

Abstract
The cardioprotective properties of inhibition of poly (ADP-ribose) synthetase (PARS) were investigated in the isolated perfused heart of the rat. Hearts were perfused in the Langendorff mode and subjected to 23 min total global ischaemia and reperfused for 60 min.
Left ventricular function was assessed by means of an intra-ventricular balloon. High energy phosphates were measured by 31P-NMR spectroscopy. Intracellular levels of NAD were measured by capillary electrophoresis of perchloric acid extracts of hearts at the end of reperfusion.
Reperfusion in the presence of the PARS inhibitor 1,5 didroxyisoquinoline (ISO, 100 μM) attenuated the mechanical dysfunction observed following 1 h of reperfusion; 27±13 and 65±8% recovery of preischaemic rate pressure product for control and 100 μM ISO, respectively.
This cardioprotection was accompanied by a preservation of intracellular high-energy phosphates during reperfusion; 38±2 vs 58±4% (P<0.05) of preischaemic levels of phosphocreatine (PCr) for control and 100 μM ISO respectively and 23±1 vs 31±3% (P<0.05) of preischaemic levels of ATP for control and 100 μM ISO respectively.
Cellular levels of NAD were higher in ISO treated hearts at the end of reperfusion; 2.56±0.45 vs 4.76±1.12 μmoles g−1 dry weight (P<0.05) for control and ISO treated.
These results demonstrate that the cardioprotection afforded by inhibition of PARS activity with ISO is accompanied by a preservation of high-energy phosphates and cellular NAD levels and suggest that the mechanism responsible for this cardioprotection may involve prevention of intracellular ATP depletion.
Keywords: ATP, creatine phosphate, global ischaemia, NMR, reperfusion injury, PARS, poly (ADP-ribose) polymerase, reactive oxygen species
Introduction
It is now well accepted that reactive oxygen species (ROS) such as superoxide anions (O2−), hydroxyl radicals (OH·), and hydrogen peroxide (H2O2) as well as peroxynitrite (ONOO−) contribute to reperfusion injury of the previously ischaemic myocardium (see Kukreja & Hess, 1992; Wang & Zweier 1996). The generation of ROS during either ischaemia or reperfusion has been directly demonstrated using electron paramagnetic resonance spectroscopy (Zweier et al., 1987), chemiluminescence (Henry et al., 1990) and spin trapping (Bolli et al., 1988). Interventions which either attenuate the generation or reduce the effects of ROS protect the heart against ischaemia-reperfusion injury (Jolly et al., 1984; Manning et al., 1984; Ambrosio et al., 1986). ROS cause cell injury by peroxidation of membrane lipids, denaturation of proteins such as enzymes and ion channels, and DNA injury. In addition, it has been demonstrated that depletion of intracellular high energy phosphates contributes to ROS-mediated cell damage (Schraufstatter et al., 1986a). For instance, exposure of cultured cells to ROS including H2O2, ONOO− or O2− results in single strand breaks in DNA and subsequent activation of the DNA repair enzyme, poly (ADP-ribose) synthetase (PARS, also known as poly (ADP-ribose) polymerase, PARP, or ADP ribosyltransferase, ADP-RT) (Schraufstatter et al., 1986a). Once activated, PARS catalyzes the poly (ADP ribosyl)ation of nuclear proteins by transfer of (ADP-ribose) groups from nicotinamide adenine dinucleotide (NAD) to the proteins with concomitant formation of nicotinamide. Under conditions of oxidant stress, DNA injury causes excessive activation of the PARS enzyme resulting in a fall in the intracellular levels of its substrate NAD (Schraufstatter et al., 1986a). The nicotinamide formed when PARS is activated can be recycled to NAD in a reaction that consumes ATP (Carson et al., 1986). This futile cycle of NAD utilization and synthesis may deplete intracellular levels of ATP. In addition, if the depletion of intracellular levels of NAD is sufficiently severe this may also contribute to depletion of ATP through interference with mitochondrial function. A decline in the intracellular levels of ATP results in severe cellular dysfunction and ultimately cell death (Schraufstatter et al., 1986a). This pathway of cell death has been termed the ‘PARS Suicide Hypothesis' (Berger et al., 1985). Inhibition of the activity of PARS e.g. with 3-aminobenzamide, prevents the NAD and ATP depletion caused by oxidant stress, and improves cell survival (Schraufstatter et al., 1986b).
We, and others, have recently discovered that inhibition of PARS activity attenuates myocardial reperfusion injury in rabbits (Thiemermann et al., 1997) and in rats (Zingarelli et al., 1997). The administration, upon reperfusion, of inhibitors of PARS activity attenuate (i) the degree of necrosis caused by regional ischaemia and reperfusion in the anaesthetized rabbit and (ii) attenuate the contractile dysfunction caused by global ischaemia and reperfusion in the isolated, perfused heart of the rabbit (Thiemermann et al., 1997). Thus, we proposed that (i) activation of PARS contributes to reperfusion injury and (ii) that inhibition of PARS activity may be useful in the therapy of reperfusion injury of the heart. The exact mechanism(s) by which PARS inhibitors cause the observed reduction in infarct size is not clear, but may be due to inhibition of PARS activity and preservation of high energy phosphates. Myocardial ischaemia and reperfusion causes a reduction in high energy phosphates, post-ischaemic contractile dysfunction and infarction (Jennings et al., 1978; Vary et al., 1979).
