

Abstract
The effect of the ether lipid 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphorylcholine (ET-18-OCH3) on the intracellular free Ca2+ concentration ([Ca2+]i) in Madin Darby canine kidney (MDCK) cells was studied using fura-2 as the Ca2+ probe. In Ca2+ medium, ET-18-OCH3 induced a significant rise in [Ca2+]i at concentrations between 10–100 μM with a concentration-dependent delay of 45–175 s. The [Ca2+]i signal was composed of a gradual rise and a sustained plateau.
In Ca2+-free medium, ET-18-OCH3 (10–100 μM) induced a Ca2+ release from internal Ca2+ stores with a concentration-dependent delay of 45–175 s. This discharge of internal Ca2+ triggered capacitative Ca2+ entry in a concentration-dependent manner. This capacitative Ca2+ entry was not inhibited by econazole (25 μM), 1-[β-[3-(4-methoxyphenyl)propoxy]-4-methoxyphenethyl]-1H-imidazole hydrochloride (SKF96365; 50 μM), nifedipine (10 μM), verapamil (10 μM), diltiazem (10 μM) and cadmium (0.5 μM).
Methyl 2-(phenylthio)ethyl-1,4-dihydro-2,4,6-trimethylpyridine-3,5-dicarboxylate (PCA-4248), a platelet-activating factor (PAF) receptor antagonist, inhibited 25 μM ET-18-OCH3-induced [Ca2+]i rise in a concentration-dependent manner between 1–20 μM, with 20 μM exerting a complete block.
The [Ca2+]i rise induced by ET-18-OCH3 (25 μM) was not altered when the production of inositol 1,4,5-trisphosphate (IP3) was suppressed by the phospholipase C inhibitor U73122 (2 μM), but was partly inhibited by the phospholipase D inhibitor propranolol (0.1 mM) or the phospholipase A2 inhibitor aristolochic acid (20–40 μM).
In Ca2+-free medium, pretreatment with 25 μM ET-18-OCH3 completely depleted the endoplasmic reticulum Ca2+ pump inhibitor thapsigargin-sensitive Ca2+ store. In contrast, pretreatment with thapsigargin abolished 0.1 mM ATP-induced [Ca2+]i rise without altering the ET-18-OCH3-induced [Ca2+]i rise. This suggests that ET-18-OCH3 depleted thapsigargin-sensitive Ca2+ stores and also released Ca2+ from thapsigargin-insensitive stores. The thapsigargin-insensitive stores involve mitochondria because the mitochondria uncoupler carbonylcyanide m-chlorophenylhydrazone (CCCP; 2 μM) induced a release of mitochondrial Ca2+ which was abolished by pretreatment with 25 μM ET-18-OCH3.
ET-18-OCH3 (25 μM) induced a significant Mn2+ quench of fura-2 fluorescence at 360 nm excitation wavelength confirming that ET-18-OCH3 induced capacitative Ca2+ entry. La3+ (0.1 mM) or Gd3+ (50 μM) abolished the ET-18-OCH3-induced Mn2+ quench and [Ca2+]i rise.
Our data imply that ET-18-OCH3 induced a [Ca2+]i rise in MDCK cells by activating PAF receptors leading to an internal Ca2+ release followed by capacitative Ca2+ entry. Phospholipase D and phospholipase A2, but not phospholipase C, might be involved in mediating the capacitative Ca2+ entry. La3+ abolished the ET-18-OCH3-induced [Ca2+]i rise presumably by inhibiting PAF receptors.
Keywords: ET-18-OCH3, MDCK cells, Ca2+ signalling, capacitative Ca2+ entry, La3+
Introduction
A number of synthetic ether lipids with structure similar to platelet-activating factor (PAF) have been reported to be DNA-noninteractive anti-tumour drugs (Berdel, 1991). Among these drugs, 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphorylcholine (ET-18-OCH3) is the most commonly used for the investigation of effects of this category of either lipids at the cellular level (Mollinedo et al., 1993). The primary cellular target for ET-18-OCH3 was previously thought to be the plasma membrane proteins (Paltauf, 1994; Boggs et al., 1995). For example, ET-18-OCH3 inhibits Na+-K+-ATP pump in bovine brain (Zheng et al., 1990), protein kinase C in HL-60 cells (Berkovic et al., 1994), and phosphatylcholine synthesis in macrophage-like cells (Boggs et al., 1995). Additionally, ET-18-OCH3 also increases intracellular free Ca2+ concentration ([Ca2+]i) in normal and tumour cells (Seewald et al., 1990; Lohmeyer & Workman, 1993; Bergmann et al., 1994; Brinkmeier et al., 1996; Alonso et al., 1997), and induces apoptosis in several human leukaemia cells (Mollinedo et al., 1993; 1997; Diomede et al., 1993). But how exactly ET-18-OCH3 acts to trigger so many different cellular events is not clear at all.
Efforts have been exerted to investigate whether the effect of ET-18-OCH3 on [Ca2+]i is correlated to its effect on apoptosis. The idea is that a prolonged elevation in [Ca2+]i could lead to apoptosis (McConkey & Orrenius, 1996). Two studies performed in several normal and tumour cells suggest that these two effects of ET-18-OCH3 appear to be dissociated (Brinkmeier et al., 1996; Alonso et al., 1997), i.e. under the experimental conditions that the ET-18-OCH3-induced [Ca2+]i rise is suppressed, the apoptosis effect of this drug is not affected.
The mechanism underlying the ET-18-OCH3-induced [Ca2+]i rise is not completely understood. Alonso and coworkers (1997) showed that in human neutrophils ET-18-OCH3 induces a robust [Ca2+]i transient with a rapid rise and decline through stimulation of PAF receptors, because the [Ca2+]i rise is suppressed by a PAF receptor antagonist. The authors also demonstrated that internal Ca2+ release and external Ca2+ influx both contribute to the ET-18-OCH3-induced [Ca2+]i rise, but neither the source of the internal Ca2+ nor the identity of the Ca2+ influx pathway was investigated. In contrast, in a neuroblastoma cell line Brinkmeier et al. (1996) observed that ET-18-OCH3 induces a Ca2+ signal with a delayed rise and a persistently elevated plateau, which is abolished by pretreatment with La3+ or Gd3+. Based on the lanthanide inhibition, the authors conclude that ET-18-OCH3 induces a [Ca2+]i rise by exclusively opening plasma membrane Ca2+ channels. However, it is not known which types of Ca2+ channels are involved. Further, the possibility that ET-18-OCH3 might release internal Ca2+ is not examined.
