

Abstract
Chemical signalling is the main mechanism by which biological function is controlled at all levels, from the single cell to the whole organism. Chemical recognition is the function of receptors, which, in addition to recognising endogenous chemical signals, are also the target of many important experimental and therapeutic drugs. Receptors, therefore, lie at the heart of pharmacology. This article describes the way in which the receptor concept originated early in the 20th century, and evolved through a highly innovative stage of quantitative theory based on chemical kinetics, to the point where receptors were first isolated and later cloned, until we now have a virtually complete catalogue of all the receptors present in the genome. Studies on signal transduction are revealing great complexity in the events linking ligand binding to the physiological or therapeutic response. Though some simple quantitative rules of ‘receptor theory' are still useful, the current emphasis is on unravelling the pathways that link receptors to responses, and it will be some time before we know enough about them to embark on the next phase of ‘receptor theory'.
Keywords: Drug receptors
Introduction
The receptor concept is to pharmacology as homeostasis is to physiology, or metabolism to biochemistry. They provide the basic framework, and are the ‘Big Ideas' without which it is impossible to understand what the subjects are about. Try to imagine a pharmacology course that made no mention of receptors.
Pharmacology as a scientific discipline was born in the mid-19th century, amid the great biomedical resurgence of that period (see also Cuthbert, this issue). The world's first pharmacology department was set up by Buchheim in 1847, in recognition of the need to understand how therapeutic drugs and poisons produced their effects. The inadequacy (or, more precisely, ‘inverse adequacy' to use today's receptor parlance) of therapeutic drugs at the time was summed up in Oliver Wendell Holmes' comment in 1860: “… if the whole materia medica as now used could be sunk to the bottom of the sea, it would be all the better for mankind – and the worse for the fishes”. The challenge for pharmacology was clear. It had, then as now, to apply scientific principles to make medicines more effective and less dangerous.
Pharmacology took up this challenge before anything was known about chemical structure, and when ‘drugs' were all either natural products of uncertain composition or inorganic substances such as mercury or arsenic salts. It was only after Kekulé discovered the structure of the benzene ring in 1865, and the now familiar 2-dimensional representations of the structure of organic molecules began to appear – the first in 1868 – and the chemical structures of natural products began to be defined, that the idea of specific ‘lock-and-key' relationships between drugs and their receptors could emerge from the shadows. It had actually been envisaged, in a philosophical way, centuries earlier. For example, John Locke in his Essay concerning human understanding (1690) wrote:
Did we but know the mechanical affections of the particles of rhubarb, hemlock, opium and a man…we should be able to tell beforehand that rhubarb will purge, hemlock kill and opium make a man sleep…
‘Mechanical affections' are, we can now see, what pharmacology is all about, though we might prefer to call them chemical interactions.
This article describes how the receptor concept evolved to become a central theme in the thinking of pharmacologists, and assesses its present status, finishing with some speculations on where the concept may lead in the future. Space precludes more than a superficial and selective overview, and readers wishing to go deeper may wish to consult reviews by Jenkinson (1996), Colquhoun (1998) and Kenakin (1997).
The idea takes shape
Credit for first suggesting (in 1878) the existence of a physiological substance or substances with which pilocarpine and atropine form ‘compounds' belongs to the Cambridge physiologist, J.N. Langley, based on his experiments on salivary secretion in the dog. In 1905, Langley used the term ‘receptive substance' (as distinct from the ‘contractile substance') to explain the actions of nicotine and curare on skeletal muscle. It fell to the mathematically-minded A.V. Hill, a student working in Langley's laboratory, to express the receptor idea quantitatively in terms of a bimolecular reaction following the law of mass action. Hill's paper, published in 1909, was remarkably prescient, anticipating by many years the emergence of ‘receptor theory' and its acceptance by pharmacologists. Hill's focus was on the time-course of the contraction of the frog rectus abdominis muscle produced by nicotine, but along the way he showed that the equilibrium concentration–effect curve (in one experiment!) fitted the equation
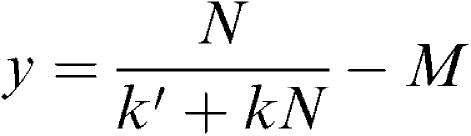 |
where y is the response height, N is the nicotine concentration, M is a threshold, and k and k′ are constants. Hill noted: “This is exactly of the form required … and is very strong evidence in favour of a combination between nicotine and some constituent of the muscle”. Apart from the gratuitous M, Hill's equation is, of course, the familiar equilibrium binding equation now known as the Hill–Langmuir equation. Langmuir was an eminent physical chemist, who derived the equation in 1918 as one possible description of the adsorption of gases as monolayers on metal surfaces. It was only recently that Hill was given the credit he deserved.