This study demonstrates that administration during reperfusion of the PARS inhibitor, 1,5-dihydroxyisoquinoline reduces both the contractile dysfunction caused by global ischaemia and reperfusion of the isolated, perfused heart of the rat. The mechanism of action is related to preservation of high energy phosphates.
Methods
Perfusion of isolated hearts
All experiments conformed to the guidelines set out by the Canadian Council on Animal Care regarding the care and use of experimental animals and were approved by the local Animal Care Committee of the National Research Council of Canada.
Male Sprague-Dawley rats (300–350 g) were obtained from Charles River and acclimatized to our animal holding facilities for 1 week prior to use. The rats were anaesthetized with sodium pentobarbitone (120 mg kg−1 i.p.). The hearts were excised, arrested in ice cold buffer and rapidly perfused in the Langendorff mode at 36.5°C. After insertion of an apical drain in the left venticle to vent the drainage from the Thebesian veins, a water filled compliant balloon was inserted into the left ventricle via the mitral valve. The balloon was connected to a pressure transducer (Stratham P23Db) to monitor left ventricular pressure and heart rate. Perfusion pressure was monitored by means of a pressure transducer (Stratham P23Db) connected to a side arm from the aortic perfusion line. Perfusion rate was monitored by means of an in-line ultrasonic flow probe (Transonics Inc). The left ventricular end diastolic pressure was adjusted to 5–10 mmHg by inflating the balloon. The balloon volume was then kept constant throughout the course of the experiment. Mechanical function was assessed as the heart rate, end diastolic pressure, developed pressure, the rate pressure product (RPP, the product of heart rate times left ventricular developed pressure), +dP dt−1 and −dP dt−1. Functional data were acquired and analysed using a PC based system (Digi-Med®, Louisville, KY, U.S.A.). Time to ischaemic contracture was measured as the time for the end diastolic pressure to increase by 10 mmHg during ischaemia. The hearts were perfused at a constant pressure of 70 mmHg with a modified Krebs'-Henseleit (K-H) buffer containing (mM) NaCl 118, KCl 4.7, CaCl2 1.75 (free Ca2+≈1.1 mM), MgSO4 1.2, EDTA 0.5, NaHCO3 25 and glucose 11, equilibrated with 95% O2/5% CO2 to achieve a pH of 7.4. Hearts were placed in a 20 mm NMR tube. Perfusate was aspirated by means of a suction line positioned just above the level of the aortic cannula, such that the heart was totally immersed in a bath of perfusate. This, in conjunction with the variable temperature unit in the magnet, ensured that the temperature throughout the experiment was maintained in the range 36–37°C (as determined by means of a thermocouple inserted in the left ventricle during initial experiments in which no NMR data were acquired).
NMR spectroscopy
NMR experiments were performed on a Bruker AM-360 spectrometer equipped with a broadband probe. Field homogeneity was adjusted by shimming on the sodium signal from the sample yielding line widths of 10–15 Hz. 31P NMR spectra were acquired at 145.8 MHz with a time resolution of 2.5 min by summing 72 free induction decays obtained using a 60° pulse and a recycle time of 2.3 s. The sweep width was 10 kHz and 4096 data points were collected. Spectra were processed by Fourier transformation following exponential multiplication (LB, 20 Hz). Spectra were processed using 1D WINNMR (Bruker). A capillary tube containing phenylphosphonic acid was placed next to the heart and acted as an internal reference for the NMR experiments.
Experimental protocol
Hearts were allowed to stabilize for a period of 15 min, during which time the magnet was shimmed and the NMR probe was tuned. The mechanical performance at the end of this stabilization period was taken as the pre-ischaemic function and upon which post-ischaemic recovery of function was based. The hearts were subjected to 23 min total global ischaemia by clamping the aortic perfusion line. Reperfusion was conducted under conditions of constant pressure by removing the clamp from the aortic perfusion line and was continued for 60 min. The PARS inhibitor, 1,5-dihydroxyisoquinoline (Aldrich) was infused as a concentrated stock solution by means of a pump driven syringe. This delivered the drug through an infusion line connected to the main perfusion line just above the aortic cannula. The drug was administered at 1/60th of the perfusion rate to achieve a final concentration of 100 μM. Infusion of 1,5-dihydroxyisoquinoline was started at reperfusion and continued for the first 45 min of reperfusion. 1,5-dihydroxyisoquinoline was initially dissolved in DMSO and then diluted with water to achieve a concentration of 6 mM and a DMSO concentration of 10%. Following the 60 fold dilution in the perfusion buffer this resulted in a final drug concentration of 100 μM in the presence of 0.15% DMSO. The rate of drug infusion was altered to match changes in the rate of perfusion to maintain the desired drug concentration. Control hearts were treated in similar fashion with DMSO to achieve a final concentration of 0.15% DMSO. Care was taken to continually adjust the infusion rate of drug or vehicle according to the direct read-out of rate of perfusion on the flow meter. This was considered important to ensure that all hearts were exposed to identical concentrations of DMSO thereby overcoming concerns that any observed cardioprotection may have been due to the radical scavenging properties of DMSO rather than the PARS inhibitory effects of ISO.
Determination of cellular NAD levels
At the end of reperfusion hearts from the 100 μM ISO and control groups were freeze clamped using aluminum tongs cooled to the temperature of liquid nitrogen and immediately plunged into liquid nitrogen. Perchloric acid extracts of samples of left ventricular tissue (≈100 mg) were neutralized and subjected to capillary zone electrophoresis for determination of total NAD (Grune & Perret, 1994). Dry : wet weight ratios were determined on aliquots of left ventricular tissue by drying to constant weight at 70°C. NAD determinations were expressed as μmoles gm−1 dry weight.