In the present study we investigated the effect of ET-18-OCH3 on [Ca2+]i in Madin Darby canine kidney (MDCK) cells, a non-tumour cell line, in more detail than the previous studies. We found that ET-18-OCH3 induced a rise in [Ca2+]i by activating PAF receptors leading to a release of Ca2+ from both thapsigargin-sensitive and -insensitive internal Ca2+ stores. This discharge of internal Ca2+ triggered capacitative Ca2+ entry (Putney & Bird, 1993) which was blocked by lanthanides. Lanthanides might also directly inhibit PAF receptors. Phospholipase D and phospholipase A2, but not phospholipase C, might be involved in mediating the [Ca2+]i rise.
Methods
Cell culture
MDCK cells obtained from American Type Culture Collection (CRL-6253, MD, U.S.A) were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% heat-inactivated foetal bovine serum, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin at 37°C in 5% CO2-containing humidified air.
Solutions
Ca2+ medium (pH 7.4) contained (in mM): NaCl 140; KCl 5; MgCl2 1; CaCl2 2; HEPES 10; glucose 10. Ca2+-free medium contained no Ca2+ plus 1 mM EGTA. ET-18-OCH3 was dissolved in dimethyl sulphoxide (DMSO) as a 10 mM stock solution. PCA-4248 and thapsigargin were dissolved in ethanol as a 20 mM stock solution. The experimental solution contained 0–1% of solvent (DMSO or ethanol) which did not affect [Ca2+]i (n=3).
Fluorescence measurements
Trypsinized cells (106 ml−1) were loaded with 2 μM 1-[2-(5-carboxyoxazol-2-yl ) -6-aminobenzofuran-5-oxy] -2- (2′-amino-5′-methylphenoxy)-ethane-N,N,N,N-tetraacetic acid pentaacetoxymethyl ester (fura-2/AM) for 30 min at 25°C in DMEM. Cells were washed and resuspended in Ca2+ medium and were washed every hour during experiments to minimize extracellular dye. Fura-2 fluorescence measurements were performed in a water-jacketed cuvette (25°C) with continuous stirring; the cuvette contained 1 ml of medium and 0.5 million of cells. Fluorescence was monitored with a Shimadzu RF-5301PC spectrofluorophotometer (Japan) by continuously recording excitation signals at 340 and 380 nm and emission signal at 510 nm at 1-s intervals. Maximal and minimal fluorescence values were obtained by adding TX-100 (0.1%) and EGTA (20 mM) sequentially at the end of an experiment. The ratio of excitation signals at 340 and 380 nm was used to calculate [Ca2+]i as described previously (Grynkiewicz et al., 1985). Mn2+ quench experiments were performed in Ca2+ medium containing MnCl2 (50 μM) by recording excitation signal at 360 nm and emission signal at 510 nm continuously at 1-s intervals. Our previous studies have shown that trypsinized cells prepared by our protocol respond to stimulation with ATP (Jan et al., 1998a), bradykinin (Jan et al., 1998b) or thapsigargin (Jan et al., 1999c) similarly to cells attached to coverslips. We decided to use trypsinized cells because this procedure is easier and less time-consuming. All experiments were performed at room temperature (25°C).
Materials
The reagents for cell culture were from Gibco (NY, U.S.A.). Fura-2/AM was from Molecular Probes (OR, U.S.A.). ET-18-OCH3, (±)1-O-hexadecyl-2-O-methylglycero-3-phosphorylcholine (2-O-methyl PAF), and PCA-4248 were from Biomol (Plymouth Meeting, PA, U.S.A.). The other reagents were from Sigma (MO, U.S.A.).
Statistics
All values are reported as means±s.e.mean of 3–4 experiments. Statistical comparisons were determined by using the Student's paired t-test, and significance was accepted when P<0.05.
Results
ET-18-OCH3 at concentrations between 10–100 μM induced a delayed, gradual rise in [Ca2+]i in the presence of extracellular Ca2+. A concentration of 1 μM had no effect. Representative traces are shown in Figure 1A. The concentration-response plot is shown in Figure 1B. The response did not saturate at 100 μM. Because the 100 μM ET-18-OCH3-induced [Ca2+]i rise reached a peak value as great as ∼1.5 μM followed by a sustained plateau, higher concentrations were not tested to avoid cell damage. The rise of the Ca2+ signal was slower (i.e. delay time was shorter) in response to lower concentrations of ET-18-OCH3, and the relationship between concentration and the delay time before [Ca2+]i rises is plotted in Figure 1C. For example, in the [Ca2+]i rise induced by 100 μM ET-18-OCH3, the delay time was 48±3 s (n=3; P<0.05), while that induced by 10 μM ET-18-OCH3 had a delay time of 171±4 s (n=3; P<0.05). At a concentration of 25 μM, 2-O-methyl PAF, another PAF agonist, induced a rise in [Ca2+]i with a similar shape and magnitude as that induced by 50 μM ET-18-OCH3 (n=5; data not shown).
Figure 1.
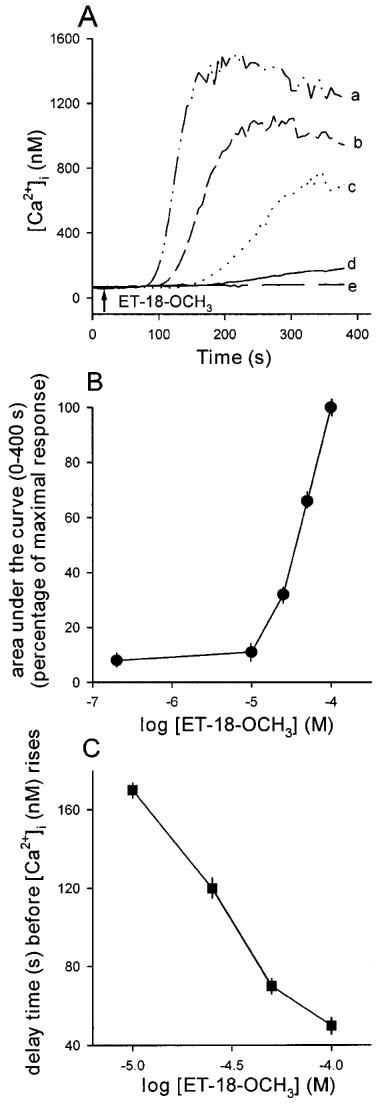
(A) Concentration-dependent effects of ET-18-OCH3 on [Ca2+]i. Concentration of ET-18-OCH3 was 100 μM in trace a, 50 μM in trace b, 25 μM in trace c, 10 μM in trace d and zero in trace e. ET-18-OCH3 was applied at 30 s. The experiments were performed in Ca2+ medium. Traces are typical of 3–4 experiments. (B) A concentration-response plot of the ET-18-OCH3-induced Ca2+ signals shown in (A). The y axis is the area under the curve (percentage of maximum response). The x axis is the concentration of ET-18-OCH3 in logarithmic scale. The data are means±s.e.mean of 3–4 experiments. (C) Relationship between the delay time (s) before [Ca2+]i (nM) rises. The data are means±s.e.mean of 3–4 experiments.