Hill lost interest in this area after this early student project, but became famous later for his work on haemoglobin and skeletal muscle biophysics. In these early days, of course, neither the British Pharmacological Society nor its journal existed, and these seminal pharmacological studies were published in the Journal of Physiology. In Germany or the United States, where the emancipation of pharmacology from physiology happened much earlier than it did in Britain, things might have been different.
It is interesting that Henry Dale, whose classical studies on the adrenaline reversal phenomenon, and on the muscarinic and nicotinic actions of acetylcholine, were conceptually very similar to those of Langley, was never inclined to explain his results in terms of receptors as we do now. He was, if anything, somewhat scornful of the idea, which he viewed as speculative and a cloak for ignorance, rather than as a useful theoretical framework for understanding drug action.
Paul Ehrlich, a contemporary of Langley working in Frankfurt, came to the idea of receptors from his interest in the immunology and chemotherapy of infectious diseases. His idea was that bacterial toxins combine with nutrient-capturing structures of cells (‘sidechains'), thus starving them. The cells respond by making more of these sidechains, some of which escape into the circulation as ‘antibodies' that combine with the toxin and make it harmless. He later suggested that the sidechains of bacteria differed from those of the host, and went on to study synthetic molecules, based on aniline dyes, that might act selectively on these bacterial sidechains, an endeavour that ended triumphantly with the discovery of Salvarsan in 1909, the first effective treatment for syphilis.
After these early beginnings, nothing much happened until 1926, when A.J. Clark and J.H. Gaddum – polymaths whose interests covered anything and everything pharmacological – published almost simultaneously key papers on the on the actions of acetylcholine and atropine on the frog's isolated heart (Clark, 1926a, 1926b), and the actions of adrenaline and ergotamine on the rabbit uterus (Gaddum, 1926). Clark and Gaddum believed in measuring things and checking whether the data fitted quantitatively with predictions derived from particular physicochemical hypotheses. They were rarely satisfied with qualitative descriptions, and were the first to introduce the log concentration–effect curve, which has become an icon of pharmacology; they would have approved warmly of the current BPS logo. In the first of these two papers (Clark, 1926a) Clark followed Hill (though he does not say so) in making the bold step of relating the hyperbolic shape of the dose–response curve for acetylcholine to the equilibrium binding equation. Figure 1a shows a figure from his 1933 monograph, ‘The mode of action of drugs on cells', where he concludes cautiously: “The hypothesis that the concentration-action curve of acetylcholine expresses an adsorption process of the type described by Langmuir appears to involve fewer improbable assumptions than any alternative hypothesis” – of which, it should be added, he considered many.
Figure 1.

A.J. Clark's early contributions to the receptor concept. (a) Concentration–effect curves for acetylcholine on (A) frog heart, (B) frog rectus abdominis muscle. Continuous curves fitted to the Hill–Langmuir equation (from Clark, 1926a, 1933). (b) Antagonism of acetylcholine by atropine on frog heart. Ordinate: % inhibition of contraction (y), plotted as log10[y/(100−y)]. Abscissa: Acetylcholine concentration (log10M). Successive lines (I–VII) represent atropine concentrations from zero to 10−3 M (from Clark, 1926b).
In the second of these two papers, Clark (1926b) made a quantitative study of the antagonism of acetylcholine by atropine (Figure 1b). Both Clark and Gaddum described the now-familiar ‘parallel shift' of the log concentration–effect curve produced by a competitive antagonist. Clark's data covered an enormous 105-fold concentration range, rarely attempted by present-day pharmacologists. In retrospect, it is surprising that neither Hill nor Clark pursued the idea of competitive antagonism by deriving the very simple equations for the binding of two mutually exclusive compounds at the same population of sites. This was performed for the first time by Gaddum (1937) in a short communication to the Physiological Society. Clark was actually put off the idea of competitive antagonism for quite the wrong reason. He argued that recovery from atropine ought to be accelerated in the presence of acetylcholine if the two were acting at a common site, and found that it was not. So Clark concluded: “Atropine and acetylcholine, therefore, appear to be attached to different receptors in the heart cells, and their antagonism appears to be an antagonism of effects rather than of combination”. In fact, the simple competitive model does not predict the effect that Clark failed to see.
As a measure of antagonism, Clark estimated the ratio of the concentrations of acetylcholine and atropine needed to produce a given level of response. It was an early example of the use of a null method, which has proved such a valuable principle for pharmacologists. Clark's [agonist] : [antagonist] ratio was actually a hair's breadth away from the agonist dose ratio metric, devised by Schild (see below); it was, however, a purely empirical metric, and a blind alley in relation to competitive antagonism. Clark was a very active founder member of the British Pharmacological Society. He died suddenly in 1941 at the age of 56, 5 years before the Society's journal came into being.