Statistical analysis
Data are expressed as mean±s.e.mean. Statistical analyses were performed using the statistical package Statistica. One way ANOVA's between groups for all time points was used as an exploratory analysis to determine the time of drug effectiveness. A repeated measures ANOVA between groups was then used over the time intervals 30–60 min reperfusion for mechanical function and 20–60 min reperfusion for energetics data. Duncan's test was used to determine statistical significance between the vehicle and drug treated groups. Pearson correlation coefficients were determined using the Systat package.
Results
Mean baseline values (Pre-Ischaemia) for all the parameters studied were similar in the control and drug treated groups (Figures 123; P>0.05). Thus, there was no difference in the basal function of the hearts between groups. Although we did not specifically test it in this study, we have previously demonstrated that the mechanical function in this model is stable over the time frame of this protocol, with the developed pressure decreasing 20% following 2 h of normoxic perfusion (Musat-Marcu et al., 1999).
Figure 1.
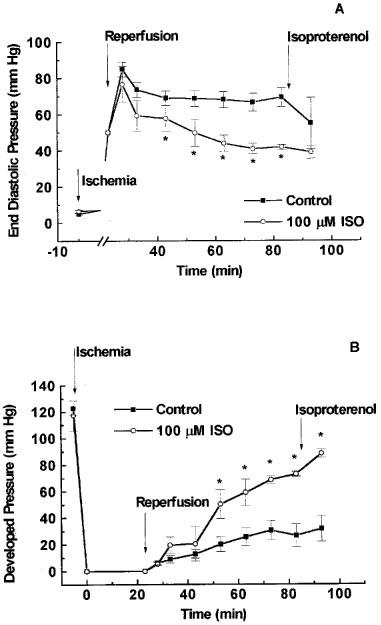
Effect of 1,5-dihydroxyisoquinoline (ISO) on diastolic and systolic function during reperfusion of the ischaemic heart. Rat hearts were subjected to 23 min of total global ischaemia and reperfused in the presence of 100 μM 1,5-dihydroxyisoquinoline or its vehicle (0.15% dimethyl sulphoxide). The drug or its vehicle was present throughout the initial 45 min of reperfusion. At the end of 60 min reperfusion the mechanical function of the heart was stimulated by infusion of isoproterenol (0.1 μM). (A) Shows the effects on end diastolic pressure and (B) shows the data for left ventricular developed pressure. *P<0.05 when compared with vehicle treated hearts.
Figure 2.
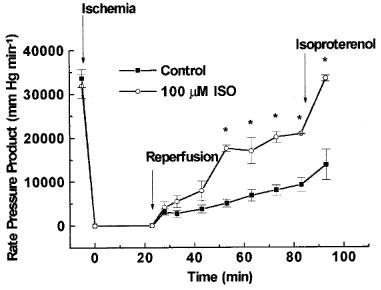
Protective effects of 1,5-dihydroxyisoquinoline (ISO) on total cardiac work during reperfusion of the ischaemic heart. Cardiac work was assessed as Rate Pressure Product (heart rate×developed pressure). Conditions were as described in Figure 1.
Figure 3.
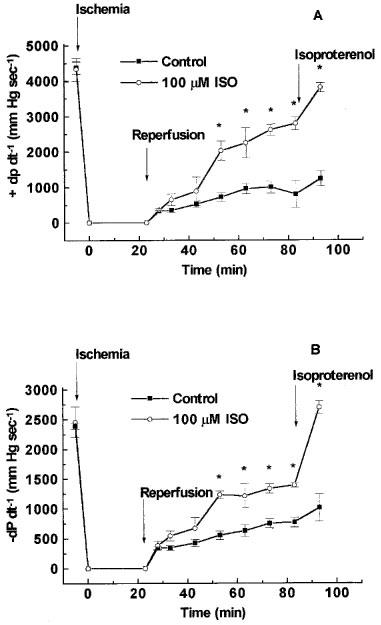
Effect of 1,5-dihydroxyisoquinoline (ISO) on contractility (+dP dt−1), shown in (A) and rate of relaxation (−dP dt−1) shown in (B) during reperfusion. Conditions were as described in Figure 1.
In hearts subjected to global myocardial ischaemia, the mean time to the onset of the development of ischaemic contracture was similar in both groups (Control: 15.7±0.4 min; 100 μM ISO: 15.6±0.5 min) indicating that the severity of ischaemia was similar in the two groups.
In hearts treated with vehicle (0.15% DMSO, control), reperfusion following 23 min of total global ischaemia caused a significant contractile dysfunction. Diastolic function was impaired as indicated by an increase in end diastolic pressure to approximately 70 mmHg at the end of reperfusion (Figure 1A), and developed pressure was reduced to 22% of the pre-ischaemic level at the end of reperfusion (Figure 1B). In addition, the recovery of the rate pressure product during reperfusion was markedly impaired resulting in a recovery to 27% of pre-ischaemic function at the end of reperfusion (Figure 2). Similarly, the recovery of +dP dt−1 as well as −dP dt−1 were impaired during the reperfusion period (Figure 3). Stimulation with isoproterenol (0.1 μM) at the end of the reperfusion period caused moderate (if any) increases in developed pressure (Figure 1) rate pressure product (Figure 2), +dP dt−1 and −dP dt−1 (Figure 3).
