

Abstract
A yeast mutant lacking the two major cytosolic sources of NADPH, glucose-6-phosphate dehydrogenase (Zwf1p) and NADP+-specific isocitrate dehydrogenase (Idp2p), has been demonstrated to lose viability when shifted to medium with acetate or oleate as the carbon source. This loss in viability was found to correlate with an accumulation of endogenous oxidative byproducts of respiration and peroxisomal β-oxidation. To assess effects on cellular protein of endogenous versus exogenous oxidative stress, a proteomics approach was used to compare disulfide bond-containing proteins in the idp2Δzwf1Δ strain following shifts to acetate and oleate media with those in the parental strain following similar shifts to media containing hydrogen peroxide. Among prominent disulfide bond-containing proteins were several with known antioxidant functions. These and several other proteins were detected as multiple electrophoretic isoforms, with some isoforms containing disulfide bonds under all conditions and other isoforms exhibiting a redox-sensitive content of disulfide bonds, i.e., in the idp2Δzwf1Δ strain and in the hydrogen peroxide-challenged parental strain. The disulfide bond content of some isoforms of these proteins was also elevated in the parental strain grown on glucose, possibly suggesting a redirection of NADPH reducing equivalents to support rapid growth. Further examination of protein carbonylation in the idp2Δzwf1Δ strain shifted to oleate medium also led to identification of common and unique protein targets of endogenous oxidative stress.
Keywords: NADPH, Isocitrate dehydrogenase, Glucose-6-phosphate dehydrogenase, Disulfide bonds, Reactive thiols, Carbonylation
INTRODUCTION
Aerobic organisms have evolved antioxidant enzymatic systems to protect against reactive oxygen species (e.g., superoxide, hydroxyl radical, and hydrogen peroxide) generated as byproducts of metabolism or resulting from environmental insults [1,2]. Significant sources of endogenous reactive oxygen species are mitochondrial respiration, in which incomplete reduction of oxygen results in the generation of superoxide anion, and peroxisomal β-oxidation, in which hydrogen peroxide is a stoichiometric product of the first enzymatic reaction. The superoxide anion can be converted to hydrogen peroxide by superoxide dismutases, and hydrogen peroxide can be converted to water either by cytosolic or peroxisomal catalases or by thiol-based peroxidases [2]. These peroxidases are present in various subcellular compartments [3–5], and their redox-active glutathione or thioredoxin cofactors are maintained in reduced states by NADPH-dependent reductases [5–8].
In genetic studies using the yeast Saccharomyces cerevisiae [9–12], we determined that the major enzymatic sources of NADPH for antioxidant functions are two cytosolic enzymes: glucose-6-phosphate dehydrogenase (Zwf1p) which catalyzes the first step in the oxidative branch of the pentose phosphate pathway, and NADP+-specific isocitrate dehydrogenase isozyme 2 (Idp2p) which catalyzes the oxidative decarboxylation of isocitrate to form α -ketoglutarate. Others previously demonstrated that disruption of the yeast ZWF1 gene, which is constitutively expressed, produced only mild growth phenotypes including methionine auxotrophy and sensitivity to exogenous hydrogen peroxide in cells grown with glucose as the carbon source [13,14]. We similarly showed that disruption of the IDP2 gene, which is expressed during growth on non-fermentable carbon sources, produced no significant growth phenotypes [15]. However, we found that co-disruption of ZWF1 and IDP2 genes resulted in an inability to grow under conditions of rapid respiratory metabolism (with acetate as the carbon source) and/or β-oxidation (with a fatty acid as the carbon source) [10,11]. Furthermore, shifting the idp2Δzwf1Δ mutant strain from permissive carbon sources (glucose or glycerol) to acetate or oleate medium resulted in a loss of cellular viability (Fig. 1), suggesting that metabolism of the latter carbon sources is lethal for this strain.
Fig. 1.
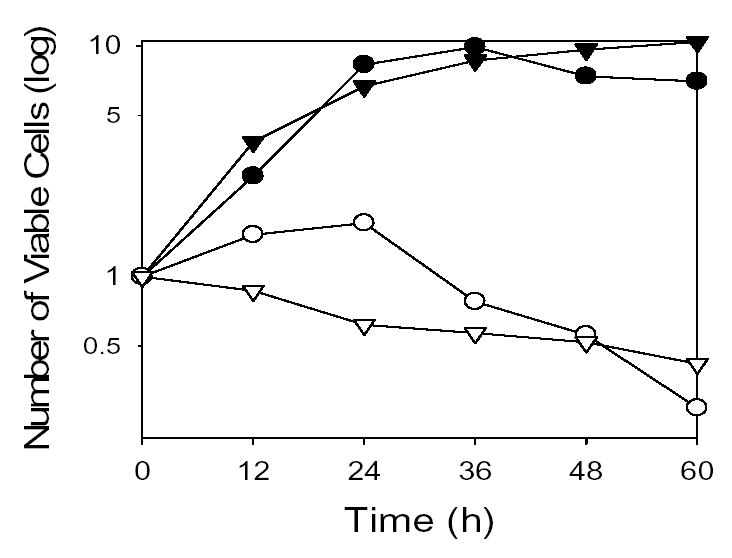
Growth and viability of yeast strains following carbon source shifts. Parental (filled symbols) and Δidp2Δzwf1 mutant (open symbols) strains were shifted from medium containing glucose to medium with acetate (circles) as the carbon source, or from medium with glycerol to medium with oleate (triangles) as the carbon source. The number of viable cell was determined by plating dilutions from the cultures onto YP glucose plates and counting colonies after 3 days of growth at 30° C. Media shifts were initiated with 2–3 x 106 viable cells/ml; these numbers were normalized to one for comparison. These data were previously presented [11], and are shown here with permission of the publisher.
Several lines of evidence suggest that the loss in viability of the idp2Δzwf1Δ mutant strain shifted to acetate or oleate medium is due to low cellular levels of NADPH. (a) The decrease in viability was shown to correlate with a significant increase in levels of intracellular oxidants [10,11]. (b) Supplementation with high levels of glutathione and dithiothreitol (DTT) during a shift to oleate medium was found to mitigate the loss of viability of the idp2Δzwf1Δ mutant strain [11]. (c) No loss in viability was observed during similar media shifts of yeast mutants lacking cytosolic catalase and/or the major mitochondrial peroxidase [11], suggesting that these peroxidative enzymes do not provide crucial antioxidant functions under these conditions. (d) Direct measurements of cellular NADP+ and NADPH levels [12] indicated a rapid cycling of NADPH in wild-type cells during such shifts (i.e., the ratio of NADP+/NADPH increased ~three-fold without a major change in total levels of cofactor). In the shifted idp2Δzwf1Δ mutant strain, levels of NADPH also decreased. However, levels of NADP+ increased dramatically (~five-fold relative to preshift and parental strain levels), suggesting that the mutant cells respond by producing more cofactor to compensate for the lack of reduced cofactor for antioxidant functions.