Quantitative pharmacologists in these early days badly wanted to understand the relationship between the amount of drug taken up by tissues and the pharmacological effect produced. The approach taken by Clark and several of his contemporaries was to estimate uptake by using a very small volume of drug solution and either applying it consecutively to several assay preparations until its effect decreased because of uptake of the drug, or by comparing the effects of the same concentration applied in a large or a very small volume. This method, the forerunner of drug binding measurements, was imprecise at best, and could not distinguish between specific and nonspecific uptake, or allow for inactivation of the drug by the tissues. Nevertheless, Clark calculated by this method that the amount of acetylcholine taken up by the frog heart in producing a 50% maximal effect was about 6 pmol/mg tissue, sufficient to cover <1% of the membrane area. He realised that this was almost certainly an overestimate of the number of receptors, and later measurements showed that it was actually about 1000 times too high.
So, though the ideas were in place by the early 1930s, it needed two more breakthroughs before the receptor concept could take hold as a major theme in pharmacology. One was the analysis of competitive antagonism, which led directly to one of the key problems with which we still struggle, namely why some drugs are agonists and others antagonists. The second breakthrough was the direct measurement of drug binding, which led on to the isolation and cloning of receptors, revealing the biochemical reality of what had hitherto been an entirely abstract concept.
The theory evolves
As we have seen, neither Hill nor Clark, having clearly established the physico-chemical basis for analysing drug–receptor interactions, took the theory one simple stage further to explain drug antagonism in terms of competition between agonist and antagonist molecules for the same receptors, though both had thought about this possibility. Gaddum (1937) derived for the first time the equation describing the binding of two drugs at the same receptor. This idea was further developed in an important paper by Schild (1947) published appropriately in a very early volume of the BJP. In this paper, Schild introduced the use of the ‘dose ratio' – the factor by which the agonist concentration must be increased in order to produce the same level of equilibrium occupancy as the concentration of antagonist is increased – a null method ostensibly very similar to the empirical [agonist] : [antagonist] ratios that Clark and others had used, but much more informative – and described the now-familiar Schild Plot of log(dose ratio –1) versus log [antagonist]. If the simple theory of competitive binding is obeyed, the dose ratio should increase linearly as a function of the antagonist concentration, and the slope of this line provided a measure, for the first time, of the affinity of a drug (the antagonist, not the agonist) for its receptors, a method that has been used countless times since. The dose ratio metric was important for two reasons. In the case of competitive antagonism, the dose ratio does not vary with the level of response at which it is measured; in other words, the log concentration–effect curve for the agonist has the same slope and maximum when the antagonist is present, merely being shifted in a parallel manner to the right along the log [agonist] axis. So the familiar ‘parallel shift' picture came to be seen as the defining feature of a competitive antagonist. Secondly, measurement of dose ratios (unlike the metric that Clark had used earlier) allows the affinity of the antagonist for the receptors to be estimated, usually expressed as an equilibrium dissociation constant, KD. The use of antagonist KD values measured in this way has played a key role in receptor classification and drug discovery.
If, as these quantitative studies clearly implied, agonists and antagonists bind to the same site, the question clearly arises: Why do agonists produce a response but antagonists do not? – a question that has exercised pharmacologists from that day to this. In the mid-1950s, the existence of partial agonists was described, both by Ariens and his colleagues in Nijmegen, and by Stephenson in Edinburgh (Figure 2). Stephenson's analysis led him to the concept of efficacy, a characteristic of the drug that describes its ability to activate receptors, distinguishable from its affinity for the receptors. Critical to Stephenson's thinking was the idea that a maximal tissue response (i.e. of smooth muscle contracting in an organ bath) did not necessarily correspond to 100% receptor occupancy, but (with an agonist of high efficacy), could occur when only a small proportion of the receptors was occupied. From this evolved the concept of ‘spare receptors', and the abandonment of Clark's idea that agonist concentration–effect curves represent receptor saturation curves. Furchgott's studies with haloalkylamine antagonists, which bind irreversibly (and therefore ‘noncompetitively') to α-adrenoceptors, confirmed the existence of spare receptors by showing that progressive inactivation of the receptors caused agonist log concentration–effect curves to shift to the right before the slope or maximum was reduced. Stephenson's idea, that binding and activation are independent processes, reflecting affinity and efficacy, made an immediate impact, and it came to be accepted that, to understand structure-activity relationships of agonists, both parameters had to be measured, since a change in agonist potency could reflect a change in either or both. In fact (as Colquhoun, 1998 explains clearly) Stephenson had failed to appreciate that, for an agonist, binding – in the sense of receptor occupancy – and activation are inextricably linked, and that ‘agonist affinity' measured in the way that he proposed (or for that matter by Furchgott's method based on irreversible antagonists) depends on the characteristics of both the initial binding reaction and the resulting activation.