Reperfusion of the hearts with buffer containing the PARS inhibitor 1,5-dihydroxyisoquinoline attenuated the contractile dysfunction during reperfusion (Figures 123). Preliminary experiments performed in attempts to determine an optimally effective concentration of ISO demonstrated a trend towards cardioprotection in the presence of 50 or 75 μM ISO (data not shown). The highest concentration of 1,5-dihydroxyisoquinoline employed (100 μM) had a significant effect on all of the functional parameters studied (Figures 123). For instance, reperfusion of hearts with buffer containing 100 μM of 1,5-dihydroxyisoquinoline caused a fall in end-diastolic pressure (from elevated towards normal values) during the reperfusion period (Figure 1). The recoveries of developed pressure and the rate-pressure product were also significantly improved in the presence of 1,5-dihydroxyisoquinoline (Figures 1 and 2). The recovery of contractility during reperfusion, assessed as the rate of development of pressure (+dP dt−1) and the rate of relaxation (−dP dt−1) were also significantly improved by treatment with the PARS inhibitor (Figure 3). Concentrations of ISO greater than 100 μM were not tested due to the limited water solubility of this compound. Administration of higher concentrations of ISO would only have been possible in the presence of unacceptably high levels of DMSO.
In hearts treated with 100 μM 1,5-dihydroxyisoquinoline, stimulation with isoproterenol further enhanced both developed pressure and rate-pressure product at the end of reperfusion. For example, in hearts treated with 100 μM of 1,5-dihydroxyisoquinoline, isoproterenol increased developed pressure from 62% of the pre-ischaemic value to 75% and increased the rate-pressure product from 65 to 106% (Figures 1 and 2). The latter effect was largely due to a profound increase in heart rate in the presence of isoproterenol. The recoveries of +dP dt−1 and −dP dt−1 in these hearts were also enhanced in the presence of isoproterenol (to pre-ischaemic values). Although developed pressure and rate pressure product were dramatically improved in the presence of isoproterenol, end diastolic pressure did remain elevated under these conditions.
Treatment with the PARS inhibitor had no significant effect on coronary flow demonstrating that the beneficial effects of this intervention are not secondary to preservation of vascular function (Table 1).
Table 1.
Coronary flow pre and post ischaemia

Examples of the NMR spectra obtained from a drug treated heart are shown in Figure 4. The spectra consist of three peaks corresponding to the three phosphates of ATP, the PCR peak, a small inorganic phosphate (Pi) peak and the reference phenylphosphonic acid. In addition to information on intracellular energetics these spectra may also be used to measure the intracellular pH from the pH dependent change in the chemical shift of Pi. There was no difference in the intracellular pH between the two groups (data not shown). In hearts subjected to global ischaemia and reperfusion and treated with vehicle (0.15% DMSO, control), there was a limited recovery of intracellular levels of high-energy phosphates during reperfusion (Figure 5). The beneficial effects of the PARS inhibitor 1,5-dihydroxyisoquinoline on recovery of mechanical function were accompanied by a partial preservation of energy status during reperfusion. The PARS inhibitor significantly increased the recovery of PCr levels at the end of reperfusion with this beneficial effect becoming apparent following 15 min of reperfusion (Figure 5). Similarly, infusion of 1,5-dihydroxyisoquinoline preserved intracellular ATP with this effect becoming apparent following 15–20 min of reperfusion (Figure 5).
Figure 4.
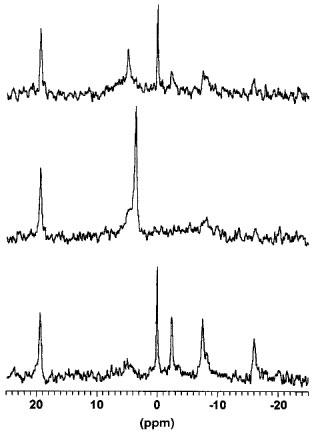
31P NMR spectra of a heart isolated from a rat and treated with 100 μM 1,5-dihydroxyisoquinoline during reperfusion following 23 min of total global ischaemia. The bottom trace shows a spectrum obtained at baseline prior to ischaemia, the middle trace is a spectrum obtained during the interval 20.5–23 min ischaemia and the top trace was obtained between 7.5 and 10 min of reperfusion. The peaks, from left to right are; the phenylphosphonic acid standard (at 19.4 p.p.m.), inorganic phosphate (Pi, initially at 5 p.p.m.), phosphocreatine (0 p.p.m.), the γ phosphate of ATP (at −2.5 p.p.m.), the α phosphate of ATP (at −7.5 p.p.m.) and the β phosphate of ATP (at −16 p.p.m.). The bottom trace demonstrates a well oxygenated heart with excellent energetics and very little Pi. The trace obtained at the end of ischaemia reveals an almost complete lack of high energy phosphates, a very large Pi peak and a change in the chemical shift of this Pi peak indicative of severe intracellular acidosis. The spectrum obtained during reperfusion demonstrates a partial recovery of energetics and a restoration of normal intracellular pH.
Figure 5.