We believe that the idp2Δzwf1Δ mutant yeast strain is a unique experimental model for analysis of the effects of oxidative stress caused by normal byproducts of endogenous metabolism. We previously reported evidence for generic damage to nuclear DNA and elevated levels of protein oxidation in this strain following shifts to non-permissive media [11]. In the current study, we have utilized a two-dimensional electrophoresis/proteomics approach to identify specific proteins that are altered in the idp2Δzwf1Δ strain following shifts to non-permissive media. We employed a fluorescence method for detection of disulfide bond-containing proteins, since the reduction status of cellular proteins should be particularly sensitive to the availability of reducing equivalents in the form of NADPH. This screen was designed to identify proteins with cysteine side chains participating in disulfide bonds in vivo at the time of sampling. This form of cysteine oxidation would encompass intramolecular or intermolecular disulfide bonds. The latter would include S-thiolation, the formation of a disulfide bond between a protein and a low molecular weight cofactor like glutathione [16,17]. It has been suggested that this type of disulfide bond formation may protect proteins during transient oxidative stress until levels of reactive oxygen species decline and reducing conditions are restored [17]. Cysteine side chains can also undergo oxidation to sulfinic or sulfonic acids, oxidative products that are not reduced by normal cellular thiol-based antioxidant systems [16,18] and that would not be detected in our disulfide bond screen.
With the goal of directly comparing our results with effects of exogenous oxidants, we also performed experiments on the parental strain using media shifts conducted in the presence of sufficient exogenous hydrogen peroxide to produce effects on viability equivalent to those observed for the idp2Δzwf1Δ strain following shifts to oleate or acetate media. Finally, to compare metabolic oxidative damage to specific proteins in the idp2Δzwf1Δ mutant strain with damage reported by others due to exogenous stress, we also used a screen for carbonylated proteins.
EXPERIMENTAL PROCEDURES
Yeast strains and growth conditions
The parental haploid yeast strain used in this study was S173-6B (MATα leu2-3,112 his3-1 ura3-52 trp1-289) [19]. A derivative of this strain, the Δidp2Δzwf1 mutant strain containing a deletion/URA3 insertion in the IDP2 locus and a kanMX4 insertion in the ZWF1 locus, was constructed as previously described [10,15].
Yeast strains were cultivated in rich YP medium (1% yeast extract, 2% Bactopeptone) with 2% glucose, 3% glycerol, 2% acetate, or 0.1% oleic acid plus 0.2% Tween 40 as the carbon source. For carbon source shift experiments, strains were cultivated overnight in YP glucose or YP glycerol medium and respectively diluted into YP acetate or YP oleate medium to an optical density at A600nm (OD600) = 0.3. As measured using the parental strain, an OD600 value of 1.0 is approximately equivalent to 5 × 106 cells/ml. For studies examining disulfide bonds, control cultures grown in YP medium with glucose or glycerol as the carbon source were harvested at an OD600 = 1.0. For some experiments, glutathione and DTT were added as previously described (11) to reduce lethality of the Δidp2Δzwf1 mutant strain during a shift to YP oleate medium. To examine parental strain sensitivity to hydrogen peroxide, similar media shifts were conducted in the presence of hydrogen peroxide (concentrations ranging from 0.1 to 2.0 mM during shifts to acetate medium, and from 1.0 to 3.5 mM during shifts to oleate medium). Viable cell numbers were determined at 12 h intervals by plating dilutions of the hydrogen-peroxide challenged parental strain onto YP glucose plates and counting colonies after 3 days growth at 30° C. Concentrations of hydrogen peroxide were chosen that produced an approximate 50% reduction in parental cell numbers after 12 h but that did not limit growth at later time points.
Detection of disulfide bonds
All experimental steps were performed as previously described [18] in a dimly-lit room using 1.5 ml amber polypropylene tubes. Harvested yeast cells were lysed by vortexing with glass beads [20] using deaerated buffer (4° C) containing 100 mM sodium phosphate (pH 8.0), 0.5 mM MgCl2, 1.0 mM EDTA, 150 mM iodoacetamide, 3.0 M urea, and 1.0 ml/g cells of a yeast protease inhibitor cocktail (Sigma P2815). Nucleic acids were removed by streptomycin sulfate precipitation. Urea was added to the cleared lysates to a final concentration of 8.0 M, and samples were incubated at 37° C for 30 min. Following precipitation with 20% trichloroacetic acid (TCA), protein pellets were washed five times with ethanol/ethyl acetate (1:1) to remove any residual iodoacetamide. The pellets were dissolved in 10 mM sodium phosphate buffer (pH 8.0) containing 8.0 M urea and divided into two samples. DTT was added to one sample to 2.0 mM, and no addition was made to the other sample. Following incubation for 1 h at 37° C, 6-iodoacetamidofluorescein (6-IAF) was added to 100 μM, and the incubation was repeated. The TCA precipitation and washes were repeated to remove any residual 6-IAF, and the protein pellets were dissolved in Rehydration/Sample buffer (Bio-Rad, Hercules, CA). Protein concentrations were determined using the Bradford method [21]. Protein samples (200 μg) were applied to Bio-Rad pH 3-6, 5-8, or 7–10 immobilized pH gradient (IPG) strips by passive rehydration, and electrophoresis was conducted using a Bio-Rad Protein IEF cell according to the manufacturer’s instructions. Following isoelectric focusing, the IPG strips were laid on 11 cm 8–16% gradient polyacrylamide/sodium dodecyl sulfate gels (Criterion, Bio-Rad) for electrophoresis. Proteins labeled with 6-IAF were visualized using an FX Pro Plus Molecular Imager (Bio-Rad) prior to staining of total proteins with SYPRO Ruby (Bio-Rad).