We next examined whether ET-18-OCH3 releases Ca2+ from internal stores. Figure 2A shows that in Ca2+-free medium (no added Ca2+ plus 1 mM EGTA) 10–100 μM ET-18-OCH3 induced significant [Ca2+]i rises. In MDCK cells, a release of Ca2+ from the endoplasmic reticulum (ER) store often induces capacitative Ca2+ entry (Jan et al., 1998a1998b1998c; 1999a1999b1999c1999d). Thus, we next examined whether ET-18-OCH3 triggers capacitative Ca2+ entry. Figure 2A shows that CaCl2 (3 mM) added at the time point of 360 s when the internal Ca2+ stores had been substantially discharged by ET-18-OCH3 pretreatment induced significant capacitative Ca2+ entry with a magnitude proportionally correlating to the concentration of ET-18-OCH3, i.e. a larger concentration of ET-18-OCH3 induces a larger capacitative Ca2+ entry. We examined the effects of several Ca2+ channel blockers on the ET-18-OCH3-induced capacitative Ca2+ entry. The blocker was added 30 s prior to CaCl2. These blockers were 10 μM of nifedipine, diltiazem and verapamil, 25 μM econazole, 50 μM SKF96365, and 0.5 mM cadmium. None of the drugs tested had any inhibition (n=3; data not shown). The effect of La3+ (0.1 mM) on capacitative Ca2+ entry could not be tested using this experimental protocol because La3+ is chelated by EGTA. Addition of EGTA (1 mM) is needed for measuring capacitative Ca2+ entry because the ET-18-OCH3-induced [Ca2+]i rise remained elevated at ∼400 nM for a sustained period of time (>6 min) in nominally Ca2+-free (no EGTA) medium, thus addition of 3 mM CaCl2 did not induce a further rise in [Ca2+]i (data not shown). The concentration-response plot of the capacitative Ca2+ entry suggests that the response saturates at a concentration of 50 μM (Figure 2B) with an EC50 of about 32 μM. Note that the [Ca2+]i rises induced by all concentrations of ET-18-OCH3 have a similar magnitude in terms of the area under the curve and the net peak value (∼40 nM). However, these responses are different in the delay time before [Ca2+]i rises. Thus, similar to the responses observed in the presence of extracellular Ca2+, in the absence of extracellular Ca2+ the ET-18-OCH3-induced internal Ca2+ release also has a delay time depending on the concentration of ET-18-OCH3, i.e. a larger concentration of ET-18-OCH3 induces a Ca2+ release with a shorter delay time. The relationship between delay time and concentration is plotted in Figure 2C.
Figure 2.
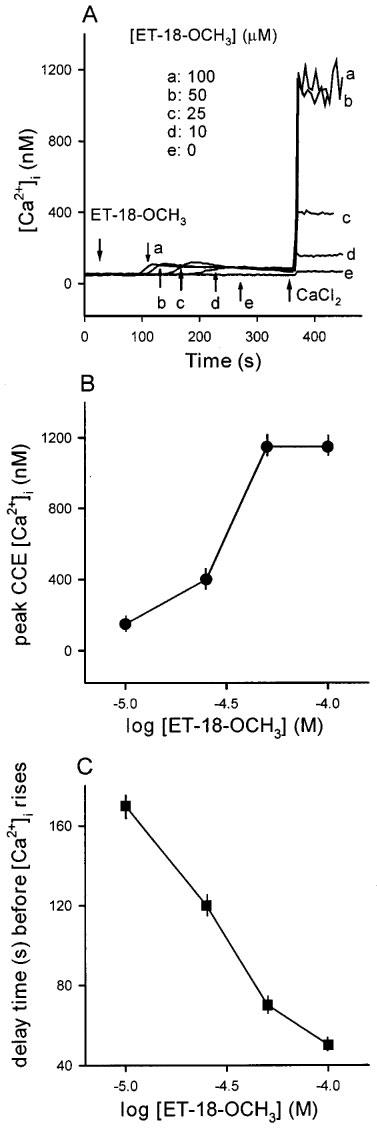
(A) ET-18-OCH3-induced capacitative Ca2+ entry. These experiments were performed in Ca2+-free medium. Capacitative Ca2+ entry was induced by depleting intracellular Ca2+ stores with different concentrations of ET-18-OCH3 (10–100 μM) in Ca2+-free medium (no added Ca2+ plus 1 mM EGTA) followed by addition of 3 mM CaCl2. Trace e is the control CaCl2 effect without ET-18-OCH3 pretreatment. (B) A concentration-response plot of the ET-18-OCH3-induced capacitative Ca2+ entry shown in (A). The y axis is the peak height of the [Ca2+]i rise induced by addition of 3 mM CaCl2. The x axis is the concentration of ET-18-OCH3 in logarithmic scale. The data are means±s.e.mean of 3–4 experiments. (C) Relationship between delay time (s) before [Ca2+]i rises and the concentration of ET-18-OCH3. The data are means±s.e.mean of 3–4 experiments.
Because ET-18-OCH3 is a synthetic analogue of platelet-activating factor (PAF), we tested whether the ET-18-OCH3-induced [Ca2+]i rise is downstream to activation of PAF receptors. We used PCA-4248, a PAF receptor antagonist (Ortega et al., 1990), to see if it could affect the ET-18-OCH3-induced [Ca2+]i rise. Figure 3A shows that pretreatment with PCA-4248 for 30 s inhibited the 25 μM ET-18-OCH3-induced [Ca2+]i rise in a concentration-dependent manner between 1–20 μM. Figure 3B shows the concentration-inhibition plot of the PCA-4248 effect. The areas under the curves of the five traces in Figure 3A were computed and compared. Maximum inhibition occurs at 20 μM PCA-4248 which inhibited 95±5% (n=3; P<0.05) of the control ET-18-OCH3 response (in the absence of PCA-4248). The plot indicates an IC50 of about 2.5 μM.
Figure 3.