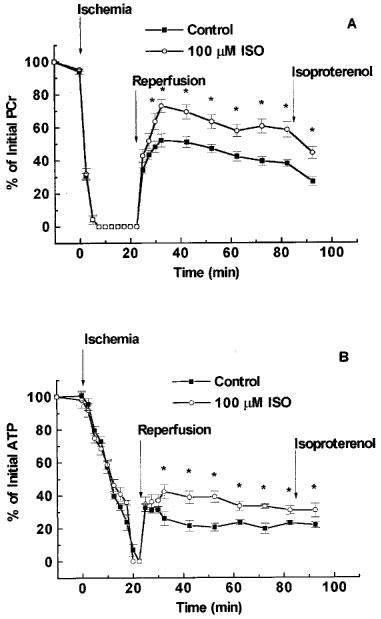
1,5-dihydroxyisoquinoline (ISO) preserves the intracellular levels of high energy phosphates during reperfusion of ischaemic hearts. PCr (A) and ATP (B) levels were determined by 31P NMR spectroscopy of hearts isolated from rats. The hearts were subjected to 23 min of total global ischaemia and reperfused for 60 min. ISO (100 μM) or its vehicle (0.15% dimethyl sulphoxide) was present for the initial 45 min of reperfusion. Following 60 min of reperfusion the mechanical function of the hearts were stimulated by infusion of isoproterenol (0.1 μM). This stimulation of work was accompanied by a decrease in PCr levels. Values for ATP and PCr are expressed as per cent of pre-ischaemic levels.
There was a good association between functional recovery (EDP, DP and RPP) and cardiac energetics (ATP, PCr) at the end of reperfusion. This analysis was performed using data obtained from 28 hearts used in the study, including hearts used in the preliminary experiments designed to determine an optimally effective concentration of 1,5-dihydroxyisoquinoline. PCr levels at the end of reperfusion correlated with EDP, DP and RPP with Pearson correlation coefficients of −0.707, 0.646 and 0.753 (P<0.001 in all cases) respectively. The Pearson correlation coefficients for ATP with EDP, DP and RPP were −0.616, 0.680 and 0.699 (P<0.001 for all) respectively. The plots for PCr vs RPP and also for ATP vs EDP are shown in Figure 6 as examples of these analyses.
Figure 6.
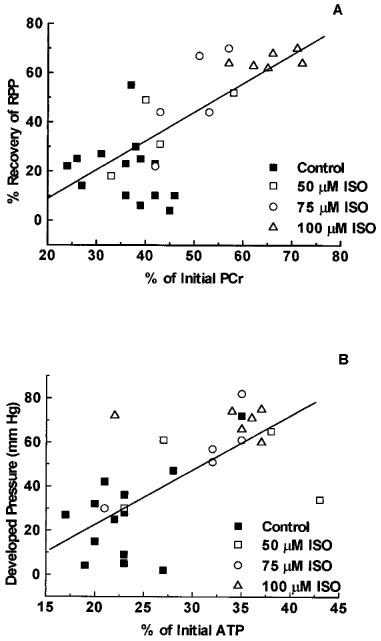
The relationship between preservation of high energy phosphates and restoration of function following reperfusion of the ischaemic heart. Hearts isolated from rats were subjected to 23 min total global ischaemia and reperfused for 60 min. Data are presented for all hearts (n=28) used in the present study and includes hearts reperfused in the presence of vehicle (0.15% DMSO, control) or 50, 75 or 100 μM 1,5-dihydroxyisoquinoline (ISO). (A) Shows the association between the recovery of PCr (expressed as per cent of the pre-ischaemic level of PCr) following 60 min of reperfusion and the rate pressure product of the heart (heart rate×developed pressure) at the end of reperfusion. (B) Shows the relationship between recovery of ATP levels at 60 min of reperfusion and the developed pressure at the end of reperfusion.
Total cellular levels of NAD in hearts freeze clamped at the end of reperfusion were also higher in hearts treated with 100 μM ISO (4.75±1.12 μmoles gm−1 dry weight) compared to control hearts (2.56±0.45 μmoles gm−1 dry weight; P<0.05). This recovery of NAD compares favourably to baseline levels of 4.82±1.0 μmoles gm−1 dry weight in the perfused rat heart (Humphrey et al., 1987).
Discussion
In January 1997, we have reported that the administration, upon reperfusion, of several chemically distinct inhibitors of PARS activity including 1,5-dihydroxyisoquinoline, 3-aminobenzamide, benzamide and nicotinamide reduce the infarct size caused by regional myocardial ischaemia and reperfusion in the anaesthetized rabbit (Thiemermann et al., 1997). In November 1997, Zingarelli and colleagues reported that the PARS-inhibitor 3-aminobenzamide also reduces the myocardial injury caused by regional myocardial ischaemic and reperfusion in the anaesthetized rat (Zingarelli et al., 1997). The exact mechanism of the cardioprotective effects of these PARS inhibitors is still unclear, but may involve a direct protection of cardiomyocytes against reperfusion injury. This study demonstrates that the PARS inhibitor 1,5-dihydroxyisoquinoline attenuates the contractile dysfunction caused by global ischaemia and reperfusion of the isolated, buffer-perfused heart of the rat. Most notably, administration of isoproterenol to hearts which had been subjected to global ischaemia and treated with 1,5-dihydroxyisoquinoline (100 μM), the rate-pressure product as well as +dP dt−1 and −dP dt−1 returned to values which were not significantly different from those measured prior to the onset of myocardial ischaemia.