Figure 2.

Concentration–effect curves for a series of alkyl trimethylammonium compounds on guinea-pig ileum (from Stephenson, 1956).
Though Stephenson's suggestion that affinity and efficacy are separable properties is misleading (unless, it should be pointed out, ‘affinity' is defined in relation to the initial binding reaction only. Affinity as measured by Stephenon's or Furchgott's methods, or by binding studies, does not distinguish between the initial binding step and subsequent ‘activation' steps), his recognition that response does not directly reflect occupancy, and that with agonists of high efficacy maximal tissue responses occur at low levels of occupancy, were very important. It could be said that he made partial agonists respectable.
What is efficacy?
Stephenson's definition of efficacy was strictly operational, and he avoided any mechanistic speculation. Significantly, his 1956 paper includes no hypothetical reaction scheme. In principle, there are two ways of thinking about efficacy (Figure 3), namely graded activation (Figure 3a) and the two-state model (Figure 3b). Graded activation implies that agonists (A1, A2, A3, etc) can induce different degrees of conformational change in the receptor, and thus different levels of response. The two-state model suggests that the receptor can switch between a resting state (R) and an activated state (R*). In the original formulation by del Castillo & Katz (1957) based on their studies of the action of acetylcholine at the motor endplate, this transition was assumed to occur only when acetylcholine was bound, with the corollary that acetylcholine was unable to dissociate from the active (R*) conformation. Realisation that this was an arbitrary and unnecessary assumption led to the fully reversible representation of the two-state model (Figure 3c), which accounts neatly and plausibly for variations in efficacy between different agonists. The model suggests that, even in the absence of any ligand, there is a conformational equilibrium between R and R*. An agonist is a ligand with a preferential affinity for R* over R, which means that the ratio AR*/AR will be greater than the ratio R*/R. The greater the selectivity of the ligand for the R* conformation, the greater will be its efficacy. A ligand that binds equally well to both conformations will occupy the receptors without shifting the conformational equilibrium, and so will act as a competitive antagonist. Under conditions where the conformational equilibrium of the unliganded receptor lies strongly in favour of R (i.e. there is very little ‘constitutive activation') the model is perfectly compatible with the earlier formulations of Schild and Stephenson, and notwithstanding the many discoveries about receptor function that have emerged since, it continues to provide a very useful basis for interpreting agonist and antagonist effects. Moving on from Stephenson's ‘black-box' concept of efficacy to the idea that it simply reflects selective binding affinity for the pre-existing resting and active states was a major advance. Coming as it did, at the same time that binding studies were developed as a viable technique for studying drug-receptor interactions (see below), it seemed that it might be possible to measure directly the binding parameters that determined agonist efficacy – misguided optimism as it turned out, for reasons that are discussed below.
Figure 3.

Hypothetical models of receptor activation by agonists (see text). (a) Graded activation model, (b) Simple 2-state model, (c) Reversible 2-state model, (d) Ternary complex model. The ternary complex model (d) includes complex formation between the receptor (R) and a G-protein (G).
The two-state model originated from observations on ligand-gated ion channels (see also Colquhoun, this issue). Early observations on the action of cholinergic agonists on the motor endplate (Jenkinson, 1960) and the eel electroplax (Changeux & Podleski, 1968) and of GABA on crayfish muscle (Takeuchi & Takeuchi, 1969) showed the phenomenon of ‘cooperativity' (i.e. at low response levels the increase in membrane conductance varied, not linearly as predicted by the Hill–Langmuir equation, but roughly as the square of the agonist concentration). In this respect, agonist effects were similar to many enzyme–substrate interactions, and to the binding of oxygen by haemoglobin, phenomena that had been interpreted in terms of a concerted transition of the subunits of oligomeric proteins between two distinct conformational states (Monod et al., 1965). Ligand-gated ion channels seemed to behave in a very similar way, and most of the pharmacological data relating to agonist and antagonist effects could be explained on the basis of the two-state model (Colquhoun, 1973).