The reproducibility of SYPRO Ruby staining and 6-IAF labeling were established by comparing results from different experiments in which parental and mutant cells were grown and harvested as described above. Other than slight variations due to electrophoresis being conducted on different days, the protein patterns were essentially superimposable for each pH range, and identities of specific proteins determined as described below were confirmed using duplicate or triplicate samples from different gels.
Detection of carbonylated proteins
Yeast cells were lysed as described above in 10 mM sodium phosphate buffer (pH 7.0) containing 50 mM DTT. Protein samples (200 μg) were treated with 2,4-dinitrophenylhydrazine to derivatize carbonylated proteins as described in the OxyBlotTM Protein Oxidation Detection Kit (Chemicon International, Temecula, CA). Proteins were precipitated using TCA and washed as described above. Pellets were dissolved in Rehydration/Sample buffer and applied to Bio-Rad pH 4-7 and 7–10 IPG strips. Isoelectric focusing and denaturing gel electrophoresis were conducted as described above. Duplicate gels were run for each sample, one for protein staining with SYPRO Ruby prior to protein spot excision, and one for transfer to a polyvinylidene difluoride membrane for immunoblot analysis. Carbonylated proteins were visualized by autoradiography following immunoreaction with anti-dinitrophenyl-horse radish peroxidase antiserum (Dako, Carpinteria, CA) [22] and a Super Signal West Fempto Maximum Sensitivity Kit (Pierce Biotechnology, Inc., Rockford, IL).
Protein identification
Protein spots on SYPRO Ruby-stained gels were imaged using an FX Pro Plus Molecular Imager (Bio-Rad) and PDQuest 8.0 software (Bio-Rad). Proteins of interest that corresponded to proteins labeled with 6-IAF or to carbonylated proteins were excised robotically using a Proteome Works spot cutter (Bio-Rad) and digested in situ with trypsin according to standard protocols based on the initial work of Mann and co-workers [23]. Briefly, protein spots were excised from the gel and destained in 50% acetonitrile/40 mM ammonium bicarbonate, pH 7.4. Gel plugs were dehydrated in 100% acetonitrile, rehydrated with 5–10 ng/μ l trypsin (Promega) in 40 mM ammonium bicarbonate, and incubated overnight at 37° C. The resulting digests were analyzed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF/MS) using an Applied Biosystems Voyager-DE STR (Framingham, MA) operated in reflection mode. Samples (1.0 μ l) were applied to the target and allowed to partially dry. Matrix solution (1.0 μ l) consisting of a saturated solution of 2,5-dihydroxybenzoic acid (Sigma) in 50% acetonitrile/0.1% trifluoroacetic acid was added to the partially dried spot, and all solvent was allowed to dry at room temperature.
The peptide maps produced by MALDI-TOF/MS were searched against published databases using Mascot (Matrix Science; in-house license) to provide information about the identity of the protein(s) in each spot. Additional sequence information/characterization of selected digests was accomplished with capillary-HPLC-electrospray tandem mass spectrometry (HPLC-ESI-MS/MS) on a Thermo Finnigan LCQ ion trap mass spectrometer coupled to a Michrom BioResources Paradigm MS4 micro HPLC by means of a home-built microspray interface. Capillary on-line HPLC separation of tryptic peptides was conducted using the following conditions: column, New Objective PicoFrit (75 μ m i.d.) packed to 10 cm with C18 adsorbent (Vydac 218MSB5); mobile phase A, 0.5% acetic acid/0.005% trifluoroacetic acid in deionized water; mobile phase B, 90% acetonitrile/0.5% acetic acid/0.005% trifluoroacetic acid in water; gradient, 2% B to 72% B in 30 min; flow rate, 0.4 μ l/min. A data-dependent acquisition protocol was employed in which one survey scan was acquired followed by four collision-induced dissociation (CID) spectra. The CID spectra were searched against the Swiss-Prot database using Mascot. Variable modifications considered included methionine oxidation and cysteine carbamidomethylation. A 95% confidence level threshold was used for Mascot protein scores (MALDI-TOF/MS) or peptide scores (ESI-MS/MS).
RESULTS
Assays for disulfide bonds
As illustrated in Fig. 1, we have found that a Δidp2Δzwf1 mutant strain of yeast that lacks two major cytosolic sources of NADPH loses viability when shifted to medium with acetate or a fatty acid (oleate) as the carbon source, and that this loss in viability correlates with an increase in levels of intracellular oxidants [10,11]. To assess effects of endogenous metabolic oxidative stress due to rapid rates of mitochondrial respiration and/or peroxisomal β-oxidation, we chose initially to employ a new method to analyze the disulfide bond status of cellular proteins [18]. This was based on the expectation that the oxidative status of cysteine residues would be a particularly sensitive index for changes in cellular levels of NADPH. We chose time points before and 48 h following a shift from glucose to acetate medium, and before and 24 h following a shift of mutant cells from glycerol to oleate medium. At the latter times in both shifts, there is a ~50% loss in viable cell numbers for the Δidp2Δzwf1 strain. These time points were chosen because of obvious effects on viability; however, the remaining viable cells can grow if transferred to permissive conditions at this point [10,11], suggesting that any macromolecular changes caused by oxidative stress are largely reversible. Similar shifts to acetate and oleate media were conducted with the parental strain, which exhibits rapid growth under these conditions (Fig. 1).
A major goal of these studies was to determine if protein targets of endogenous metabolic oxidants in the Δidp2Δzwf1 strain are similar to protein targets of exogenous oxidants in a wild-type strain. Therefore, we empirically determined times and levels of challenge of the parental strain with hydrogen peroxide that produced similar losses (~50%) in viable cell numbers during media shifts. These conditions involved treatment of the parental strain with 0.75 mM hydrogen peroxide for 12 h following a shift to acetate medium and treatment with 2.0 mM hydrogen peroxide for 12 h following a shift to oleate medium. At later time points (24–72 h in these shifts) following detoxification of the hydrogen peroxide, the parental strain resumes growth with acetate or oleate carbon sources, again suggesting that many macromolecular changes in viable cells are reversible. As controls, we also sampled the Δidp2Δzwf1 and parental strains grown on glucose and glycerol prior to the media shifts.