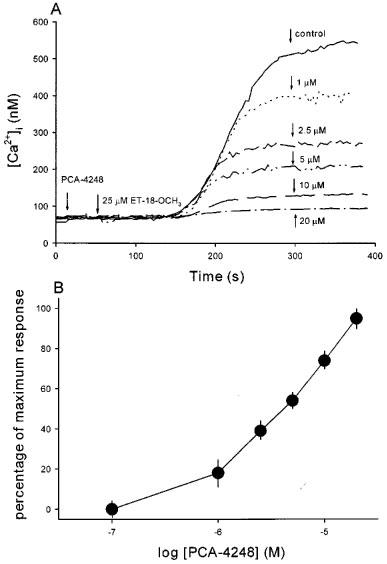
Effect of PCA-4248 on the ET-18-OCH3-induced [Ca2+]i rise. (A) ET-18-OCH3 (25 μM) was added at 50 s. PCA-4248 at a concentration between 1–20 μM was added at 20 s. Control trace represents the ET-18-OCH3 response without PCA-4248 pretreatment. The experiments were performed in Ca2+ medium. Traces are typical of 3–4 experiments. (B) A concentration-response plot of the PCA-4248 inhibition of the ET-18-OCH3-induced Ca2+ signal shown in (A). The y axis is the percentage of maximum inhibition which is exerted by 20 μM PCA-4248 (95±5% inhibition of control ET-18-OCH3 response in terms of the area under the curve). The x axis is the concentration of PCA-4248 in logarithmic scale. The data are means±s.e.mean of 3–4 experiments.
We next examined whether ET-18-OCH3 releases internal Ca2+ by first elevating cytosolic levels of IP3. We have previously shown that the phospholipase C inhibitor U73122 blocks IP3 production leading to an inhibition of ATP- or bradykinin-induced release of Ca2+ from thapsigargin-sensitive ER Ca2+ stores (Jan et al., 1998c). Because U73122 induces significant Ca2+ influx (Jan et al., 1998c), the following experiments were performed in Ca2+-free medium. Shown in Figure 4A is the [Ca2+]i transient induced by ATP (0.1 mM). Figure 4B (solid trace) shows that pretreatment with U73122 (2 μM) for 320 s did not alter the resting [Ca2+]i, but substantially reduced the ATP-induced [Ca2+]i rise by 91±5% in net peak height (31±5 vs 350±16 nM; n=3; P< 0.05). This result most likely suggests that the IP3 production via phospholipase C was significantly inhibited under this condition. When ET-18-OCH3 (25 μM) was added subsequently at 440 s there occurred a [Ca2+]i rise which was indistinguishable from the control ET-18-OCH3 response (without U73122/ATP pretreatment; dashed trace). We also examined whether phospholipase D and phospholipase A2 are involved in mediating the ET-18-OCH3-induced [Ca2+]i rise. We used propranolol to inhibit phospholipase D (Billah, 1989) and aristolochic acid to inhibit phospholipase A2 (Rosenthal et al., 1989). Table 1 shows that in the presence of extracellular Ca2+, propranolol (0.1 mM) and aristolochic acid (20–40 μM) both significantly inhibited the [Ca2+]i rise induced by 25 μM ET-18-OCH3. However, these two inhibitors did not alter the [Ca2+]i rise induced by ET-18-OCH3 in the absence of extracellular Ca2+ (n=3; data not shown).
Figure 4.
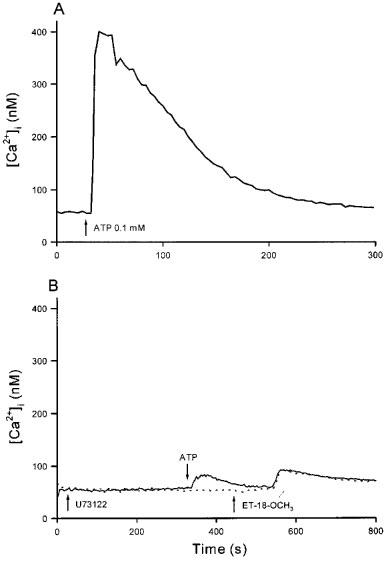
Effect of inhibition of IP3 production on the ET-18-OCH3-induced internal Ca2+ release. (A) 0.1 mM ATP was added at 30 s. (B) U73122 (2 μM) was added at 30 s followed by ATP (0.1 mM) and ET-18-OCH3 (25 μM) at 340 s and 440 s, respectively. These experiments were performed in Ca2+-free medium. Traces are typical of 3–4 experiments.
Table 1.
Effects of propranolol and aristolochic acid on the [Ca2+]i rise induced by ET-18-OCH3 (25 μM)
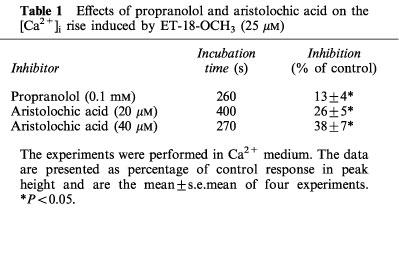
We went on to investigate whether ET-18-OCH3 releases Ca2+ from the thapsigargin-sensitive ER store, the major internal Ca2+ store in MDCK cells (Jan et al. 1998a1998b1998c; 1999a1999b1999c1999d). Thapsigargin is a substance known to cause a passive leak of Ca2+ from the ER store by inhibiting the ER Ca2+ pump (Thastrup et al., 1990). Figure 5A shows that in the absence of extracellular Ca2+, after pretreatment with 25 μM ET-18-OCH3 for 320 s thapsigargin hardly induced a rise in [Ca2+]i (see Figure 5B for control thapsigargin response). This suggests that ET-18-OCH3 completely discharged the thapsigargin-sensitive ER Ca2+ store. However, other Ca2+ stores also contribute to the ET-18-OCH3 response because, as shown in Figure 5B, after thapsigargin had completely depleted the thapsigargin-sensitive ER Ca2+ store (evidenced by the observation that 0.1 mM ATP added after thapsigargin did not increase [Ca2+]i), ET-18-OCH3 still induced a rise in [Ca2+]i normally (compared to Figure 5A).
Figure 5.
Effect of ET-18-OCH3 on internal Ca2+ stores. (A) ET-18-OCH3 (25 μM) was added at 30 s followed by thapsigargin (1 μM) at 350 s. (B) Thapsigargin (1 μM) was added at 30 s, followed by ATP (0.1 mM) and ET-18-CH3 (25 μM) at 630 s and 760 s, respectively. These experiments were performed in Ca2+-free medium. Traces are typical of 3–4 experiments.