Not surprisingly, the simple two-state model could not explain everything about receptor function, and as more phenomena and mechanisms were discovered further elaboration of the model was needed. One of these phenomena was that of the rapid receptor desensitisation commonly seen with ligand-gated ion channels, first analysed by Katz & Thesleff (1957). It was suggested that receptor desensitisation involved at least one (and possibly more than one) ‘desensitised' state in addition to resting and activated states, an idea supported by studies with nicotinic receptor antagonists that bind selectively to receptors in this (R′) state (Rang & Ritter, 1970). It is now known that several different mechanisms, operating on different time scales, contribute to desensitisation at ligand-gated ion channels and G protein-coupled receptors (GPCRs), so the ‘desensitised state' model as originally conceived is a considerable oversimplification. With GPCRs, the discovery that coupling of the activated receptor with G-protein is the first step of the signal transduction pathway (see also Milligan & Kostenis, this issue) gave rise to a further elaboration of the two-state model, known as the ‘ternary complex' model (Figure 3d), involving the G-protein as well as the ligand and the receptor in its resting and active states. Weiss et al. (1996) published a detailed analysis of the properties of this model, and De Lean et al. (1980) showed that it accounted well for the ligand binding to the β-adrenoceptor.
An important new finding, originally from a study of opioid receptor function (Costa & Herz, 1989) was that receptors may be constitutively active. This property, subsequently observed for many GPCRs, is easily explained in terms of the two-state model if it is supposed that the equilibrium distribution of R and R* is not heavily biased in favour of R. It also led naturally to the concept of inverse agonism, since a ligand that binds selectively to R will reduce the population of R*, whereas a conventional agonist, favouring R*, has the opposite effect.
Getting to grips with receptors as molecules
The striking success of simple quantitative models based on the Hill–Langmuir equation in explaining the actions of agonists and competitive antagonists spurred several attempts in the 1960s to measure drug binding directly, in an effort to understand what kind of molecules these mysterious ‘receptors' really were. By analogy with enzymes, it was generally assumed that they must be proteins, or some kind of protein–lipid complex, but there was no evidence for this. Several unsuccessful attempts by biochemists to isolate acetylcholine receptors from electric tissue were reported in the late 1950s, but the first clear evidence that drug binding to receptors could be directly measured came from the beautiful autoradiographic studies of [14C]-calabash curare binding to the endplate region of mouse diaphragm (Figure 4a, Waser, 1960). These experiments required months of exposure of the autoradiographic film, and quantitative measurements were very uncertain, but they clearly showed the localised binding of curare to the endplate region of the diaphragm, which disappeared and became diffuse following denervation of the muscle, and was prevented by addition of curare-like drugs, but not cholinesterase inhibitors, to the bathing medium.
Figure 4.

Early studies of receptor binding. (a) Autoradiographic image of [14-C] calabash curarine to endplate regions of mouse diaphragm (from Waser, 1960). (b) [3H]-Atropine binding to longitudinal muscle of guinea-pig ileum. Analysis of the full binding curve revealed two saturable binding sites, plus a linear component. Binding site 1 represents binding to muscarinic ACh receptors (from Paton & Rang, 1965).
Around this time, liquid scintillation counters became available, as well as tritium labelling of compounds by catalytic exchange methods, making it possible to carry out quantitative drug binding experiments. Possible, but not easy, since the specific activity and radiochemical purity of these tritiated compounds was low, and scintillation counters primitive and subject to many artefacts. Curve-fitting had to be performed, not at the touch of a button, but by programming a mainframe computer half a mile down the road, inputting data laboriously with punched paper tape, and waiting days in a queue for scarce computer time. Nevertheless, the first studies on atropine uptake by guinea-pig ileum smooth muscle (Paton & Rang, 1965) showed clearly the existence of saturable binding with the characteristics of muscarinic receptors (Figure 4b), heralding the ‘grind-and-bind' era of the 1970s and 1980s. It took a full year to make the binding measurements, carry out the necessary controls and estimate the binding parameters – a task now routinely performed by a technician in less than a day. We followed-up the atropine binding experiments by preparing and labelling an irreversible atropine-like compound, benzilylcholine mustard (Gill & Rang, 1966), and tried unsuccessfully to use this to purify the solubilised receptor protein, an endeavour later achieved by Birdsall and others (Birdsall & Hulme, 1976). Around this time also, nicotinic acetylcholine receptors were first successfully labelled with α-bungarotoxin (Miledi et al., 1971), and β-adrenoceptors with alprenolol (Alexander et al., 1975), leading to the purification, sequencing and cloning of all these receptors and several others. Consequently, during the 1980s and 1990s, receptor cloning became a major preoccupation of molecular pharmacologists until by the end of the century the task was virtually complete and pharmacology had entered a new era.
It is often mistakenly believed that binding studies, by measuring directly the level of receptor occupancy as a function of drug concentration, provides an estimate of affinity that is independent of efficacy. However, it is easy to show, on the basis of the two-state model shown in Figure 3c that although the overall level of occupancy is hyperbolic in form as expected, the apparent KD calculated from the binding curve does not represent the drug's affinity for either R or R*, but lies somewhere in between; in other words, the calculated KD value is a function of both affinity and efficacy. Irrespective of the particular model used, it is, in principle, not possible to use equilibrium binding measurements to separate the two without methods (not yet invented) for distinguishing between molecules that are bound to the different receptor conformations.