For proteomic analysis of changes in disulfide bond formation, cellular protein extracts were prepared as shown schematically in Fig. 2 in the presence of excess iodoacetamide (6-IAA) and urea to covalently modify all free sulfhydryl side chains and to inhibit formation of non-native disulfide bonds [18]. The precipitated protein samples were then dissolved and divided into two portions for incubation in the presence or in the absence of dithiothreitol (DTT). After reduction of residual disulfide bonds, newly exposed sulfhydryl side chains were reacted with 6-iodoacetamidofluorescein (6-IAF). Proteins containing cysteine residues that had been participating in disulfide bonds in vivo were then detected by two-dimensional gel electrophoresis and fluorographic imaging. Isoelectric focusing with three different pH ranges was employed. Spots corresponding to proteins that were specifically labeled with 6-IAF after treatment with DTT were visualized by SYPRO Ruby staining of the same gel. The stained proteins of interest were excised, digested with trypsin, and identified by mass spectrometry. Examples of pH 5–8 gels used for such an analysis of parental strain proteins following a shift to acetate medium in the presence of hydrogen peroxide are shown in Fig. 3: gel A shows the general baseline fluorescence obtained in the absence of incubation with DTT; gel B shows proteins specifically labeled with 6-IAF following treatment with DTT to reduce disulfide bonds; gel C shows SYPRO Ruby-stained proteins. Comparison of gels B and C shows that only a subset of cellular proteins were labeled with 6-IAF. These patterns were found to be highly reproducible in independent experiments.
Fig. 2.
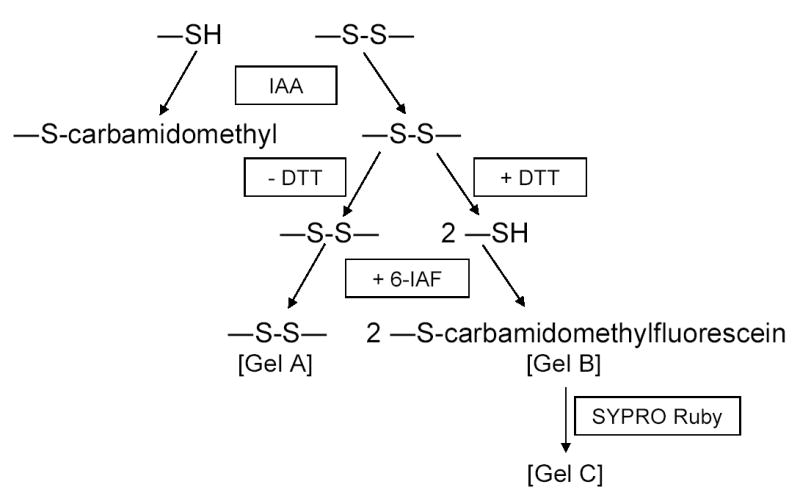
Method for comparison of disulfide bond labeling in cellular protein samples. Cellular protein extracts were treated with iodoacetamide (IAA) to block sulfhydryl side chains. The extract was divided in two: residual disulfide bonds were left intact in one sample (- DTT) and reduced in the other (+ DTT). Both samples were incubated with 6-iodoacetamidofluorescein (6-IAF) and subsequently separated using two-dimensional gel electrophoresis. The gel containing 6-IAF-labeled proteins was stained with SYPRO Ruby. Examples of gels A, B, and C are shown in Fig. 3.
Fig. 3.
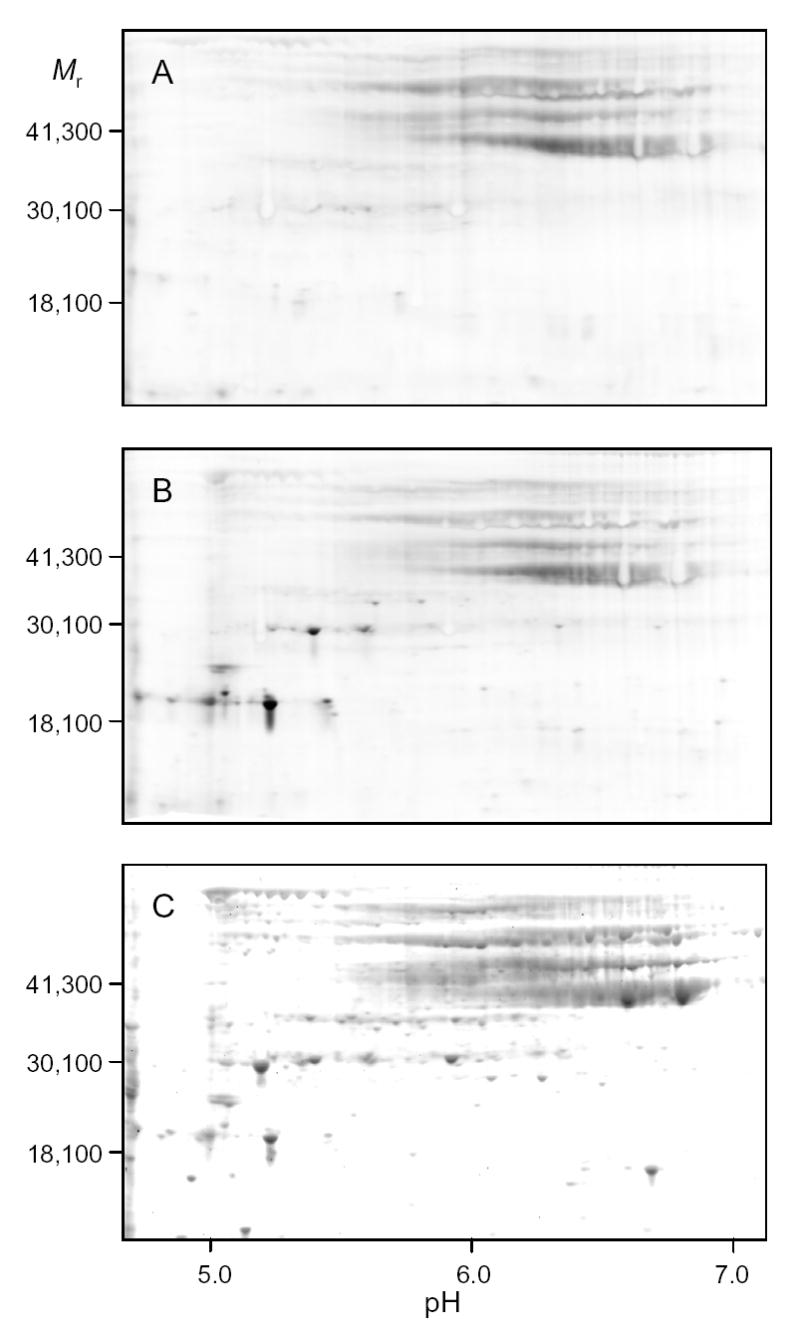
Comparison of disulfide bond labeling in cellular proteins using two-dimensional electrophoresis. Cellular protein samples from the parental strain following a shift to acetate medium in the presence of hydrogen peroxide were taken as indicated in Fig. 2. Samples were electrophoresed using isoelectric focusing in the pH 5–8 range. Fluorographs of gels are shown in panels A (− DTT) and B (+ DTT). Total proteins in Gel B were subsequently stained with SYPRO Ruby (C).