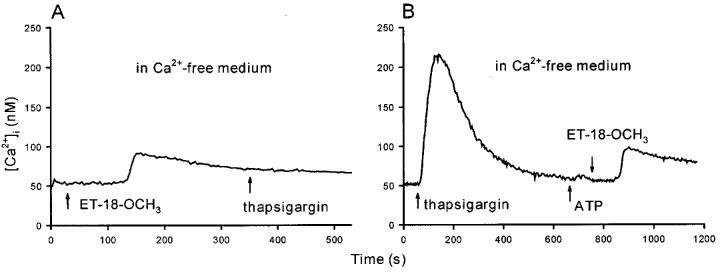
The thapsigargin-insensitive Ca2+ store in MDCK cells which could be examined by pharmacological tools is mitochondria. We have previously shown that the mitochondrial uncoupler carbonylcyanide m-chlorophenylhydrazone (CCCP; 2 μM) induced a rise in [Ca2+]i (Jan et al., 1998c; 1999b), suggesting that in this cell mitochondria contain a significant amount of Ca2+ which could be released when mitochondria are uncoupled by CCCP. Thus, we investigated whether ET-18-OCH3 releases Ca2+ from mitochondria. Figure 6A shows that in the absence of extracellular Ca2+, pretreatment with 25 μM ET-18-OCH3 prevented CCCP (2 μM) from inducing a rise in [Ca2+]i. However Figure 6B clearly demonstrates that CCCP induced a significant rise in [Ca2+]i with a net peak height of 51±7 nM (n=4; P<0.05). Thus, these results suggest that ET-18-OCH3 depletes the mitochondrial Ca2+ store. Figure 6B also shows that the ET-18-OCH3-induced rise in [Ca2+]i was not much altered after the mitochondrial Ca2+ had been depleted.
Figure 6.
Effect of ET-18-OCH3 on mitochondrial Ca2+ stores. (A) In Ca2+-free medium, ET-18-OCH3 (25 μM) was added at 30 s followed by CCCP (2 μM) at 410 s. (B) Similar to (A) except that CCCP was added at 30 s followed by ET-18-OCH3 at 390 s. Traces are typical of 3–4 experiments.
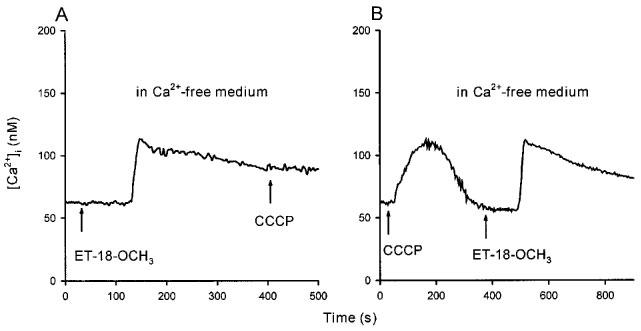
To confirm that ET-18-OCH3 induced capacitative Ca2+ entry, we next measured whether ET-18-OCH3 caused Mn2+ quench of fura-2 fluorescence. Mn2+ enters cells through similar pathways as Ca2+, but quenches fura-2 fluorescence at all excitation wavelengths (Merritt et al., 1989). Thus, Ca2+ influx can be estimated by Mn2+ quench of fura-2 fluorescence at the Ca2+-insensitive excitation wavelength of 360 nm and emission wavelength of 510 nm. Figure 7A illustrates that 25 μM ET-18-OCH3 induced a significant Mn2+ quench of fura-2 fluorescence after a delay of 120±8 s (n =3; P<0.05) (control; solid trace) which closely matches the delay time observed in Figure 1C. This result suggests that ET-18-OCH3 induced considerable Ca2+ influx, in keeping with our data in Figure 2 that ET-18-OCH3 induced significant capacitative Ca2+ entry. Because we have observed that La3+ is a potent blocker of capacitative Ca2+ entry in MDCK cells (Jan et al., 1998a, 1998c; 1999c, 1999d), we next examined the effect of La3+ on the ET-18-OCH3-induced Mn2+ quench of fura-2 fluorescence. Figure 7A shows that the ET-18-OCH3-induced Mn2+ quench of fura-2 fluorescence was abolished by pretreatment with 0.1 mM La3+ (dashed trace). Consistently, Figure 7B shows that pretreatment with 0.1 mM La3+ for 20 s totally suppressed 25 μM ET-18-OCH3-induced [Ca2+]i rise (trace f). La3+ (0.1 mM) also abolished the [Ca2+]i rise induced by 25 μM 2-O-methyl PAF (n=3; data not shown). When La3+ is added during the ET-18-OCH3 response, the inhibition of La3+ depends on the time it is added. Thus, when added at time points of 180 s (trace e), 205 s (trace d), 220 s (trace c) and 265 s (trace b), La3+ immediately stopped the rise of [Ca2+]i and maintained the [Ca2+]i at a persistently elevated phase. However, when added at the late phase of the [Ca2+]i rise, for example at 475 s, La3+ had little effect on the [Ca2+]i rise (trace a). Substitution of 50 μM Gd3+ for 0.1 mM La3+ produced identical results (data not shown).
Figure 7.
(A) The ET-18-OCH3-induced Ca2+ influx detected by Mn2+ quench measurements. The experiments were performed in Ca2+ medium plus 50 μM MnCl2. Solid trace: control response of ET-18-OCH3 (25 μM), applied at 30 s. Dashed trace: La3+ (0.1 mM) was added at 0 s followed by ET-18-OCH3 (25 μM) at 30 s. Excitation signal at 360 nm and emission signal at 510 nm were recorded. (B) Effect of La3+ on the ET-18-OCH3-induced [Ca2+]i rise. ET-18-OCH3 was added at 30 s in traces a–f and La3+ (0.1 mM) was added at 475 s in trace a, 265 s in trace b, 220 s in trace c, 205 s in trace d, 180 s in trace e and 0 s in trace f. Traces are typical of 3–4 experiments.
Discussion
Although ET-18-OCH3 has been found to cause a rise in [Ca2+]i in a number of cells, its effect on Ca2+ signalling is not fully characterized and the precise underlying mechanisms are not completely clear. In the present study we investigated how ET-18-OCH3 affects [Ca2+]i in MDCK cells. Our results suggest the following mechanism for the ET-18-OCH3-induced [Ca2+]i rise: ET-18-OCH3 activates PAF receptors leading to a release of Ca2+ from both thapsigargin-sensitive and -insensitive Ca2+ stores. This discharge of internal Ca2+ subsequently activates capacitative Ca2+ entry which is blocked by lanthanides. Lanthanides might also directly inhibit PAF receptors. Phospholipase D and phospholipase A2, but not phospholipase C, might be involved in triggering the [Ca2+]i rise.
