed from the chamber (arrow).
Table 1.
Time to the peak of the current response (in seconds)
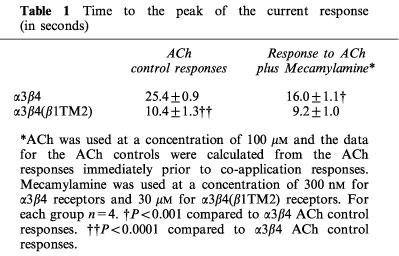
The rate of recovery of α3β4 receptors from the inhibition produced by the co-application of 100 μM ACh and 10 μM mecamylamine was determined by repeatedly administering 100 μM ACh at 5 min intervals (not shown). The data were fit by an exponential function with a time constant for recovery of approximately 16 min. Although the induction of this form of inhibition was use dependent (see above), it did not appear that recovery could be enhanced by repeated agonist application. There was no significant difference in the amount of recovery over a 20 min period between cells that received four ACh applications at 5 min intervals and cells that received a single ACh application after a 20 min wash (data not shown).
Addition of neuronal β4TM2 sequence to muscle α1β1γδ combinations promotes inhibition by mecamylamine
Compared to neuronal nicotinic AChR, muscle-type nicotinic AChR are relatively insensitive to mecamylamine. As shown in Figure 3, the average response to the co-application of 1 μM mecamylamine with 10 μM ACh to α1β1γδ receptors was 83±6% of the amplitude of the control responses to 10 μM ACh alone. In contrast, the average response to the co-application of 1 μM mecamylamine with 10 μM ACh to α1β1(β4TM2)γδ receptors were reduced to 53±3% of the ACh controls, suggesting a nearly 3 fold increase in inhibition associated with the presence of the β4TM2 domain. After a 5 min washout of mecamylamine, control responses of α1β1γδ receptors were within 5% of the original control amplitude, and the responses of α1β1(β4TM2)γδ receptors had recovered to 83±6% of the control values.
Figure 3.
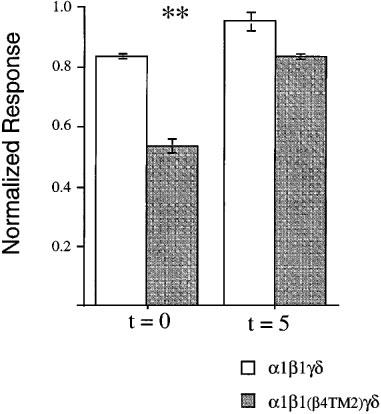
Mecamylamine effects on α1β1γδ and α1β1(β4TM2)γδ receptors. The effects of 1 μM mecamylamine, co-applied with 10 μM ACh were measured during the co-application response (t=0) and for a subsequent response to 10 μM ACh alone (t=5). Experimental responses were normalized to the cells' initial control ACh responses. The co-application of 1 μM mecamylamine produced a significant decrease in the responses of α1β1(β4TM2)γδ receptors (P<0.01). Each bar represents the average normalized response of four cells (±s.e.mean).
The muscle β1TM2 can regulate the sensitivity of neuronal receptors to prolonged inhibition by nicotine
Nicotine concentration-response experiments with α3β4 receptors showed maximal peak currents that were less than could be obtained with ACh (Figure 4), suggesting that nicotine may only be a partial agonist for these receptors (Hussy et al., 1994). However, our data also suggest that maximal responses to nicotine might be limited by a secondary antagonist activity, such as has been previously described for α4β2 receptors (de Fiebre et al., 1995). At concentrations above 100 μM, responses to nicotine showed accelerated times to peak compared to the ACh control responses, and clearly evident relaxation from inhibition was seen when the nicotine was washed from the chamber (Figure 5A). Due to this fast washout of inhibitory effects, nicotine produced very little residual inhibition of α3β4 receptors (Figure 4B).
Figure 4.
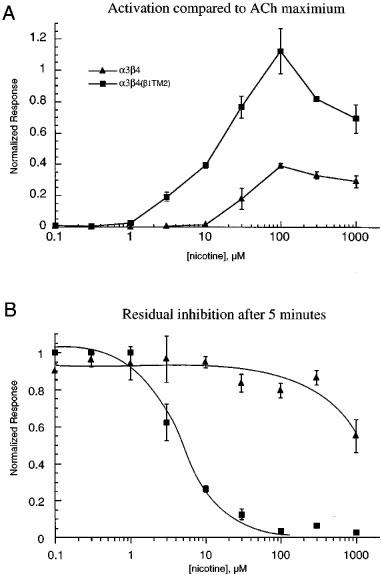
Concentration-response curves for the activation and residual inhibition of α3β4 and α3β4(β1TM2) receptors by nicotine. (A) The concentration-response relationship for nicotine's activation of currents in cells expressing α3β4 or α3β4(β1TM2) receptors. Data from each oocyte were first normalized to that cell's response to 100 μM ACh alone and then adjusted for the ratio between the 100 μM ACh response and the ACh maximum, as measured in separate experiments (not shown) and as previously reported (Papke et al., 1997). (B) Nicotine's residual effects on ACh responses of α3β4 and α3β4(β1TM2) receptors. The data plotted represent the responses to 100 μM ACh when it was applied 5 min after nicotine had been applied at the indicated concentrations. Data were normalized to each cell's prior control response to 100 μM ACh alone. Each point represents the average normalized response of 4–7 cells (±s.e.mean).
Figure 5.