Representative fluorographic images of 6-IAF-labeled proteins from two-dimensional gels conducted under different isoelectric focusing conditions are shown in Figs. 4A–C. In these examples, protein samples were obtained from the parental strain following the shift to acetate medium in the presence of hydrogen peroxide (upper panels) and from the Δidp2Δzwf1 strain following the shift to acetate medium (lower panels). As stated above, similar analyses (data not shown) were conducted with parental and mutant strains prior to the medium shifts, and with these strains prior to and following shifts to oleate medium (plus hydrogen peroxide for the parental strain). Isoelectric focusing was conducted in the pH 3–6 range (Fig. 4A), in the pH 5–8 range (Fig. 4B), and in the pH 7–10 range (Fig. 4C), prior to second dimensional sodium dodecylsulfate gel electrophoresis. Proteins identified by mass spectrometry are indicated in the figures. Descriptions of all proteins identified in this and other experiments described below, and the number of independent peptides used for identification, are listed in Table 1.
Fig. 4.
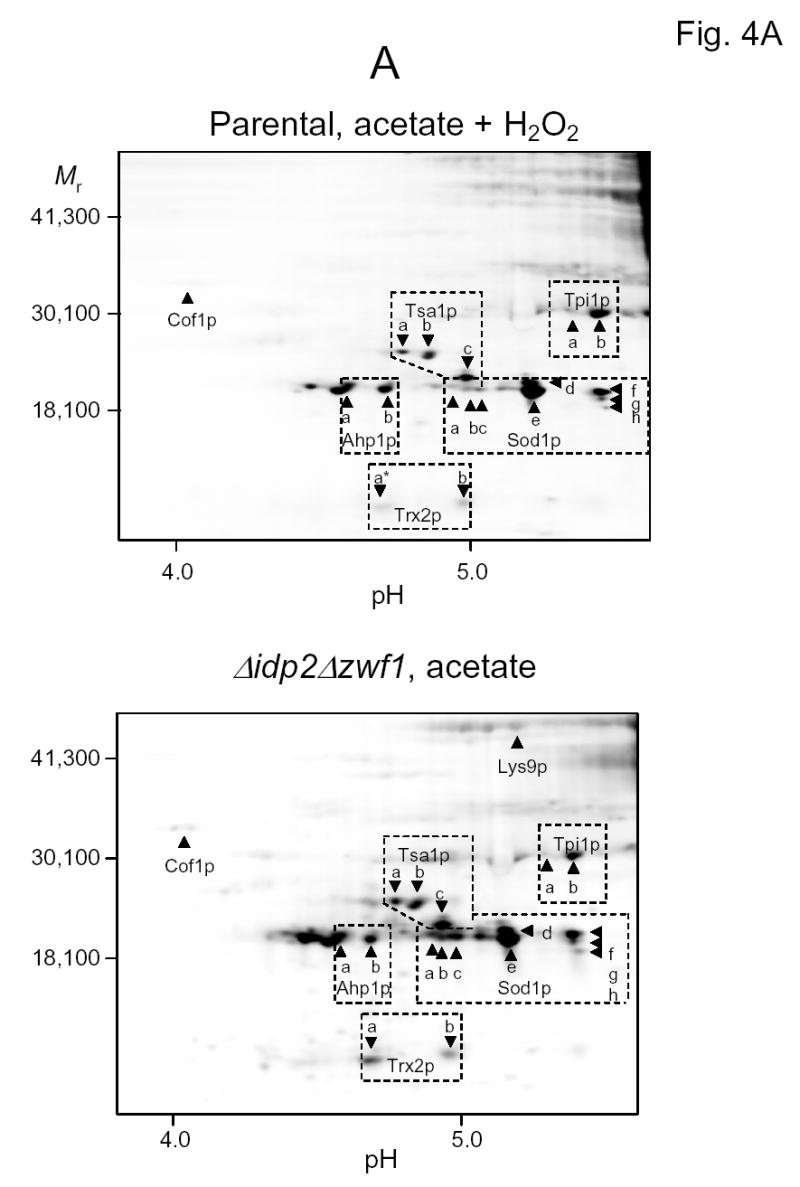
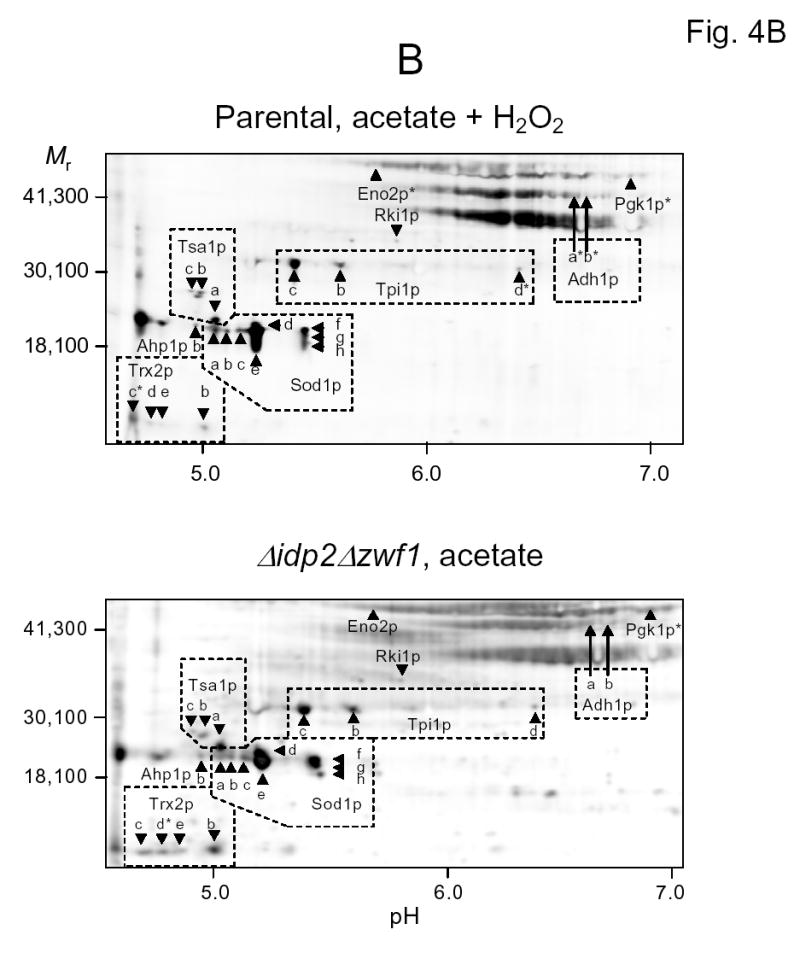
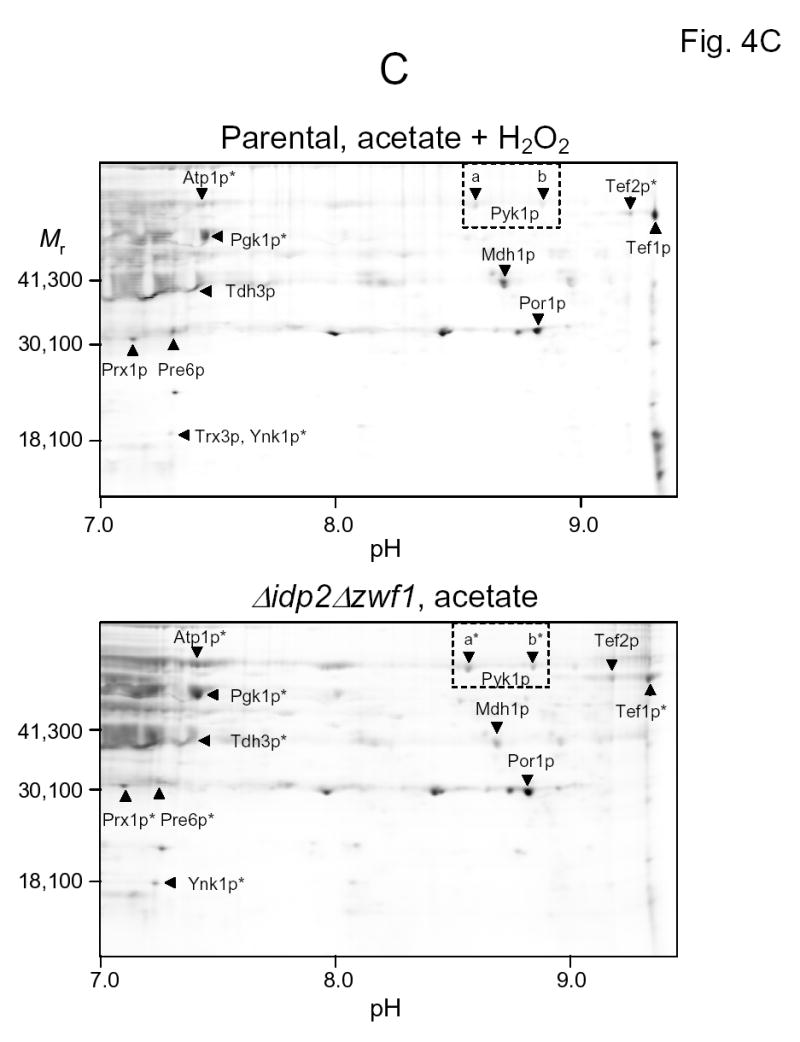
Two-dimensional gel electrophoresis of 6-IAF-labeled proteins from the parental strain shifted to acetate medium in the presence of hydrogen peroxide (top panels) and from the Δidp2Δzwf1 mutant strain shifted to acetate medium (bottom panels). Isoelectric focusing was conducted in the pH 3–6 range (A), in the pH 5–8 range (B), or in the pH 7–10 range (C). The indicated proteins were identified by mass spectrometry as described in Experimental Procedures. An asterisk indicates that labeling of a spot with 6-IAF was not above background in a sample.
Table 1.
Identities and Properties of Modified Proteins
| Protein1 | Function | Accession # | MW (Da) | pI | #Peptides |
|---|---|---|---|---|---|
| HEAT SHOCK, CHAPERONE, AND OXIDATIVE STRESS | |||||
| Ahp1p | Alkyl hydroperoxidase | P38013 | 19,115 | 4.87 | 8 |
| Cpr1p | Peptidyl-prolyl cis trans isomerase | P14832 | 17,391 | 7.51 | 5 |
| Hsp26 | Heat shock chaperone | P15992 | 23,879 | 5.22 | 13 |