The advent of receptor cloning had a major impact on pharmacology, and many of the articles in this volume describe advances in molecular and cellular pharmacology that have followed in its wake.
The status of ‘receptor theory'
We have moved on a long way from the early days when receptors were theoretical entities invoked to allow drug effects to be explained in simple quantitative terms by applying the principles of chemical kinetics.
The basic ideas, as formulated by Hill, Gaddum, Schild, Stephenson and elaborated by Black, Leff and Kenakin (see below) form the basis of what came to be known by pharmacologists as ‘receptor theory'. However, as detailed information is gained about the molecular events that result from the binding of a ligand to its receptor, ‘receptor theory' is becoming increasingly inadequate as an overall framework for interpreting and analysing drug effects. At one level, it is self-evident that the laws of chemical kinetics must apply at every stage of the chain of events, but this can be said of anything that happens in the physical universe. Is there anything about drug-receptor interactions that distinguishes them, as a class, from other kinds of biochemical goings-on, that might justify the use of the specific term ‘receptor theory'? Not obviously. This author, for one, would be happy to see the phrase pass into oblivion. Nevertheless, receptor theory undoubtedly provided the foundation on which the study of receptor function at the biochemical and molecular level was based, so its importance in the early stages cannot be doubted. Moreover, Schild's approach to receptor classification, based on pA2 measurements, provided the basis for some major therapeutic discoveries, most famously Black's discovery of β-blockers and H2 receptor antagonists (see also Parsons & Ganellin, this issue). Many other GPCR-based therapeutic drugs have subsequently been developed by following much the same approach, so the usefulness of this type of quantitative analysis in drug discovery is also beyond question.
Recognition of the complexity of the molecular and physiological events that intervene between the initial step of receptor activation and the response that is measured led Black & Leff (1983) to develop an ‘operational model' for agonist action. Following Clark and others, they assumed that receptor activation as a function of agonist concentration followed a hyperbolic saturation curve. Since, empirically, concentration–effect curves are also usually hyperbolic, it followed that all the intermediate steps could also be described by a hyperbolic saturation curve, defined by a system-specific equilibrium constant KE.
The assumptions and limitations of the operational model are clear. Being independent of the actual molecular events involved in agonist action, it throws no light on mechanism, nor does it resolve the problem, referred to earlier, that affinity and efficacy are inextricably linked. Nevertheless, it has undoubtedly proved useful as a basis for describing agonist action in quantitative terms, though to the current generation of molecular pharmacologists concerned to unravel the intricacies of signal transduction pathways, the operational model has little relevance.
So far, nearly all of the quantitative theoretical modelling of receptor function has centred on ligand-gated ion channels and GPCRs. In the case of ion channels, the use of single channel recording has been important as a method for observing the behaviour of single receptor molecules in real time at a high level of temporal resolution. As Colquhoun describes in this issue, fitting kinetic data to theoretical models can allow mechanistic conclusions to be drawn with some confidence. It gives access to the intramolecular events that cause a channel to open when an agonist binds, but of course neglects the manifold functional effects that result. With GPCRs, techniques for observing receptor function directly are less well developed, though the use of fluorescence techniques to investigate agonist-induced conformational changes may have considerable potential (see Gether & Kobilka, 1998; Gether, 2000). In most cases, researchers have to infer what they can from measurements of binding, and a variety of downstream functional changes, such as GDP/GTP exchange, alterations in enzyme activity, protein phosphorylation, levels of intracellular second messengers, membrane currents, changes in gene expression, etc, Such studies have resulted in many useful flow charts representing postulated pathways and interactions, but assigning values to the various rate and equilibrium constants to allow quantitative modelling is rarely possible. The same limitation applies, in general, to studies of nuclear receptors and receptor protein kinases.
Future prospects
In recent years, the study of receptor function has taken full advantage of the molecular biology revolution, and many discoveries are being made that challenge the simple quantitative analyses introduced by pioneers such as Hill, Stephenson, Schild and others (which, it should be emphasised, were crucial in laying the foundations of the receptor concept in pharmacology).
Some recent discoveries, particularly in the GPCR field, pose a real challenge to those seeking to construct more elaborate general theories of receptor function.
Different agonists acting on the same GPCR activate different signal transduction pathways, a phenomenon sometimes described as ‘protean agonism' (Kenakin, 2001; 2002), implying that different activated states are favoured by different agonists (see Perez & Karnik, 2005). The model begins to look more like the graded activation model (Figure 3a). With ligand-gated ion channels, the situation seems to be simpler, as there are few if any examples where different ligands acting on the same receptor open channels with different conductance or selectivity characteristics. Nuclear receptors are also activated differently by different ligands (see Nettles & Greene, 2005).