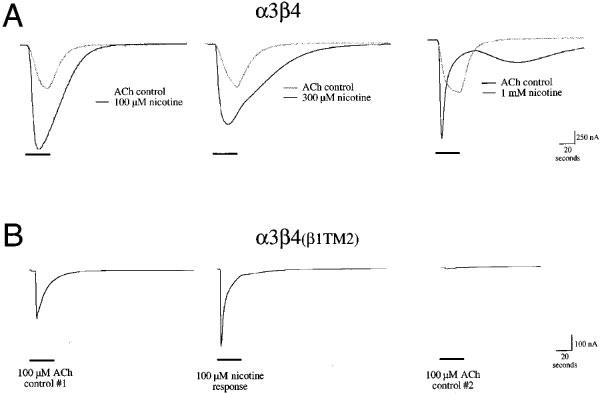
Representative traces illustrating the effect of nicotine on α3β4 and α3β4(β1TM2) receptors. (A) Sample responses of α3β4 expressing cells to the application of nicotine at the indicated concentration, shown along with respective control ACh responses obtained 5 min earlier from the same cells. Note that at concentrations above 100 μM, nicotine fails to produce further increases in peak currents compared to ACh controls. However, nicotine does produce a protracted or late-phase current that can be associated with secondary inhibitory activity by this compound. Recovery from this inhibition is relatively rapid, as can be seen from the relaxation out of the inhibited state, clearly visible after the application of 1 mM nicotine. (B) The responses of an α3β4(β1TM2) receptor-expressing oocyte to the initial control application of 100 μM ACh (left-most trace), the application of 100 μM nicotine (middle trace), and a subsequent 100 μM ACh control response (right-most trace).
When nicotine was applied to α3β4(β1TM2) receptors, there was also an accelerated time to peak, even when nicotine was applied at relatively low concentrations. However, with this receptor subtype no relaxation from the inhibition was observed. In fact, the application of nicotine to α3β4(β1TM2) receptors produced profound inhibition that could be measured as decreases in the response to control ACh applications after a 5 min wash period (Figure 4B). The IC50 for the residual inhibition of α3β4(β1TM2) receptors by nicotine was approximately 5 μM, while there was less than 50% residual inhibition of α3β4 receptors following the application of 1 mM nicotine (the highest concentration tested).
The rate of recovery of α3β4(β1TM2) receptors from the inhibition produced by the application of 30 μM nicotine was determined by repeatedly administering 100 μM ACh at 5 min intervals (not shown). The data was fit by an exponential function with a time constant for recovery of approximately 51 min.
It is interesting to note that channel block by acetylcholine itself was apparently also enhanced in α3β4(β1TM2) receptors compared to the wild-type α3β4. This is suggested by the consistent observation that the peak responses of the α3β4(β1TM2) receptors to control applications of ACh were accelerated compared to responses of wild-type α3β4 receptors (Table 1). Note in Figures 2 and 5 that the peak in the response of α3β4(β1TM2) receptors to the application of ACh occurred before solution exchange was completed. An increased inhibitory effect of ACh might also explain why the relative efficacy of nicotine compared to ACh appears increased in the α3β4(β1TM2) receptors (Figure 4A), such that compared to ACh, nicotine appears to be a full agonist for these receptors. When ACh was applied to α3β4(β1TM2) receptors at concentrations greater than 100 μM, subsequent ACh control responses were also decreased. The inhibition produced by the application of high (1 mM) ACh concentrations showed a time constant recovery of approximately 7 min (not shown).
The regulatory effects of TM2 sequence on receptor inhibition can be generalized to other neuronal receptor combinations
In order to determine whether the reciprocal effects of the beta subunit TM2 domain could be generalized to neuronal nicotinic AChR containing other alpha subunits, we co-expressed the α2 subunit with either β4 or β4(β1TM2). As shown in Figure 6, 1 μM mecamylamine produced a strong inhibition of α2β4 receptors but not α2β4(β1TM2) receptors. Likewise, 5 min after the application of 100 μM nicotine, α2β4 receptors showed very little residual inhibition of control ACh responses, while the responses of α2β4(β1TM2) receptors were reduced to only 10% control levels, suggesting that they remained inhibited by approximately 90%. We were not able to obtain functional receptors from the co-expression of α4 with the β4(β1TM2) chimera. However, since the α4 and α2 subunits have identical sequence through the entire TM2 domain, it is likely that this important effect of the beta subunit TM2 in regulating sensitivity to mecamylamine and nicotine may be generalized for all beta subunit-containing nicotinic AChR.
Figure 6.
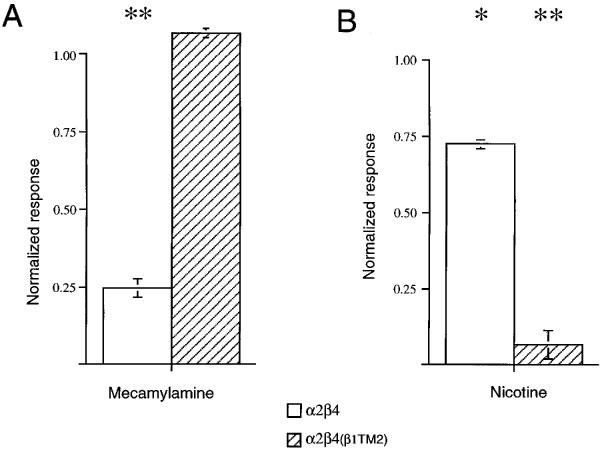
The effects of mecamylamine and nicotine on α2β4 and α2β4(β1TM2) receptors. (A) The effects of 1 μM mecamylamine co-applied with 30 μM ACh on α2β4 and α2β4(β1TM2) receptors. Mecamylamine caused a strong inhibition of α2β3 receptor mediated-responses (P<0.001), while there was no significant effect of mecamylamine on α2β4(β1TM2) receptor mediated-responses. (B) Control ACh responses measured 5 min after the application of 100 μM nicotine to α2β4 and α2β4(β1TM2) receptors. Nicotine caused a large inhibition of α2β4(β1TM2) receptor responses (P<0.001) and a small decrease in α2β4 responses (P<0.05). Each bar represents the average normalized responses of 4–7 cells (±s.e.mean).