What, then, is the mechanism by which the PARS inhibitor 1,5-dihydroxyisoquinoline reduces the contractile dysfunction caused by global ischaemia-reperfusion of the heart? Reperfusion of the previously ischaemic myocardium leads to the generation of ROS including superoxide anions, hydrogen peroxide, hydroxyl radicals as well as peroxynitrite (Zweier et al., 1987; Wang et al., 1996). All of these radicals cause DNA strand breaks in cultured cells (Schraufstatter et al., 1986b; Okamoto et al., 1996; Thies & Autor, 1991). There is good evidence that inhibitors of PARS activity do not reduce the degree of DNA damage suggesting that these agents do not function as radical scavengers (Schraufstatter et al., 1986a,1986b). Indeed, the PARS inhibitor 1,5-dihydroxyisoquinoline does not scavenge superoxide anions (generated in vitro) (Bowes et al., 1999). Thus, it appears unlikely that the beneficial effects of 1,5-dihydroxyisoquinoline observed here are due to the scavenging of ROS. We have recently demonstrated that exposure of rat or human cardiac myoblasts in culture to hydrogen peroxide causes an increase in PARS activity (within 30 min) followed by cell necrosis (within 4 h). Treatment of these cells with several PARS inhibitors including 1,5-dihydroxyisoquinoline attenuated the increase in PARS activity and reduced the cell injury/necrosis caused by hydrogen peroxide in these cells (Bowes et al., 1998; 1999). Accumulation of poly (ADP-ribose) has recently been demonstrated in ischaemic and reperfused hearts (Walles et al., 1998a) suggesting that PARS activity is indeed stimulated in the intact heart under the conditions used in the present study.
Recent studies utilizing PARS knockout mice have lent support to the notion that activation of PARS at reperfusion contributes to myocardial reperfusion injury. A fibroblast cell line derived from PARS knockout mice was protected against the suppression of mitochondrial respiration in response to peroxynitrite or reoxygenation, as compared to cells derived from wild type mice (Gilad et al., 1997). Similar protection was observed following hypoxia-reoxygenation of isolated working hearts, with improved functional recovery in hearts from PARS knock out mice as compared to wild type (Grupp et al., 1999). Studies on ischaemia-reperfusion injury in vivo in PARS knockout mice suggested that this cardioprotection may be–in part–due to a decreased recruitment of neutrophils secondary to an inhibition of the expression of P-selectin and intercellular adhesion molecule 1 (Zingarelli et al., 1998). It should be noted that the latter study does not rule out additional direct effects of the PARS inhibitors used on the myocardium. Indeed, isolated, buffer-perfused hearts from PARS knockout mice were similarly protected against the injury/dysfunction caused by total global ischaemia (Walles et al., 1998b). In this preliminary report, the improved functional recovery was accompanied by a preservation of cardiac energetics.
In the present study treatment with 1,5-dihydroxyisoquinoline at reperfusion significantly improved the recovery of NAD and high energy phosphates during reperfusion. It is likely that this beneficial effect on cardiac energetics contributes to the observed attenuation of contractile dysfunction following reperfusion of the ischaemic myocardium. There remains some controversy regarding the correlation between the recovery of function and intracellular ATP levels following myocardial ischaemia-reperfusion. Thus, the studies of Neely & Grotyohann (1984) suggested that there is no relation between intracellular ATP levels and functional recovery following ischaemia-reperfusion. On the other hand, studies by Rovetto (1979) suggested that there is indeed a correlation between heart ATP content and work during reperfusion. In the present study we did observe a correlation between cardiac energetics at the end of reperfusion and functional recovery. This correlation, while significant, was quite modest and this appears reasonable given the multi-factorial nature of ischaemia-reperfusion injury. A stronger correlation between recovery of energetics and function would be observed only if energetic status were the only determinant of the degree of ischaemia-reperfusion injury. The correlation between recovery of energetics and function lends support to the notion that the cardioprotective effects of PARS inhibition are, at least in part, due to preservation of energetic status.
In the present study, stimulation with isoproterenol at the end of the reperfusion period increased the functional recovery in the hearts treated with the PARS inhibitor, 1,5-dihydroxyisoquinoline to pre-ischaemic levels. This is likely to be a result of increased Ca2+ influx overcoming the decreased sensitivity of the myofilaments to Ca2+ (Kusuoka et al., 1987; Carrozza et al., 1992). The isoproterenol-enhanced recovery of function was not observed in the vehicle-treated hearts. These results suggest that the ischaemia-reperfusion insult to the untreated hearts resulted in irreversible damage and/or insufficient energy reserves to respond to the isoproterenol stimulation. The degree of irreversible damage in the 1,5-dihydroxyisoquinoline-treated hearts appeared to be attenuated and sufficient high energy phosphates remained to allow the hearts to respond to the isoproterenol stimulation. Preservation of high energy phosphates during the reperfusion period appears to be involved in the cardioprotective properties of 1,5-dihydroxyisoquinoline. This preservation of high-energy phosphates is very likely secondary to the observed enhancement in NAD levels at the end of reperfusion.
In conclusion, this study demonstrates that the PARS inhibitor 1,5-dihydroxyisoquinoline reduces the contractile dysfunction and improves the contractile reserve (upon challenge with isoproterenol) caused by global ischaemia and reperfusion in the isolated, buffer-perfused heart of the rat. In addition, administration of this PARS inhibitor improved the recovery in NAD and also ATP and PCr during the reperfusion period. The ‘PARS Suicide Hypothesis' states that the excessive activtion of PARS under conditions of oxidant stress causes cell death by depletion of NAD and ATP (Berger et al., 1985). Our findings support the view that the cardioprotective effects of the PARS inhibitor 1,5-dihydroxyisoquinoline are (at least in part) due to the preservation of high energy phosphates.