We have found that ET-18-OCH3 induces a significant rise in [Ca2+]i between 10–100 μM. A similar effective concentration range was found for a neuroblastoma cell line (Brinkmeier et al., 1996) but a lower range was effective in human neutrophils (10 nM–10 μM) (Alonso et al., 1997). This discrepancy might be due to differences in cell type. We propose that ET-18-OCH3 acts through activating PAF receptors because the ET-18-OCH3-induced [Ca2+]i rise is inhibited by PCA-4248, a PAF receptor antagonist, in a concentration-dependent manner. Our results are supported by the observations that 2-O-methyl PAF, another PAF receptor agonist, also induces a [Ca2+]i rise in MDCK cells similarly to ET-18-OCH3 (data not shown), and that in U937 cells and human neutrophils WEB-2170, a PAF receptor antagonist (Heuer et al., 1990), also blocks the ET-18-OCH3-induced [Ca2+]i rise (Alonso et al., 1997).
One of the unique characteristics of the ET-18-OCH3-induced [Ca2+]i rise in MDCK cells is that it has a concentration-dependent delay before the [Ca2+]i starts to rise, which is not observed in the [Ca2+]i responses induced by other agents such as ATP (Jan et al., 1998a), bradykinin (Jan et al., 1998b), the phospholipase C inhibitor U73122 (Jan et al., 1998c), econazole (Jan et al., 1999a), SKF96365 (Jan et al., 1999b) and thapsigargin (Jan et al., 1998c) in MDCK cells. This delay time is not altered by removal of extracellular Ca2+ (compare Figure 1C to Figure 2C), implying that this delay time is needed for Ca2+ to be released from internal stores in response to ET-18-OCH3 stimulation. Because the ET-18-OCH3-induced [Ca2+]i rise is blocked by lanthanides and PCA-4248, the possibility that ET-18-OCH3 induces a [Ca2+]i rise by entering the cell and directly acting on intracellular sites is unlikely.
Our findings suggest that activation of phospholipase D and phospholipase A2, but not phospholipase C, might be involved in mediating the [Ca2+]i rise induced by ET-18-OCH3 because the [Ca2+]i rise is partly inhibited by the phospholipase D inhibitor propranolol and the phospholipase A2 inhibitor aristolochic acid, but not the phospholipase C inhibitor U73122. Indeed, in Swiss 3T3 fibroblasts ET-18-OCH3 was found to inhibit the IP3 production induced by platelet-derived growth factor (PDGF) and [AlF4]1− (Seewald et al., 1990). We found that phospholipase D and phospholipase A2 appear to be involved in the regulation of the ET-18-OCH3-induced extracellular Ca2+ influx instead of intracellular Ca2+ release because propranolol and aristolochic acid only inhibit the [Ca2+]i rise induced by ET-18-OCH3 in the presence, but not in the absence, of extracellular Ca2+. How ET-18-OCH3 releases internal Ca2+ is not clear.
We have previously shown that U73122 (Jan et al., 1998c), econazole (Jan et al., 1999a) and SKF96365 (Jan et al., 1999b) all release Ca2+ from the thapsigargin-sensitive ER Ca2+ store presumably via inhibition of the ER Ca2+ pump. However, the action of ET-18-OCH3 differs from that of these three substances at least in two aspects. First, the latter three substances are not known to act through a receptor on the plasma membrane. Second, ET-18-OCH3 not only depletes the thapsigargin-sensitive ER Ca2+ store but also releases Ca2+ from mitochondria and possibly other stores, because in the absence of extracellular Ca2+, pretreatment with ET-18-OCH3 prevents thapsigargin or CCCP from releasing Ca2+. Consistently, the [Ca2+]i rise induced by ET-18-OCH3 is not altered by pretreatment with either thapsigargin or CCCP, suggesting that both the thapsigargin-sensitive ER store and the CCCP-sensitive mitochondrial store contribute to the internal Ca2+ release induced by ET-18-OCH3. The involvement of other stores cannot be excluded.
It is interesting that econazole (25 μM) and SKF96365 (50 μM) do not inhibit the capacitative Ca2+ entry induced by ET-18-OCH3. We have recently shown that econazole partly inhibits the capacitative Ca2+ entry induced by thapsigargin (Jan et al., 1999c), cyclopiazonic acid (Jan et al., 1999a) and U73122 (Jan et al., 1998c); and that SKF96365 partly inhibited the capacitative Ca2+ entry induced by thapsigargin (Jan et al., 1999c) and 2,5-di-tert-butylhydroquinone (Jan et al., 1999d). Thus, the capacitative Ca2+ entry induced by ET-18-OCH3 and that induced by the other agents appear to be different in nature.
Brinkmeier and coworkers (1996) also found a 10–25 s delay time before [Ca2+]i starts to rise in response to ET-18-OCH3 in a neuroblastoma cell line, but whether this delay depends on concentration is not shown. However, there is no delay time in the ET-18-OCH3-induced [Ca2+]i rise in other cells such as HL-60 cells, U937 cells and human neutrophils (Alonso et al., 1997). This discrepancy may be caused by differences in cell type. Brinkmeier and coworkers (1996) proposed that the delay time is required for ET-18-OCH3 or its derivative(s) to integrate into the plasma membrane and open a Ca2+ channel based on the fact that La3+ and Gd3+ abolish the ET-18-OCH3-induced [Ca2+]i rise. However, the authors did not investigate whether ET-18-OCH3 could release internal Ca2+ by removing extracellular Ca2+ as we did in this study. As we previously discussed, La3+ is unlikely to enter MDCK cells (Jan et al., 1998a), and because we have demonstrated that La3+ abolishes the capacitative Ca2+ entry induced by ATP (Jan et al., 1998a), thapsigargin (Jan et al., 1999c), 2,5-di-tert-butylhydroquinone (Jan et al., 1999d) and U73122 (Jan et al., 1998c) in MDCK cells, it is reasonable to believe that La3+ might also abolish the ET-18-OCH3-induced capacitative Ca2+ entry. But this cannot explain why La3+ completely inhibits the ET-18-CH3-induced Mn2+ entry and [Ca2+]i rise because La3+ would not be expected to inhibit internal Ca2+ release. The inhibition of La3+ is not specific for ET-18-OCH3 alone because La3+ also abolishes the [Ca2+]i rise induced by another PAF agonist, 2-O-methyl PAF (data not shown). Thus, one possible interpretation is that La3+ might inhibit PAF receptors. This hypothesis gains support from the fact that La3+ might also inhibit ATP receptors in MDCK cells (Jan et al., 1998a).
Figure 7B shows that the La3+-sensitive capacitative Ca2+ entry is triggered early during the ET-18-OCH3-induced Ca2+ signal because when added 40 s after the [Ca2+]i starts to rise, La3+ is able to inhibit the rise of the signal. We have found a similar effect of La3+ on the phospholipase C inhibitor U73122-induced [Ca2+]i rise in MDCK cells (Jan et al., 1999c). These results suggest that capacitative Ca2+ entry is initiated when internal Ca2+ stores are only partly depleted.