The voltage dependence of inhibition by nicotine and mecamylamine
The requirement for channel activation prior to the onset of noncompetitive (i.e. use-dependent) inhibition confounds the estimation of the inhibitory effectiveness of mecamylamine when it is initially applied. Therefore, the measurement of residual inhibition may be taken as a more accurate estimate of the degree of drug equilibration during the complete course of the co-application. For that reason, we chose to measure the extent to which this residual inhibition was influenced by the holding potential used when either mecamylamine or nicotine was applied. This measurement of the suppression of subsequent control responses had the additional advantage that the equivalent measurement could be made following the administration of nicotine to α3β4(β1TM2) receptors, giving an indication of inhibitory activity separate from the agonist activity of the drug.
When 300 nM mecamylamine was co-applied with ACh at a holding potential of 0 to −20 mV, subsequent control responses to ACh applied at the standard holding potential of −50 mV were suppressed by less than 20%. However, when 300 nM mecamylamine was co-applied with ACh at a holding potential of −60 to −80 mV, subsequent control responses to ACh applied at the standard holding potential of −50 mV were suppressed by more than 50%. This is in contrast to our previously reported observation that inhibition of α3β4 receptors by the bis-tetramethylpiperidine compound BTMPS was equally effective at both positive and negative holding potentials (Francis et al., 1998).
In order to quantify the effect of voltage on inhibition by mecamylamine and nicotine, the data were transformed by the method of Woodhull (1973, see Methods). Inhibition by mecamylamine showed an e-fold increase for a 42 mV change in voltage, suggesting that the binding site for mecamylamine would be approximately 60% into the membrane's electric field. In contrast to this clear voltage dependence for mecamylamine, there was no apparent effect of voltage on the inhibition produced by nicotine, suggesting that like BTMPS, it may be binding at sites external to the membrane's electric field (Figure 7).
Figure 7.
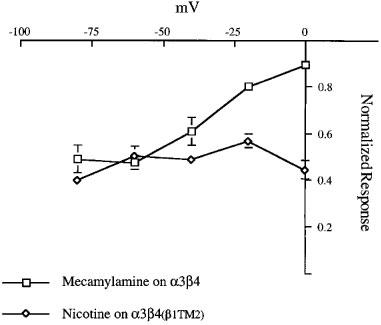
The voltage dependence of inhibition by mecamylamine and nicotine measured 5 min after application. The plot shows the amplitude of responses to control ACh applications when ACh was delivered 5 min after the application of drugs at the indicated voltages. Responses were normalized to the amplitude of the control response obtained 5 min before the application of the drugs. Mecamylamine (300 nM) was co-applied with 100 μM ACh to α3β4 expressing cells and 10 μM nicotine was applied to α3β4(β1TM2)-expressing cells. Mecamylamine and nicotine were applied at the indicated voltages, and the pre- and post-drug application control measurements were both made at the standard holding potential of −50 mV. Each point represents the average normalized response of 4–5 cells (±s.e.mean).
Analysis of point mutations in TM2
The β1 and β4 subunits differ at only four amino acids within the sequence exchanged in our chimeras, the 4′, 6′, 7′, and 10′ TM2 residues. Therefore, we made point mutations which exchanged sequence at these locations. As shown in Figure 1, compared to α3β4 wild-type receptors, α3β4(β1TM2) receptors show reduced sensitivity to mecamylamine as measured both at time of co-application and after a 5 min wash period. When the α3 subunit was co-expressed with either the β4(4′S), β4(7′A), or β4(10′T) mutants, the pattern of inhibition generally resembled that of α3β4 wild-type (Figure 8A). When α3 was co-expressed with β4(6′F), there was a trend toward decreased inhibition. However the responses of this receptor subtype were quite variable, and in fact, no significant differences were determined when compared to either wild-type or the full chimera. This would suggest that the 6′ serine maybe important for long term inhibition by mecamylamine but that this modification alone is not sufficient to reproduce the effect seen with the TM2 chimera.
Figure 8.