Acknowledgments
We thank Randy Summers for help with the statistical analyses. Jocelyn Silvester is the recipient of a Women in Science and Engineering Studentship from the NRC. Joanne Bowes was the recipient of a British Heart Foundation (BHF) PhD studentship (FS 96/015). Christoph Thiemermann is a Senior Research Fellow of the BHF (FS 96/018).
Abbreviations
- ISO
1,5 dihydroxyisoquinoline
- NMR
nuclear magnetic resonance
- PARS
poly (ADP-ribose) synthetase
- PCr
phosphocreatine
References
- AMBROSIO G., BECKER L.C., HUTCHINS G.M., WEISMAN H., WEISFELDT M.L. Reduction in experimental infarct size by recombinant human superoxide dismutase: insights into the pathophysiology of reperfusion injury. Circulation. 1986;74:1424–1433. doi: 10.1161/01.cir.74.6.1424. [DOI] [PubMed] [Google Scholar]
- BERGER N.A. Symposium; cellular response to DNA damage: the role of poly (ADP-ribose) Radiat. Res. 1985;101:4–15. [PubMed] [Google Scholar]
- BOLLI R., PATEL B.S., JEROUDI M.O., LAI E.K., MCCAY P.B. Demonstration of free radical generation in ‘stunned' myocardium of intact dogs with the use of the spin trap alpha-phenyl-N-tert-butyl nitrone. J. Clin. Invest. 1988;82:476–485. doi: 10.1172/JCI113621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOWES J., MCDONALD M.C., PIPER J., THIEMERMANN C. Inhibitors of poly (ADP-ribose) synthetase protect rat cardiomyocytes against oxidant stress. Cardiovasc. Res. 1999;41:126–134. doi: 10.1016/s0008-6363(98)00221-1. [DOI] [PubMed] [Google Scholar]
- BOWES J., PIPER J., THIEMERMANN C. Inhibitors of the activity of poly (ADP-ribose) synthetase reduce the cell death caused by hydrogen peroxide in human cardiac myoblasts. Br. J. Pharmacol. 1998;124:1760–1766. doi: 10.1038/sj.bjp.0702009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARROZZA J.P., BENTIVEGNA L.A., WILLIAMS C.P., KUNTZ R.E., GROSSMAN W., MORGAN J.P. Decreased myofilament responsiveness in myocardial stunning follows transient calcium overload during ischemia and reperfusion. Circ. Res. 1992;71:1334–1340. doi: 10.1161/01.res.71.6.1334. [DOI] [PubMed] [Google Scholar]
- CARSON D.A., SET S., WASSON B., CARRERA C.J. DNA strand breaks, NAD metabolism and programmed cell death. Exp. Cell. Res. 1986;164:273–281. doi: 10.1016/0014-4827(86)90028-5. [DOI] [PubMed] [Google Scholar]
- GILAD E., ZINGARELLI B., SALZMAN A.L., SZABO C. Protection by inhibition of poly (ADP-ribose) synthetase against oxidant injury in cardiac myoblasts in vitro. J. Mol. Cell Cardiol. 1997;29:2585–2597. doi: 10.1006/jmcc.1997.0496. [DOI] [PubMed] [Google Scholar]
- GRUNE T., PERRET D. Rapid simultaneous measurement of nucleotides, nucleosides and bases in tissues by capillary electrophoresis. Adv. Exp. Med. Biol. 1994;370:805–810. doi: 10.1007/978-1-4615-2584-4_169. [DOI] [PubMed] [Google Scholar]
- GRUPP I.L., JACKSON T.M., HAKE P., GRUPP G., SZABO C. Protection against hypoxia reoxygenation in the absence of poly (ADP-ribose) synthetase in isolated working hearts. J. Mol. Cell. Cardiol. 1999;31:297–303. doi: 10.1006/jmcc.1998.0864. [DOI] [PubMed] [Google Scholar]
- HENRY T.D., ARCHER S.L., NELSON D., WEIR E.K., FROM A.H. Enhanced chemiluminescence as a measure of oxygen-derived free radical generation during ischemia and reperfusion. Cir. Res. 1990;67:1453–1461. doi: 10.1161/01.res.67.6.1453. [DOI] [PubMed] [Google Scholar]
- HUMPHREY S.M., CARTNER L.A., HOLISS D.G. Critical early metabolite changes associated with myocardial recovery or failure after total ischemia in the rat heart. Basic Res. Cardiol. 1987;82:304–316. doi: 10.1007/BF01906863. [DOI] [PubMed] [Google Scholar]
- JENNINGS R.B., HAWKINS H.K., LOWE J.E., HILL M.L., KLOTMAN S., REIMER K.A. Relation between high energy phosphate and lethal injury in myocardial ischemia in the dog. Am. J. Pathol. 1978;92:187–214. [PMC free article] [PubMed] [Google Scholar]
- JOLLY S.R., KANE W.J., BAILIE M.B., ABRAMS G.D., LUCCHESI B.R. Canine myocardial reperfusion injury. Its reduction by combined administration of superoxide dismutase and catalase. Circ. Res. 1984;54:277–285. doi: 10.1161/01.res.54.3.277. [DOI] [PubMed] [Google Scholar]
- KUKREJA R.C., HESS M.L. The oxygen free radical system: from equations through membrane-protein interactions to cardiovascular injury and protection. Cardiovasc. Res. 1992;6:641–655. doi: 10.1093/cvr/26.7.641. [DOI] [PubMed] [Google Scholar]