| Prx1p | Mitochondrial peroxidase | P34227 | 29,496 | 9.47 | 9 |
| Sod1p* | Cu, Zn superoxidase | P00445 | 15,855 | 5.93 | 8 |
| Trx2p | Cytosolic thioredoxin | P22803 | 11,204 | 4.62 | 6 |
| Trx3p | Mitochondrial thioredoxin | P25372 | 14,432 | 9.31 | 2 |
| Tsa1p* | Cytosolic thioredoxin peroxidase | P34760 | 21,590 | 4.87 | 12 |
| PROTEIN TRANSLATION OR DEGRADATION | |||||
| Pre6p | Proteasome alpha | P40303 | 28,439 | 7.37 | 3 |
| Tef1p | EF1-alpha elongation factor | P02994 | 50,032 | 9.72 | 4 |
| Tef2p | EF1-alpha elongation factor | P02994 | 50,032 | 9.72 | 17 |
| CYTOSKELETAL OR MEMBRANE PROTEINS | |||||
| Atp1p | Alpha subunit mitochondrial ATP synthase | P07251 | 58,617 | 9.85 | 13 |
| Cof1p | Actin filament organization | Q03048 | 15,901 | 4.89 | 6 |
| Por1p | Outer mitochondrial membrane porin | P04840 | 30,428 | 8.10 | 11 |
| METABOLIC ENZYMES | |||||
| Adh1p* | Alcohol dehydrogenase | P00330 | 36,849 | 6.66 | 8 |
| Adh3p | Mitochondrial alcohol dehydrogenase | P07246 | 40,369 | 8.52 | 2 |
| Eno1p | Enolase | P00924 | 46,816 | 6.60 | 16 |
| Eno2p | Enolase | P00925 | 46,914 | 5.88 | 6 |
| Fba1p | Fructose 1.6-bisphosphate aldolase | P14540 | 39,620 | 5.65 | 6 |
| Gpm1p | Phosphoglyceromutase | P00950 | 27,608 | 9.65 | 5 |
| Lys9p* | Saccharopine dehydrogenase | P38999 | 48,917 | 4.95 | 11 |
| Mcr1p | Mitochondrial NADH cytochrome b5 reductase | P36060 | 34,138 | 9.33 | 2 |
| Mdh1p | Mitochondrial malate dehydrogenase | P17505 | 35,650 | 8.99 | 9 |
| Pdc1p | Pyruvate decarboxylase | P06169 | 61,495 | 6.12 | 2 |
| Pgk1p* | Phosphoglycerokinase | P00560 | 44,738 | 7.77 | 15 |
| Pyk1p | Pyruvate kinase | P00549 | 54,545 | 7.66 | 15 |
| Rki1p | Ribose-5-phosphate isomerase | Q12189 | 28,258 | 5.77 | 16 |
| Tdh2p | Glyceraldehyde-3-phosphate dehydrogenase | P00358 | 35,847 | 6.96 | 4 |
| Tdh3p* | Glyceraldehyde-3-phosphate dehydrogenase | P00359 | 35,746 | 6.96 | 9 |
| Tpi1p* | Triosephosphate isomerase | P00942 | 26,795 | 5.86 | 10 |
| Ynk1p | Nucleoside diphosphate kinase | P36010 | 17,167 | 9.13 | 9 |
Proteins named in bold type were identified only in carbonylation screens;
, proteins identified in screens for both disulfide bonds and carbonylation. Other proteins were identified only in the disulfide bond screens.