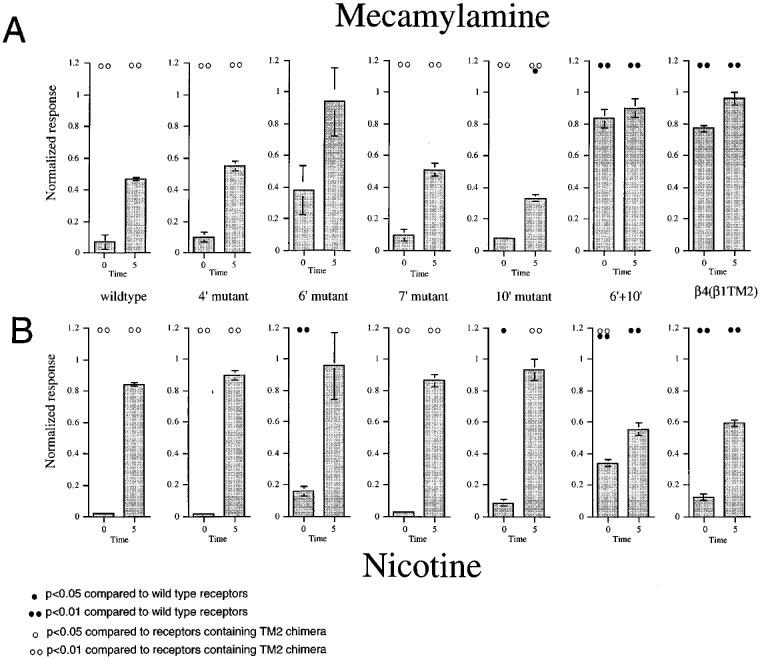
Analysis of point mutations. (A) The effects of 3 μM mecamylamine, co-applied with 100 μM ACh, to receptors containing α3 in combination with wild-type or mutant β4 subunits. (B) The effects of 10 μM nicotine applied to receptors containing α3 in combination with wild-type or mutant β4 subunits. For both (A) and (B), going from left to right, the beta subunits were wild-type β4 , β4 (4′S), β4 (6′F), β4 (7′A), β4 (10′T), β4 (6′F10′T), and β4(β1TM2). Note that the X axis label on the left-most graph is applicable for all of the plots. The zero time points represents the response during the drug application normalized to an initial control ACh response in the same cell. The 5 min bars represent the responses to control (100 μM) ACh applications obtained 5 min after the drug treatments, normalized to the earlier control response. Each bar represents the average normalized response of four cells (±s.e.mean). Statistical differences were determined based on pairwise t-tests between the responses of mutant receptors compared to wild-type receptors and receptors containing TM2 chimeric beta subunits, as indicated.
It has been previously reported that drugs that bind within the channel may interact with multiple levels of the transmembrane helix (Charnet et al., 1990). The 10′ residue is predicted to line the channel at the helical turn above the 6′ residue, and while a partial effect was suggested for the 6′ mutant, no significant reversal of mecamylamine sensitivity was seen when the 10′ residue was exchanged between the neuronal and muscle sequences. In fact, the 10′ mutant showed a slight enhancement of the inhibition measured at 5 min when compared to control (P<0.05).
To investigate the potential interactions between residues at the 6′ and 10′ sites, we made a double mutant β4(6′F10′T). Our data indicate that in fact the 10′ threonine substitution did enhance the effect obtained with the 6′ phenylalanine substitution (Figure 8), such that the double mutant showed diminished sensitivity both during the co-application response and as measured after a 5 min wash period (Figure 8) and was not statistically distinguishable from the full TM2 chimera.
The same series of mutants were examined for their sensitivity to nicotine as an agonist and as an antagonist. As shown in Figure 4, the substitution of the β1TM2 domain into β4 increased both the agonist and antagonist effects of a 10 μM nicotine application. As shown in Figure 8B, the 6′ serine to phenylalanine mutation increased the apparent agonist activity of nicotine (P<.01, pairwise t-test compared to wild-type receptors) but did not produce increased antagonism. The 10′ single mutation also had a slight effect on nicotine's agonist activity (compared to ACh) but no significant effect on residual inhibition compared to wild-type receptors. The 6′F10′T double mutation, however, produced significant increases in both agonist and antagonist activity compared to wild-type (P<0.01). Moreover, at the concentration tested, nicotine had an apparent agonist activity on the 6′F10′T double mutant that was also significantly greater than that seen with the full TM2 chimera (Figure 8).
We have previously reported that the beta subunit TM2 domain regulates the long term inhibition of neuronal nicotinic AChR produced by the use-dependent antagonist, BTMPS (Francis et al., 1998). A comparison of the responses obtained from wild-type and TM2 chimeric receptors (Figure 9) illustrates this effect. Note that since the effects of BTMPS are use-dependent, co-application responses may in fact be larger than those recorded 5 min later (see for example, α3β4 wild-type). This is because channels must begin to open before they can be inhibited, so it is typical to see a brief spike in response to co-application (Papke et al., 1994). As shown in Figure 9, mutations at the 6′ position in either β1 or β4 are essential for determining the difference in sensitivity to BTMPS that exists between wild-type receptors and receptors containing TM2 chimeras. The data also suggest that substitution at the 10′ site can further enhance the effect of the 6′ mutation (Figure 9).
Figure 9.