In HL-60 cells, Lohmeyer & Workman (1993) proposed that the ether lipid (ET-18-OCH3 and its analogue SRI 62-834)-induced [Ca2+]i rise should be transient (i.e. with an initial peak followed by a fast decline), and the previously reported elevated plateau of the [Ca2+]i rise induced by SRI 62-834 (Lazenby et al., 1990) is due to external Ca2+ influx and leakage of internal quin-2 caused by extensive cell damage. In view of our results, it is unlikely that the elevated plateau in the 25 μM ET-18-OCH3-induced [Ca2+]i rise in MDCK cells could be attributed to cell damage-induced fura-2 leakage and external Ca2+ influx, because if it were the case, the ET-18-OCH3-induced [Ca2+]i rise would not be abolished by pretreatment with La3+ or PCA-4248.
Taken together, we have found several unique characteristics about the ET-18-OCH3-induced [Ca2+]i rise which have not been reported previously: (1) The delay time before the [Ca2+]i rise is dependent on the concentration of ET-18-OCH3. A higher concentration needs a shorter delay time. (2) ET-18-OCH3 releases internal Ca2+ from both thapsigargin-sensitive ER stores and thapsigargin-insensitive, CCCP-sensitive mitochondrial stores. (3) The ET-18-OCH3-induced [Ca2+]i rise in the presence of external Ca2+ is composed of two sources. First, an IP3-independent internal Ca2+ release followed by econazole- and SKF96365-insensitive capacitative Ca2+ entry. This capacitative Ca2+ entry might be modulated by phospholipase D and phospholipase A2, but not by phospholipase C. And lastly, we suspect that lanthanides might directly inhibit PAF receptors. What remains to be answered is how ET-18-OCH3 activation of PAF receptors causes a release of internal Ca2+ via a mechanism independent of the activities of phospholipases C, D and A2. The possibility that lanthanides might inhibit PAF receptors needs further investigation.
Acknowledgments
We thank C.M. Ho for culturing the cells. This work was supported by grants from National Science Council (NSC88-2314-B-075B-003) and Veterans General Hospital-Kaohsiung (VGHKS88-32) to C.-R. Jan
Abbreviations
- ATP
(adenosine 5′-triphosphate)
- [Ca2+]i
intracellular free Ca2+ concentration
- DMEM
Dulbecco's modified Eagle medium)
- ER
endoplasmic reticulum
- ET-18-OCH3
1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphorylcholine
- fura-2/AM
1-[2-(5-carboxyoxazol-2-yl)-6-aminobenzofuran-5-oxy]-2-(2′-amino-5′-methylphenoxy)-ethane-N,N,N,N-tetraacetic acid pentaacetoxymethyl ester
- IP3
inositol 1,4,5-trisphosphate
- MDCK cells
Madin Darby canine kidney cells
- 2-O-methyl PAF
(±)1-O-hexadecyl-2-O-methylglycero-3-phosphorylcholine
- PAF
platelet-activating factor
- PCA-4248
methyl 2-(phenylthio)ethyl-1,4-dihydro-2,4,6-trimethylpyridine-3,5-dicarboxylate
- SKF96365
1-[β-[3-(4-methoxyphenyl)propoxy]-4-methoxyphenethyl]-1H-imidazole hydrochloride
- U73122
1-(6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione
References
- ALONSO M.T., GAJATE C., MOLLINEDO F., MODOLELL M., ALVAREZ J., GARCIA-SANCHO J. Dissociation of the effects of the antitumour ether lipid ET-18-OCH3 on cytosolic calcium and on apoptosis. Br. J. Pharmacol. 1997;121:1364–1368. doi: 10.1038/sj.bjp.0701271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERDEL W.E. Membrane-interactive lipids as experimental anticancer drugs. Br. J. Cancer. 1991;64:208–211. doi: 10.1038/bjc.1991.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERGMANN J., JUNGHAHN I., BRACHWITZ H., LANGEN P. Multiple effects of antitumor alkyl-lysophospholipid analogs on the cytosolic free Ca2+ concentration in a normal and a breast cancer cell line. Anticancer Res. 1994;14:1549–1556. [PubMed] [Google Scholar]
- BERKOVIC D., BERKOVIC K., FLEERE A., EIBL H., UNGER C. Inhibition of calcium-dependent protein kinase C by hexadecylphosphocholine and 1-O-octadecy1-2-O-methyl-rac-glycero-3-phosphocholine do not correlate with inhibition of proliferation of HL60 and K562 cell lines. Eur. J. Cancer. 1994;30A:509–515. doi: 10.1016/0959-8049(94)90428-6. [DOI] [PubMed] [Google Scholar]
- BILLAH M.M. , ECKEL S. , MULLMANN T.J. , EGAN R.W., SIEGEL M.I. Phosphatidylcholine hydrolysis by phospholipase D determines phosphatidate and diglyceride levels in chemotactic peptide-stimulated human neutrophils. Involvement of phosphatidate phosphohydrolase in signal transduction. Biochim. Biophys. Acta. 1989;1001:1–8. [PubMed] [Google Scholar]
- BOGGS K.P., ROCK C.O., JACKOWSKI S. Lysophosphatidylcholine and 1-O-octadecy1-2-O-methyl-rac-glycero-3-phosphocholine inhibit the CDP-choline pathway of phosphatidylcholine synthesis at the CTP: phosphocholine cytidylyltransferase step. J. Biol. Chem. 1995;270:7757–7764. doi: 10.1074/jbc.270.13.7757. [DOI] [PubMed] [Google Scholar]
- BRINKMEIER H., SCHNEIDER M., RUDEL R. Ether lipid-induced cell damage of neuroblastoma cells is only weakly correlated with increased intracellular Ca2+ levels. Cell Calcium. 1996;19:383–390. doi: 10.1016/s0143-4160(96)90111-6. [DOI] [PubMed] [Google Scholar]
- DIOMEDE L., COLOTTA F., PIOVANI B., RE F., MODEST E.J., SALMONA M. Induction of apoptosis in human leukemic cells by the ether lipid 1-octadecyl-2-methyl-rac-glycero-3-phosphocholine. Int. J. Cancer. 1993;53:124–130. doi: 10.1002/ijc.2910530123. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HEUER H.O., CASALS-STENZEL J., MUACEVIC G., WEBER K.H. Pharmacologic activity of bepafant (WEB 2170), a new and selective hetrazepinoic antagonist of platelet activating factor. J. Pharmacol. Exp. Ther. 1990;255:962–968. [PubMed] [Google Scholar]