Important general observations include: (a) There is an overall similarity between the types of 6-IAF-labeled proteins in the Δidp2Δzwf1 mutant shifted to acetate medium and those in the parental strain shifted to acetate medium in the presence of hydrogen peroxide (Figs. 4A–C), suggesting there are some common effects of endogenous and exogenous oxidants. (b) Many of these fluorescein-labeled proteins exhibit multiple isoforms (labeled with letters) with slight differences in apparent molecular weights and isoelectric points. Multiple isoforms are common in two-dimensional analyses of yeast cellular proteins [24–28]. (c) Many of the same proteins were found to contain disulfide bonds in oleate-shifted cells (gels not shown). As shown in Table 2 and as discussed in detail below, some of these isoforms exhibited different patterns of labeling with 6-IAF. (4) It should also be noted that most of the labeled proteins identified in these experiments exhibited expected mobilities based on sizes and predicted pI values listed in databases (Table 1). Due to the overlap in pH ranges used for isoelectric focusing, some proteins were detected in two of the gels analyzed (e.g., upper panels in Figs. 4A and 4B).
Table 2.
Disulfide-Containing Proteins
|
Parental1 |
idp2Δ zwf1Δ
|
Parental + H2O2 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Glu2 | Gly | Ace | Ole | Glu | Gly | Ace | Ole | Ace | Ole | ||
| OXIDATIVE STRESS 3 | |||||||||||
| cytosolic | |||||||||||
| Sod1p | a, b, cI,II | + | − | − | − | + | + | + | + | + | + |
| d, e, fI,II | + | + | + | + | + | + | + | + | + | + | |
| gI,II | nd | − | − | + | + | + | + | + | + | + | |
| hI,II | nd | − | − | − | + | + | + | + | + | + | |
| Tsa1p | aI,II | + | + | + | + | + | + | + | + | + | + |
| BI,II | + | − | − | − | nd | + | + | + | + | + | |
| CI,II | − | − | − | − | nd | + | + | + | + | + | |
| Ahp1p | aI | + | − | − | − | + | + | + | + | + | + |
| BI,II | + | + | − | + | + | + | + | + | + | + | |
| Trx2p | aI | + | − | − | − | − | + | + | − | − | + |
| BI,II | + | − | − | − | − | + | + | − | + | + | |
| CII | nd | − | − | − | nd | − | + | + | − | − | |
| DII | − | − | − | + | − | − | − | + | + | + | |
| EII | nd | − | − | − | + | + | + | − | + | + | |
| mitochondrial | |||||||||||
| Prx1p | aIII | nd | − | − | − | nd | − | − | − | + | + |
| Trx3p | aIII | − | − | − | − | − | − | − | − | + | + |
| METABOLIC | |||||||||||
| cytosolic | |||||||||||
| Tpi1p | a, b, cI,II | + | + | + | + | + | + | + | + | + | + |
| DII | − | − | − | − | − | + | + | − | − | − | |
| Pgk1p | aII,III | − | + | + | − | − | − | − | − | − | + |
| Tdh3p | aIII | − | − | + | + | − | − | − | − | + | − |
| Pyk1p | a, bIII | − | − | + | − | − | − | − | − | + | + |
| Eno2p | aII | + | − | − | + | + | + | + | − | − | − |
| Adh1p | a, bII | − | − | − | − | − | − | + | + | − | + |
| Rki1p | aII | − | − | − | − | − | − | + | − | + | + |
| Ynk1p | aIII | − | + | + | + | − | − | − | + | − | − |
| Lys9p | aI | − | − | − | − | + | − | + | + | − | − |
| mitochondrial | |||||||||||
| Mdh1p | aIII | − | − | − | − | − | − | + | + | + | − |
| Atp1p | aIII | − | − | + | − | − | − | − | − | − | − |
| CYTOSKELETON | |||||||||||
| Cof1p | aI | − | − | + | − | + | + | + | + | + | + |
| MITOCHONDRIAL MEMBRANE | |||||||||||
| Por1p | aIII | − | − | − | − | − | + | + | + | + | + |
| TRANSLATION/DEGRADATION | |||||||||||
| Tef1p | aIII | − | + | + | − | − | − | − | + | + | + |
| Tef2p | aIII | − | − | + | − | − | − | + | + | − | + |
| Pre6p | aIII | − | − | − | + | − | − | − | − | + | − |
Protein samples were from strains were grown on or shifted to various media as described in the text.
According to the Saccharomyces Genome Database (http://yeastgenome.org/), all proteins in this list contain at least one and no more than eight cysteine residues/monomer. There is also a wide range in abundance of these proteins in yeast cells grown on glucose, with Tef1p and Tef2p being the least abundant and with Sod1 being the most abundant in this group.