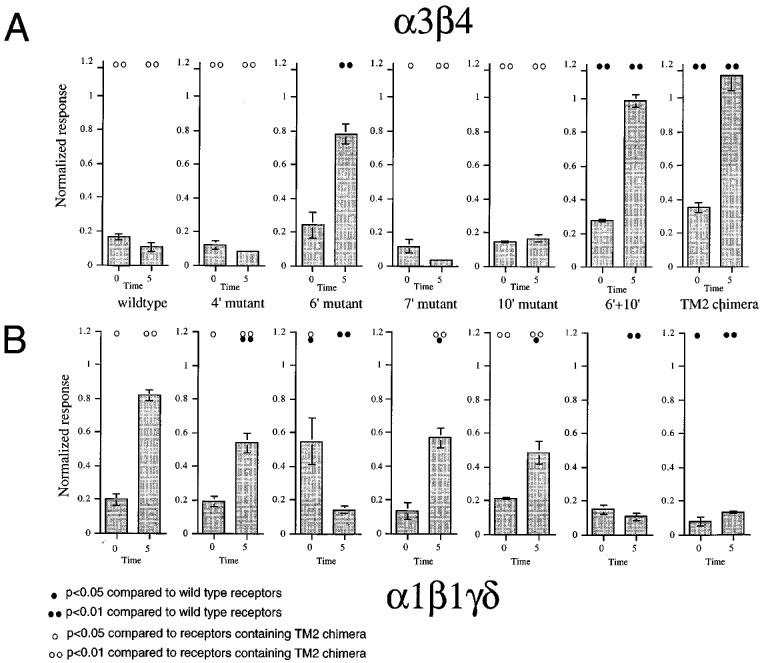
Point mutations regulate sensitivity to BTMPS. (A) The effects of 2 μM BTMPS, co-applied with 100 μM ACh, to receptors containing α3 in combination with wild-type or mutant β4 subunits. Going from left to right, the beta subunits were wild-type β4, β4 (4′S), β4 (6′F), β4 (7′A), β4 (10′T), β4 (6′F10′T), and β4(β1TM2). (B) The effects of 2 μM BTMPS, co-applied with 10 μM ACh, to receptors containing α1γδ in combination with wild-type or mutant β1 subunits. Going from left to right, the beta subunits were wild-type β1, β1 (4′C), β1 (6′S), β1 (7′V), β1 (10′A), β1 (6′S10′A), and β1(β4TM2). Note that the X axis label on the left-most graph is applicable for all of the plots. For both (A) and (B) the responses at the zero time point represents the drug application normalized to an initial control ACh response in the same cell. The 5 min bars represent the responses to control (100 μM) ACh applications obtained 5 min after the drug treatments, normalized to the earlier control response. Each bar represents the average normalized response of 4–6 cells (±s.e.mean). Statistical differences were determined based on pairwise t-tests between the responses of mutant receptors compared to wild-type receptors and receptors containing TM2 chimeric beta subunits, as indicated.
Discussion
Due to its ability to block nicotinic activity in the brain with systemic administration, mecamylamine remains probably the most commonly used of all neuronal nicotinic AChR-specific inhibitors. Nonetheless, previous reports of mecamylamine's inhibitory activity in vitro have failed to reach a consensus for explaining this drug's mechanism of action. Mecamylamine was found to produce a voltage-dependent inhibition of rat cardiac parasympathetic ganglion nicotinic AChR with an IC50 of 37 nM measured at −120 mV (Fieber & Adams, 1991). However, while Ascher et al. (1979) also found that the inhibition of parasympathetic neurons of the rat submandibular ganglion by mecamylamine showed a measurable voltage dependence, consistent with channel blocking activity, they further reported that inhibition was lessened when the drug was co-applied at high agonist concentrations. This observation was more consistent with a competitive mechanism for inhibition. Bertrand et al. (1990) reported that the effects of mecamylamine on heterologously expressed chick α4β2 receptors were persistent after the drug was co-applied with agonist, while application of the drug alone had no effect. In those experiments the inhibition measured during co-application with ACh was reported to be voltage independent.
Our data indicate that mecamylamine is a potent non-competitive inhibitor of the heterologously expressed neuronal nicotinic AChR α3β4 subtype. Our experiments give an IC50 for the inhibition of rat α3β4 receptor that is in very good agreement with the value reported by Cachelin & Rust (1995) of 190 nM as well as the value reported by Fieber & Adams (1991) for nicotinic AChR in rat cardiac ganglion neurons. In our experiments this inhibition appeared to be only slowly reversible for the wild-type α3β4 receptors, similar to the persistent inhibition of α4β2 receptors (Bertrand et al., 1990). It may seem somewhat curious that the effects of mecamylamine on chick α4β2 receptors were reportedly voltage independent. However, in addition to the potential significance of receptor subtype and species differences, there were many procedural differences between our experiments and those reported by Bertrand. Most notably, Bertrand reported voltage independence for instantaneous inhibition (i.e. during co-application), while we report a voltage dependence for the persistent inhibition measured 5 min after the antagonist was applied.
The inhibition of α3β4 receptors by mecamylamine in our experiments has the characteristics of open channel block and shows a voltage dependence similar to that of QX-222 for the block of muscle-type receptors (Neher & Steinbach, 1978). Specifically, based on single channel kinetic analysis, QX-222 was calculated to show an e-fold change in potency for each 32 mV change in potential (Neher & Steinbach, 1978), suggesting that the site of QX-222 binding was 78% through the membrane electric field. Our data suggest that in α3β4 receptors mecamylamine binds to a site approximately 60% through the membrane's electric field.
It has been proposed (Charnet et al., 1990) that binding of noncompetitive agents within the ion channel of the nicotinic AChR involves interactions with both a polar site and a hydrophobic site oriented along the axis of the membrane-spanning TM2 domain. Channel activation presumably controls the orientation of the TM2 helix so that these sites become accessible to inhibitors from the extracellular surface. The voltage dependence of inhibition would then be related to the depth of the polar site relative to the voltage drop within the channel. Site-directed mutagenesis studies indicated that the binding of QX-222 is associated with an inner polar site corresponding to the 6′ residue of TM2. It is likely that mecamylamine associates with the homologous site in neuronal α3β4 receptors, such that the primary polar interaction at the 6′ site is further stabilized by the hydrophobic character of the 10′ ring. In wild-type muscle receptors (α1β1γδ), which are relatively insensitive to mecamylamine, the β1 subunit limits the polar nature of this site with a phenylalanine residue at the 6′ position. In wild-type α3β4 receptors there would be serine residues present at this site in all five subunits. In the chimeric α3β4(β1TM2) receptors, at least two residues at this location are mutated to phenylalanine. This change in the character of the inner polar site in multiple subunits could easily account for the observed 200 fold decrease in mecamylamine potency. While inclusion of the β4TM2 sequence into the muscle β1 subunit increases the transient inhibition produced by mecamylamine, the relatively modest effect obtained may be related to the fact that the chimeric receptors were likely to be changed at a single site in the pentameric complex.