- JAN C.R., HO C.M., WU S.N., HUANG J.K., TSENG C.J. Mechanism of lanthanum inhibition of extracellular ATP-evoked calcium mobilization in MDCK cells. Life Sci. 1998a;62:533–540. doi: 10.1016/s0024-3205(97)01149-1. [DOI] [PubMed] [Google Scholar]
- JAN C.R., HO C.M., WU S.N., TSENG C.J. Bradykinin-evoked Ca2+ mobilization in MDCK cells. Eur. J. Pharmacol. 1998b;355:219–233. doi: 10.1016/s0014-2999(98)00481-6. [DOI] [PubMed] [Google Scholar]
- JAN C.R., HO C.M., WU S.N., TSENG C.J. The phospholipase C inhibitor U73122 elevates cytoplasmic calcium levels in Madin Darby canine kidney cells by activating calcium influx and releasing stored calcium. Life Sci. 1998c;63:895–908. doi: 10.1016/s0024-3205(98)00346-4. [DOI] [PubMed] [Google Scholar]
- JAN C.R., HO C.M., WU S.N., TSENG C.J. Multiple effects of econazole on calcium signaling: depletion of thapsigargin-sensitive calcium store, activation of extracellular calcium influx, and inhibition of capacitative calcium entry. Biochim. Biophys. Acta. 1999a;1448:533–542. doi: 10.1016/s0167-4889(98)00159-1. [DOI] [PubMed] [Google Scholar]
- JAN C.R., HO C.M., WU SN., TSENG C.J. Multiple effects of 1-[β-[3-(4-methoxyphenyl)propoxy]-4-methoxyphenethyl]-1H-imidazole (SKF 96365) on Ca2+ signaling in MDCK cells: depletion of thapsigargin-sensitive Ca2+ store followed by capacitative Ca2+ entry, activation of a direct Ca2+ entry, and inhibition of thapsigargin-induced capacitative Ca2+ entry. Naunyn-Schmeideberg's Arch Pharmacol. 1999b;359:92–101. doi: 10.1007/pl00005336. [DOI] [PubMed] [Google Scholar]
- JAN C.R., HO C.M., WU S.N., TSENG C.J. Mechanism of rise and decay of thapsigargin-evoked calcium signals in MDCK cells. Life Sci. 1999c;64:259–267. doi: 10.1016/s0024-3205(98)00561-x. [DOI] [PubMed] [Google Scholar]
- JAN C.R., HO C.M., WU S.N., TSENG C.J. Mechanism of rise and decay of 2,5-di-tert-butylhydroquinone-induced Ca2+ signals in MDCK cells. Eur. J. Pharmacol. 1999d;365:111–117. doi: 10.1016/s0014-2999(98)00871-1. [DOI] [PubMed] [Google Scholar]
- LAZENBY C.M., THOMPSON M.G., HICKMAN J.A. Elevation of leukemic cell intracellular calcium by the ether lipid SRI 62-834. Cancer Research. 1990;50:3327–3330. [PubMed] [Google Scholar]
- LOHMEYER M., WORKMAN P. The role of intracellular free calcium mobilization in the mechanism of action of antitumor ether lipids SRI 62-834 and ET 18-OMe. Biochem. Pharmacol. 1993;45:77–86. doi: 10.1016/0006-2952(93)90379-b. [DOI] [PubMed] [Google Scholar]
- MCCONKEY D.J., ORRENIUS S. Signal transduction pathways in apoptosis. Stem Cells. 1996;14:619–631. doi: 10.1002/stem.140619. [DOI] [PubMed] [Google Scholar]
- MERRITT J.E., JACOB R., HALLAM T.J. Use of manganese to discriminate between calcium influx and mobilization from internal stores in stimulated human neutrophils. J. Biol. Chem. 1989;264:1522–1527. [PubMed] [Google Scholar]
- MOLLINEDO F., FERNANDEZ-LUNA J.L., GAJATE C., MARTIN-MARTIN B., BENITO A., MARTINEZ-DALMAU R., MODOLELL M. Selective induction of apoptosis in cancer cells by the ether lipid ET-18-O-CH3 (Edelfosine): Molecular requirements, cellular uptake, and protection by Bcl-2 and Bcl-XL. Cancer Res. 1997;57:1320–1328. [PubMed] [Google Scholar]
- MOLLINEDO F., MARTINEZ-DALMAU R., MODOLELL M. Early and selective induction of apoptosis in human leukemic cells by the alkyl-lysophospholipid ET-18-OCH3. Biochem. Biophys. Res. Commun. 1993;192:603–609. doi: 10.1006/bbrc.1993.1458. [DOI] [PubMed] [Google Scholar]
- ORTEGA M.P., GARCIA M.C., GIJON M.A., DE CASA-JUANA M.F., PRIEGO J.G., SANCHEZ CRESPO M., SUNKEL C. 1,4-Dihydropyridines, a new class of platelet-activating factor receptor antagonists: in vitro pharmacologic studies. J. Pharmacol. Exp. Ther. 1990;255:28–33. [PubMed] [Google Scholar]
- PALTAUF F. Ether lipids in biomembranes. Chem. Phys. Lipids. 1994;74:101–139. doi: 10.1016/0009-3084(94)90054-x. [DOI] [PubMed] [Google Scholar]
- PUTNEY J.W., JR, BIRD G.S. The signal for capacitative calcium entry. Cell. 1993;75:199–201. doi: 10.1016/0092-8674(93)80061-i. [DOI] [PubMed] [Google Scholar]
- ROSENTHAL M.D. , VISHWANATH B.S., FRANSON R.C. Effects of aristolochic acid on phospholipase A2 activity and arachidonate metabolism of human neutrophils. J. Biol. Chem. 1989;264:17069–17077. doi: 10.1016/0005-2760(89)90299-3. [DOI] [PubMed] [Google Scholar]
- SEEWALD M.J., OLSEN RA., SEHGAL I., MELDER D.C., MODEST E.J., POWIS G. Inhibition of growth factor-dependent inositol phosphate Ca2+ signaling by antitumor ether lipid analogues. Cancer Res. 1990;50:4458–4463. [PubMed] [Google Scholar]
- THASTRUP O., CULLEN P.T., DROBAK B.K., HANLEY M.R., DAWSON A.P. Thapsigargin, a tumor promotor, discharges intracellular calcium stores by specific inhibition of the endoplasmic reticulum calcium ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG B., OISHI K., SHOJI K., SHOJI M. , EIBL H., BERDEL W.E. , HAJDU J., VOGLER W.R., KUO J.F. Inhibition of protein kinase C, (sodium plus potassium)-activated adenosine triphosphatase, and sodium pump by synthetic phospholipid analogues. Cancer Res. 1990;50:3025–3031. [PubMed] [Google Scholar]