Many GPCRs exist as dimers, sometimes forming heterodimers with other GPCRs (see Angers et al., 2002). They also associate with accessory proteins, such as RAMPs which alter their pharmacological properties (McLatchie et al., 1998; see also Brain & Cox, this issue). We do not yet know whether, to what extent, or how quickly, such associations are affected by ligand binding. There are parallels with many receptor tyrosine kinases, where dimerisation occurs in response to agonist binding, and is the first step in the signal transduction pathway.
Receptors often occur in clusters on the cell membrane (‘lipid rafts', Ostrom & Insel, 2004), along with other molecules involved in signal transduction, forming isolated microdomains within the cell that are only detectable by methods providing a high level of spatial resolution.
Constitutive activity and the existence of inverse agonists calls into question the original view of receptors as ‘silent' molecules that produce effects only when activated by an agonist. Rather they may serve a controlling function, which can be turned up or down by combination with different ligands, and what we choose to call ‘up' and ‘down' is quite arbitrary. Adding to the complexity, the concept of protean agonism suggests that a given ligand could turn some functions ‘up', others ‘down' and leave others unaffected, while another ligand, acting on the same receptor, could produce a quite different profile of effects.
There are, particularly among GPCRs and nuclear receptors, large numbers of ‘orphan receptors' whose endogenous ligands, if they exist at all, are not yet known.
In general, the reductionist focus made possible in recent years by applying the principles of molecular and cell biology to pharmacological problems is undoubtedly providing major new insights into how drug molecules interact with receptors. The Holy Grail, which may well come into view in the foreseeable future, will be reached when we can depict, at high resolution, exactly what happens when a ligand binds to its receptor, explain why this event leads on to, for example, channel opening or G-protein binding, and (most importantly) design ligands de novo which will act as agonists, antagonists or other sorts of modulator. This certainly won't be the end of pharmacology, for between the ‘molecular' response to a ligand, and its effect on the functioning of the cell, tissue, system or whole animal lie mechanisms of far greater complexity. Pharmacology, committed as it always has been to the improvement of therapeutic drugs, has to concern itself with drug effects on the functioning of sick human beings, receptor mechanisms being just the first step.
In future the receptor concept may come to be seen, not just as the Big Idea of pharmacology, but as one of the Big Ideas of biology in general. Living organisms are chemical machines, and rely on chemical signalling within and between cells, at long or short range, through the agency of ligands and receptors. Understanding the processes involved in these signalling pathways is therefore crucial to understanding biology. Pharmacologists can take pride in having started the ball rolling.
Glossary
- GABA
γ-aminobutyric acid
- GDP
guanosine diphosphate
- GPCR
G protein-coupled receptor
- GTP
guanosine triphosphate
- RAMP
receptor activity modifying protein
References
- ALEXANDER R.W., WILLIAMS L.T., LEFKOWITZ R.J. Identification of cardiac beta-adrenergic receptors by (−) [3H]alprenolol binding. Proc. Natl. Acad. Sci. U.S.A. 1975;72:1564–1568. doi: 10.1073/pnas.72.4.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANGERS S., SALAHPOUR A., BOUVIER M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu. Rev. Pharmacol. Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- BIRDSALL N.J., HULME E.C. Biochemical studies on muscarinic acetylcholine receptors. J. Neurochem. 1976;27:7–16. doi: 10.1111/j.1471-4159.1976.tb01536.x. [DOI] [PubMed] [Google Scholar]
- BLACK J.W., LEFF P. Operational models of pharmacological agonists. Proc. Roy. Soc. B. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- CHANGEUX J.-P., PODLESKI T. On the excitability and cooperativity of the electroplax membrane. Proc. Natl. Acad. Sci. U.S.A. 1968;59:944–950. doi: 10.1073/pnas.59.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARK A.J. The reaction between acetyl choline and muscle cells. J. Physiol. 1926a;61:530–546. doi: 10.1113/jphysiol.1926.sp002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARK A.J. The antagonism of acetyl choline by atropine. J. Physiol. 1926b;61:547–556. doi: 10.1113/jphysiol.1926.sp002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARK A.J. The mode of action of drugs on cells. London: Edward Arnold; 1933. [Google Scholar]