The chimeric α3β4(β1TM2) receptors appear to have faster kinetics of deactivation than the wild-type receptors, suggesting a decrease in the net Popen during an ACh-evoked response. This decreased Popen would subsequently reduce the likelihood of use-dependent effects. However, the relative change in the ratio of net charge to peak current for chimeric receptors, compared to wild-type, is much less than the decrease we observe in mecamylamine potency. Moreover, the greatly increased rate of recovery from mecamylamine effects in the chimeric receptor suggests that these mutations have direct effects at the mecamylamine binding site, and are not just reducing the probability of block.
The activity profile of nicotine for the various neuronal AChR is influenced by subtype specific competitive and noncompetitive effects. Consistent with reports by others (Hussy et al., 1994), our experiments indicate that nicotine's efficacy for the activation of α3β4 receptors is limited, at least in part, by readily reversible antagonist activity. While it might be argued that the increased inhibition of α3β4(β1TM2) receptors by agonist might represent accelerated desensitization, it would also be consistent with increases in secondary inhibition by the agonists themselves. Note that the wild-type α3β4 receptors show clear indication of channel block (or other form of noncompetitive inhibition) by nicotine, with a suppression of the current response during a nicotine application that is use-dependent and concentration-dependent in its onset. This would suggest that the most parsimonious explanation of the chimeric receptor's prolonged inhibition may be that nicotine is binding to some inhibitor site on both the wild-type and the chimeric receptors but with enhanced affinity for the chimeric receptors, most probably due to a decreased off-rate.
The basic concept of desensitization is that the desensitized receptor is in a non-activatable state that can persist in the absence of bound agonist, such that a new agonist-binding event is unlikely to open the channel. However, we see that the recovery from inhibition produced by the two agonists ACh and nicotine proceeds at very different rates (7 min and 51 min, respectively). To interpret this in terms of desensitization would require that these two drugs produced distinctly different forms of desensitization. This is inconsistent with the accepted concept of the Markov-type behaviour of ion channels, and would require that the channel retained some memory of which agonist produced the conformational transition to the desensitized state. Again, the more parsimonious explanation is that during the onset of inhibition, the agonist (either nicotine or ACh) binds at a specific site, not associated with activation, and that recovery from inhibition occurs at a rate derived by the dissociation rate for the specific ligand from that inhibitory site.
It is unclear whether the actual binding site for nicotine is likely to be within the channel, or as proposed for BTMPS, somewhere in the extracellular part of the protein, with the affinity for nicotine regulated by the translation of gating movements through the TM2 domain. The apparent lack of voltage-dependence for nicotine's inhibitory effect would argue against the idea that nicotine may be binding directly within the channel. However, the mutational analysis indicates that the inhibitory activity of nicotine may require both the 10′ and the 6′ substitutions. These mutations effectively create a new polar ring at the 10′ position, corresponding to the next level up on the helical wheel from where mecamylamine appears to be binding. There will be an enhanced polar quality to the intermediate ring in α3β4(β1TM2) receptors and a highly hydrophobic site at the 6′ position. It may be the case that these changes produce a binding site for nicotine that is configured so that nicotine preferentially binds with the relatively hydrophobic pyridine ring of nicotine oriented toward the intracellular end of the channel at the 6′ ring, while the charged nitrogen of nicotine may interact with the more polar residues at the 10′ position in TM2. A specific interaction of nicotine at this intermediate polar site would correspond to a reduced voltage dependence for the nicotine-induced inhibition compared to the effects of mecamylamine. It may be the case that our measurements were not sufficiently sensitive to resolve what could have been a relatively small voltage dependence.
Alternatively, nicotine may not be binding to the 10≈prime; site. Instead, what we observed may be due to how the specific configuration of polar sites within TM2 affect the translation of conformational change to other parts of the receptor, including an inhibitory site for nicotine that is above the membrane's electric field. Our previous study of bis-tetramethylpiperidine inhibitors demonstrated a similar effect, such that the neuronal beta subunit TM2 domain was important for the translation of the conformational changes associated with channel gating, regulating the accessibility or affinity of a binding site for the selective neuronal nicotinic AChR inhibitor BTMPS. The BTMPS binding site was outside of the membrane's electric field, and therefore unlikely to be located within the chimeric domain itself. While in the present study we demonstrate that the neuronal beta TM2 can also directly control the selectivity of the ganglionic blocker mecamylamine for neuronal nicotinic AChR by providing a binding site within TM2 that appears to be analogous to the binding site for local anaesthetics to the muscle–type AChR, it may be the case that nicotine produces inhibitory effects in a manner more analogous to the inhibition produced by BTMPS. However, if this is the case, then it would suggest that BTMPS and nicotine bind with high affinity to qualitatively different sites, since their use-dependent inhibitory effects have opposite requirement for TM2 sequence.