- KUSUOKA H., PORTERFIELD J.K., WEISMAN H.F., WEISFELDT M.L., MARBAN E. Pathophysiology and pathogenesis of stunned myocardium. Depressed Ca2+ activation of contraction as a consequence of reperfusion-induced cellular calcium overload in ferret hearts. J. Clin. Invest. 1987;79:950–961. doi: 10.1172/JCI112906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANNING A.S., COLTART D.J., HEARSE D.J. Ischemia and reperfusion-induced arrhythmias in the rat. Effects of xanthine oxidase inhibition with allopurinol. Circ. Res. 1984;55:545–548. doi: 10.1161/01.res.55.4.545. [DOI] [PubMed] [Google Scholar]
- MUSAT-MARCU S., GUNTER H.E., JUGDUTT B.I., DOCHERTY J.C.Inhibition of apoptosis after ischemia-reperfusion in rat myocardium by cycloheximide J. Mol. Cell. Cardiol. 1984(In Press) [DOI] [PubMed]
- NEELY J.R., GROTYOHANN L.W. Role of glycolytic products in damage to ischemic myocardium. Dissociation of adenosine triphosphate levels and recovery of function of reperfused ischemic hearts. Circ. Res. 1999;55:816–824. doi: 10.1161/01.res.55.6.816. [DOI] [PubMed] [Google Scholar]
- OKAMOTO T., ADACHI K., MURAISHI A., SEHI Y., HIDAKA T., TOSHIMA H. Induction of DNA breaks in cardiac myoblast cells by norepinephrine. Biochem. Mol. Biol. Int. 1996;38:821–827. [PubMed] [Google Scholar]
- ROVETTO M.J. Energy metabolism in the ischemic heart. Texas Reports on Biology and Medicine. 1979;39:397–407. [PubMed] [Google Scholar]
- SCHRAUFSTATTER I.U., HINSHAW D.B., HYSLOP P.A., SPRAGG R.G., COCHRANE C.G. DNA strand breaks activate poly adenosine disphosphate-ribose polymerase and lead to a depletion of nicotinamide adenine dinucleotide. J. Clin. Invest. 1986b;77:1312–1320. doi: 10.1172/JCI112436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHRAUFSTATTER I.U., HYSLOP P.A., HINSHAW D.B., SPRAGG R.G., SKLAR L.A., COCHRANE C.G. Hydrogen peroxide induced injury and its prevention by inhibitors of poly (ADP-ribose) polymerase. Proc. Natl. Acad. Sci. U.S.A. 1986a;83:4908–4912. doi: 10.1073/pnas.83.13.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THIEMERMANN C., BOWES J., MYINT F., VANE J.R. Inhibition of the activity of poly (ADP-ribose) synthetase reduces ischemia-reperfusion injury in the heart and skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 1997;94:679–683. doi: 10.1073/pnas.94.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THIES R.L., AUTOR A.P. Reactive oxygen injury to cultured pulmonary artery endothelial cells: mediation by poly (ADP-ribose) polymerase activation causing NAD depletion and altered energy balance. Arch. Biochem. Biophys. 1991;286:353–363. doi: 10.1016/0003-9861(91)90051-j. [DOI] [PubMed] [Google Scholar]
- VARY T.C., ANGELAKOS E.T., SCHAFFER S.W. Relationship between adenine nucleotide metabolism and irreversible ischemic tissue damage in isolated perfused rat heart. Circ. Res. 1979;45:218–225. doi: 10.1161/01.res.45.2.218. [DOI] [PubMed] [Google Scholar]
- WALLES T., PIEPER A.A., ZHANG J.J., SNYDER S.H., ZWEIER J.L.Demonstration that poly (ADP-ribose) accumulation occurs in the post ischemic heart and is associated with myocardial necrosis Circulation 1998a98Suppl I260(Abstract) [Google Scholar]
- WALLES T., WANG P., PIEPER A., SNYDER S., ZWEIER J.L.Mice lacking poly (ADP-ribose) polymerase gene show attenuated cellular energy depletion and improved recovery of myocardial function following global ischemia Circulation 1998b98Suppl.I-260(Abstract) [Google Scholar]
- WANG P.H., ZWEIER J.L. Measurement of nitric oxide and peroxynitrite generation in the post-ischemic heart. Evidence for peroxynitrite-mediated reperfusion injury. J. Biol. Chem. 1996;271:29223–29230. doi: 10.1074/jbc.271.46.29223. [DOI] [PubMed] [Google Scholar]
- ZINGARELLI B., CUZZOCREA S., ZSENGELLER Z., SALZMAN A.L., SZABO C. Protection against myocardial ischemia and reperfusion injury by 3 aminobenzamide, an inhibitor of poly (ADP-ribose) synthase. Cardiovasc. Res. 1997;36:205–215. doi: 10.1016/s0008-6363(97)00137-5. [DOI] [PubMed] [Google Scholar]
- ZINGARELLI B., SALZMAN A.L., SZABO C. Genetic disruption of poly (ADP-ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia/reperfusion injury. Circ. Res. 1998;83:85–94. doi: 10.1161/01.res.83.1.85. [DOI] [PubMed] [Google Scholar]
- ZWEIER J.L., FLAHERTY J.T., WEISFELDT M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]