- COLQUHOUN D.1973The relation between classical and cooperative models for drug actionIn: Drug Receptors, Rang, H.P. ed. London: Macmillan [Google Scholar]
- COLQUHOUN D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and the effects of mutating receptors. Br. J. Pharmacol. 1998;125:923–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSTA T., HERZ A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LEAN A., STADEL J.M., LEFKOWITZ R.J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- DEL CASTILLO J., KATZ B. Interaction at end-plate receptors between different choline derivatives. Proc. Roy. Soc. B. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- GADDUM J.H. The action of adrenaline and ergotamine on the uterus of the rabbit. J. Physiol. 1926;61:141–150. doi: 10.1113/jphysiol.1926.sp002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GADDUM J.H. The quantitative effects of antagonistic drugs. J. Physiol. 1937;89:6–7P. [Google Scholar]
- GETHER U. Uncovering molecular mechanisms linked to activation of G protein-coupled receptors. Endocr. Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- GETHER U., KOBILKA B.K. G protein-coupled receptors. II. Mechanism of agonist activation. J. Biol. Chem. 1998;273:17979–17982. doi: 10.1074/jbc.273.29.17979. [DOI] [PubMed] [Google Scholar]
- GILL E.W., RANG H.P. An alkylating derivative of benzilylcholine with specific and long-lasting parasympatholytic activity. Mol. Pharmacol. 1966;2:284–297. [PubMed] [Google Scholar]
- JENKINSON D.H. The antagonism between tubocurarine and substances which depolarise the motor end-plate. J. Physiol. 1960;152:308–324. doi: 10.1113/jphysiol.1960.sp006489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENKINSON D.H.1996Classical approaches to the study of drug-receptor interactions Textbook of Receptor Pharmacologyeds. Foreman, J.C. & Johansen, T. pp4–62.Boca Raton: CRC Press [Google Scholar]
- KATZ B., THESLEFF S. A study of the ‘desensitization' produced by acetylcholine at the motor end-plate. J. Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENAKIN T.1997Pharmacologic Analysis of Drug–Receptor Interactions3rd edn. New York: Lipincott-Raven [Google Scholar]
- KENAKIN T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Drug efficacy at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2002;42:349–379. doi: 10.1146/annurev.pharmtox.42.091401.113012. [DOI] [PubMed] [Google Scholar]
- MCLATCHIE L.M., FRASER N.J., MAIN M.J., WISE A., BROWN J., THOMPSON N., SOLARI R., LEE M.G., FOORD S.M. RAMPs regulate the transport and ligand specificity of the calcitonin-recetor-like-receptor. Nature. 1998;393:333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- MILEDI R., MOLINOFF P., POTTER L.T. Isolation of the cholinergic receptor protein of Torpedo electric tissue. Nature. 1971;229:554–557. doi: 10.1038/229554a0. [DOI] [PubMed] [Google Scholar]
- MONOD J., WYMAN J., CHANGEUX J.-P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- NETTLES K.W., GREENE G.L. Ligand control of coregulator recruitment to nuclear receptors. Annu. Rev. Physiol. 2005;67:309–333. doi: 10.1146/annurev.physiol.66.032802.154710. [DOI] [PubMed] [Google Scholar]
- OSTROM R.S., INSEL P.A. The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br. J. Pharmacol. 2004;143:235–245. doi: 10.1038/sj.bjp.0705930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATON W.D.M., RANG H.P. The uptake of atropine and related drugs by intestinal smooth muscle of the guinea-pig in relation to acetylcholine receptors. Proc. Roy. Soc. Lond. B. 1965;163:1–44. doi: 10.1098/rspb.1965.0058. [DOI] [PubMed] [Google Scholar]
- PEREZ D.M., KARNIK S.S. Multiple Signaling States of G-Protein-Coupled Receptors. Pharmacol. Rev. 2005;57:147–161. doi: 10.1124/pr.57.2.2. [DOI] [PubMed] [Google Scholar]
- RANG H.P., RITTER J.M. On the mechanism of desensitisation at cholinergic receptors. Mol. Pharmacol. 1970;6:357–382. [PubMed] [Google Scholar]
- SCHILD H.O. pA, a new scale for the measurement of drug antagonism. Br. J. Pharmacol. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEPHENSON R.P. A modification of receptor theory. Br. J. Pharmacol. 1956;11:379–393. doi: 10.1111/j.1476-5381.1956.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKEUCHI A., TAKEUCHI N. A study of the action of picrotoxin on the inhibitory neuromuscular junction of the crayfish. J. Physiol. 1969;205:377–391. doi: 10.1113/jphysiol.1969.sp008972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WASER P.G. The cholinergic receptor. J. Pharm. Pharmacol. 1960;12:577–594. doi: 10.1111/j.2042-7158.1960.tb12714.x. [DOI] [PubMed] [Google Scholar]
- WEISS J.M., MORGAN P.H., LUTZ M.W., KENAKIN T.P.1996The cubic ternary complex receptor occupancy model. (Series of 3 papers) J. Theoret. Biol. 178151–167.169–182, 381–397. [DOI] [PubMed] [Google Scholar]