In conclusion, the present study provides new insight into how noncompetitive inhibition by both agonists and antagonists share a dependence on specific sequence in a channel forming domain. Our data support the hypothesis that inhibition can occur, either through direct binding to sites within the channel or alternatively, through allosteric interactions associated with the translation of conformational change regulated by channel gating. The observation that conformational information may be transmitted from TM2 to a remote site is likely to be of general significance for our understanding of protein structure and function. Moreover, since the nicotinic agents presently in clinical development typically have mixed agonist/antagonist effects, our data may suggest ways to optimize the development of these drugs for specific therapeutic applications. Through the identification of the inhibitory mechanisms for specific drugs, we may seek to achieve the most effective balance of selectivity for activation and inhibition of the various neuronal nicotinic AChR subtypes.
Acknowledgments
We thank Dr Steve Heinemann and Jim Boulter for providing nicotinic AChR cDNAs. We also thank Dr Robert Oswald for helpful discussions as well as Chad Wheeler for technical assistance. This work was supported by National Institutes of Health Grant NS32888.
Abbreviations
- ACh
acetylcholine
- AChR
acetylcholine receptor
- BTMPS
bis-tetramethyl-piperidinyl sebacate
- MS222
3-aminobenzoic acid ethyl ester
- PCR
polymerase chain reaction
- TM2
second transmembrane (domain of an AChR subunit)
- TMP
tetramethylpiperidine
References
- ASCHER P., LARGE W.A., RANG H.P. Studies on the mechanism of action of acetylcholine antagonists on rat parasympathetic ganglion cells. J. Physiol. 1979;295:139–170. doi: 10.1113/jphysiol.1979.sp012958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERTRAND D., BALLIVET M., RUNGGER D. Activation and blocking of neuronal nicotinic acetylcholine receptor reconstituted in Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1993–1997. doi: 10.1073/pnas.87.5.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CACHELIN A.B., RUST G. β-subunits co-determine the sensitivity of rat neuronal nicotinic receptors to antagonists. Eur. J. Physiol. 1995;429:449–451. doi: 10.1007/BF00374164. [DOI] [PubMed] [Google Scholar]
- CHANGEUX J.-P., GIRAUDAT J., DENNIS M. The nicotinic acetylcholine receptor: molecular architecture of a ligand-regulated ion channel. Trends Pharmacol. Sci. 1987;8:459–465. [Google Scholar]
- CHARNET P., LABARCA C., LEONARD R.J., VOGELAAR N.J., CZYZYK L., GOUIN A., DAVIDSON N., LESTER H.A. An open-channel blocker interacts with adjacent turns of α-helices in the nicotinic acetylcholine receptor. Neuron. 1990;2:87–95. doi: 10.1016/0896-6273(90)90445-l. [DOI] [PubMed] [Google Scholar]
- COHEN B.N., LABARCA C., DAVIDSON N., LESTER H.A. Mutations in M2 alter the selectivity of the mouse nicotinic acetylcholine receptor for organic and alkali metal cations. J. Gen. Physiol. 1992;100:373–400. doi: 10.1085/jgp.100.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONROY W.G., VERNALLIS A.B., BERG D.K. The alpha 5 gene product assembles with multiple acetylcholine receptor subunits to form distinctive receptor subtypes in brain. Neuron. 1992;9:679–691. doi: 10.1016/0896-6273(92)90031-8. [DOI] [PubMed] [Google Scholar]
- CORRIVEAU R.A., BERG D.K. Coexpression of multiple acetylcholine receptor genes in neurons: quantification of transcripts during development. J. Neurosci. 1993;13:2662–2671. doi: 10.1523/JNEUROSCI.13-06-02662.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE FIEBRE C.M., MEYER E.M., ZOLTEWICZ J., HENRY J.C., MURASKIN S., KEM W.R., PAPKE R.L. Characterization of a family of anabaseine-derived compounds reveals that the 3-(4)-dimethylaminocinnamylidine derivative (DMAC) is a selective agonist at neuronal nicotinic α7/[125I]α-bungarotoxin receptor subtypes. Mol. Pharmacol. 1995;47:164–171. [PubMed] [Google Scholar]
- DENNIS M., GIRAUDAT J., KOTZYBA-HIBERT F., GOELDNER M., HIRTH C., CHANG J.-Y., CHANGEUX J.-P. A photoaffinity ligand of the acetylcholine binding site predominantly labels the region 179–207 of the alpha-subunit on native acetylcholine receptor from Torpedo marmorata. FEBS Lett. 1986;207:243–249. [Google Scholar]
- FIEBER L.A., ADAMS D.J. Acetylcholine-evoked curents in cultured neurones dissociated from rat parasympathetic cardiac ganglia. J. Physiol. 1991;434:215–237. doi: 10.1113/jphysiol.1991.sp018466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANCIS M.M., CHOI K.I., HORENSTEIN B.A., PAPKE R.L. Sensitivity to voltage-independent inhibition determined by pore-lining region of ACh receptor. Biophys. J. 1998;74:2306–2317. doi: 10.1016/S0006-3495(98)77940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANCIS M.M., PAPKE R.L. Muscle-type nicotinic acetylcholine receptor delta subunit determines sensitivity to noncompetitive inhibitors while gamma subunit regulates divalent permeability. Neuropharm. 1996;35:1547–1556. doi: 10.1016/s0028-3908(96)00103-7. [DOI] [PubMed] [Google Scholar]
- GIRAUDAT J., DENNIS M., HEIDMANN T., CHANG J.Y., CHANGEUX J.-P. Structure of the high affinity binding site for noncompetitive blockers of the acetylcholine receptor: Serine-262 of the delta subunit is labeled by [3H]chlorpromazine. Proc. Natl. Acad. Sci. U.S.A. 1986;83:2719–2723. doi: 10.1073/pnas.83.8.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEINEMANN S., GOLDMAN D., BOULTER J., PATRICK J.Molecular biology of the muscle and neural acetylcholine receptors Proceedings of NATO Conference on Mechanism of Action of Nicotinic Acetylcholine Receptor 1986Santorini, Greece; 359–388.In: ed. Maelicke A [Google Scholar]
- HORTON R.M., HUNT H.D., HO S.N., PULLEN J.K., PEASE L.R. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- HUSSY N., BALLIVET M., BERTRAND D. Agonist and antagonist efffects of nicotine on chick neuronal nicotinic receptors are defined by α and β subunits. J. Neurophys. 1994;72:1317–1326. doi: 10.1152/jn.1994.72.3.1317. [DOI] [PubMed] [Google Scholar]
- KATZ B., THESLEFF S. A study of the ‘desensitization' produced by acetylcholine at the motor end-plate. J. Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LABARCA C., NOWAK M.W., ZHANG H., TANG L., DESHPANDE P., LESTER H.A. Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature. 1995;376:514–516. doi: 10.1038/376514a0. [DOI] [PubMed] [Google Scholar]
- LEONARD R.J., LABARCA C.G., CHARNET P., DAVIDSON N., LESTER H.A. Evidence that the M2 membrane-spanning region lines the ion channel pore of the nicotinic receptor. Science. 1988;242:1578–1581. doi: 10.1126/science.2462281. [DOI] [PubMed] [Google Scholar]
- LUETJE C.W., PATRICK J. Both α- and β-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J. Neurosci. 1991;11:837–845. doi: 10.1523/JNEUROSCI.11-03-00837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUETJE C.W., WADA K., ROGERS S., ABRAMSON S.N., HEINEMANN S., PATRICK J. Neurotoxins distinguish between different neuronal nicotinic acetylcholine receptors. J. Neurochem. 1990;55:632–640. doi: 10.1111/j.1471-4159.1990.tb04180.x. [DOI] [PubMed] [Google Scholar]
- NEHER E., STEINBACH J.H. Local anaesthetics transiently block current through single acetylcholine receptor channels. J. Physiol. 1978;277:135–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAPKE R.L. The kinetic properties of neuronal nicotinic receptors: genetic basis of functional diversity. Prog. Neuro. 1993;41:509–531. doi: 10.1016/0301-0082(93)90028-q. [DOI] [PubMed] [Google Scholar]
- PAPKE R.L., CRAIG A.G., HEINEMANN S.F. Inhibition of nicotinic acetylcholine receptors by BTMPS, (Tinuvin® 770), an additive to medical plastics. J. Pharmacol. Exp. Ther. 1994;268:718–726. [PubMed] [Google Scholar]
- PAPKE R.L., HEINEMANN S.F. The partial agonist properties of cytisine on neuronal nicotinic receptors containing the β2 subunit. Mol. Pharm. 1994;45:142–149. [PubMed] [Google Scholar]
- PAPKE R.L., THINSCHMIDT J.S., MOULTON B.A., MEYER E.M., POIRIER A. Activation and inhibition of rat neuronal nicotinic receptors by ABT-418. Br. J. Pharmacol. 1997;120:429–438. doi: 10.1038/sj.bjp.0700930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REVAH F., BERTRAND D., GALZI J.-L., DEVILLERS-THIERY A., MULLE C., HUSSY N., BERTRAND S., BALLIVET M., CHANGEUX J.-P. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- ROZENTAL R., ARACAVA Y., SCOBLE G.T., SWANSON K.L., WONNACOTT S., ALBUQUERQUE E.X. Agonist recognition site of the peripheral acetylcholine receptor ion channel complex differentiates the enantiomers of nicotine. J. P. E. T. 1989;251:395–404. [PubMed] [Google Scholar]
- SANGER F., NICKLEN S., COULSON A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINE S.M., STEINBACH J.H. Agonists block currents through acetylcholine receptor channels. Biophys. J. 1984;46:277–284. doi: 10.1016/S0006-3495(84)84022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STONE C., TORCHIANA M., NAVARRO A., BEYER K. Ganglionic blocking properties of 3-methyl-aminoisocamphane hydrochloride (mecamylamine): a secondary amine. J. Pharmacol. Exp. Ther. 1956;117:169–183. [PubMed] [Google Scholar]
- VARANDA W., ARACAVA Y., SHERBY S.M., VANMETER W.G., ELDEFRAWI M.E., ALBUQUERQUE E.X. The acetylcholine receptor of the neuromuscular junction recognizes mecamylamine as a noncompetitive antagonist. Mol. Pharm. 1985;28:128–137. [PubMed] [Google Scholar]
- VERNALLIS A.B., CONROY W.G., BERG D.K. Neurons assemble acetylcholine receptors with as many as three kinds of subunits while maintaining subunit segregation among receptor subtypes. Neuron. 1993;10:451–464. doi: 10.1016/0896-6273(93)90333-m. [DOI] [PubMed] [Google Scholar]
- WOODHULL A.M. Ionic blockage of sodium channels in nerve. J. Gen. Phy. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]