Different electrophoretic isoforms of identified proteins are indicated by letters and are grouped according to their patterns of disulfide bond content. Roman numerals in italics indicate the pH range of the isoelectric focusing gel used for identification (I = pH 3–6, II = pH 5–8, III = pH 7–10). “+” indicates staining with 6-IAA, “-” indicates no staining above background with 6-IAA, and “nd” indicates no apparent SYPRO Ruby-stained protein or isoform detected under a given condition.
Specific proteins containing disulfide bonds
Disulfide bond-containing proteins identified by mass spectrometry are grouped in Table 2 according to general cellular function. The most consistently labeled group of proteins includes those with known functions in protection from oxidative stress. Among the most striking of these disulfide-containing proteins was cytosolic superoxide dismutase (Sod1p), which was identified in our two-dimensional gels in eight different spots, i.e., isoforms with slight differences in pI and/or molecular weight (Table 2, Figs. 4A and 4B). Three of these isoforms (Sod1p-d, e, and f) were labeled with 6-IAF under all conditions, and thus contain what we designate ‘constitutive’ disulfide bonds. These are most likely due to the normal intramolecular disulfide bond formation known to be formed posttranslationally during aerobic growth, and/or to intermolecular disulfide bond formation with its copper chaperone protein Ccs1p [29,30]. The other isoforms of Sod1p were labeled with 6-IAF in the Δidp2Δzwf1 mutant strain under all conditions and in the parental strain following shifts in the presence of hydrogen peroxide, suggesting that these isoforms contain ‘redox sensitive’ disulfide bonds, i.e. that appear particularly responsive to reduced NADPH levels in the mutant strain and to oxidative stress in the parental strain. Some isoforms of Sod1p and several other antioxidant proteins discussed below contained disulfide bonds in cells grown on glucose; it is possible that this may reflect diversion of NADPH to support rapid growth on this carbon source (see Discussion).
As also detailed in Table 2, several other proteins with antioxidant functions, especially thiol peroxidases that form transient disulfide bonds during catalytic conversion of hydrogen peroxide to water [31–35], were found to be readily labeled with 6-IAF. Tsa1p is the major cytosolic peroxidase in yeast; disruption of its corresponding gene was shown to produce a dramatic sensitivity to exogenous oxidants [36,37]. We identified three isoforms of Tsa1p (Figs. 4A and 4B). Tsa1p-a contained a constitutive disulfide bond in all cell extracts, whereas Tsa1p-b and Tsa1p-c were primarily labeled in the mutant strain grown on or shifted to nonfermentable carbon sources and in the parental strain challenged with hydrogen peroxide. Cytosolic alkyl peroxidase (Ahp1p) was similarly detected as two isoforms, Ahp1p-b, which appeared to contain an essentially constitutive disulfide bond, and Ahp1p-a, which was labeled in the challenged mutant and parental strain samples. Five isoforms with apparent pIs ranging from 4.6 to 5.0 were identified for the major cytosolic thioredoxin, Trx2p (Table 2). In general, these isoforms were labeled with 6-IAF in the parental strain challenged with hydrogen peroxide and in the Δidp2Δzwf1 mutant strain grown on nonfermentable carbon sources.
Two mitochondrial antioxidant proteins, periodoxin Prx1p and thioredoxin Trx3p, were only labeled with 6-IAF in the peroxide-challenged parental strain (Table 2). These proteins did not form disulfide bonds in response to endogenous stress in the mutant strain.
Patterns of disulfide bond content were more variable for metabolic enzymes identified in this study (Table 2). Of four isoforms of the glycolytic enzyme triose phosphate isomerase, three (Tpi1p-a, b, and c) appeared to contain constitutive disulfide bonds whereas one (Tpip-d) was labeled only in the Δidp2Δzwf1 mutant strain. Two other glycolytic enzymes, phosphoglycerokinase (Pgk1p) and glyceraldehyde-3-phosphate dehydrogenase isozyme 3 (Tdh3p), as well as Ynk1p, a nucleoside diphosphate kinase primarily localized in the cytosol [38], contained disulfide bonds in the parental strain during growth on nonfermentable carbon sources. Pyruvate kinase (Pyk1p) exhibited labeling with hydrogen peroxide challenge of the parental strain, and enolase isozyme 2 (Eno2p) was labeled under a variety of conditions.
Other non-glycolytic metabolic enzymes that appeared to demonstrate redox-sensitive disulfide bond formation, in that they were labeled in shifted mutant cells and in the parental strain shifted in the presence of hydrogen peroxide, were two isoforms of alcohol dehydrogenase isozyme 1 (Adh1p-a and b), ribose-5-phosphate isomerase (Rki1p) which functions in the non-oxidative branch of the pentose phosphate pathway, and mitochondrial malate dehydrogenase (Mdh1). In contrast, saccharopine dehydrogenase (Lys9p), which catalyzes an NADP+-dependent reaction in the lysine biosynthetic pathway [39], was labeled only in the mutant strain, and the alpha subunit of ATP synthase (Atp1p) was uniquely labeled in the parental strain cultivated with acetate as the carbon source.
Two other interesting proteins that exhibited redox-sensitive disulfide bond formation were Cof1p, a protein involved in actin filament organization [40], and Por1p, an outer-mitochondrial membrane voltage dependent anion channel [41] that may play a role in apoptosis [42].
Variable patterns of labeling were observed for two elongation factor proteins, Tef1p and Tef2p, that share overlapping functions in protein synthesis [43,44], and for Pre1p, an alpha-type proteosome subunit [45].
Specific proteins modified by carbonylation
In previous studies, we demonstrated that the addition of glutathione and DTT during a shift of the Δidp2Δzwf1 mutant strain to oleate medium prevented the loss of viability of this strain [11]. In preliminary studies using one-dimensional gel electrophoresis, we also found that the shift of the Δidp2Δzwf1 mutant strain to oleate medium resulted in a substantial increase in protein carbonylation, particularly at 72 h following the shift (data not shown). This increase in carbonylation was not observed when the shift was conducted in the presence of exogenous reducing agents. (These effects were much less pronounced with a shift of the mutant strain to acetate medium.) We used two-dimensional electrophoresis to examine carbonylated proteins in the mutant strain shifted to oleate in the absence or presence of glutathione and DTT, with the goal of comparing results with our data on disulfide bond-containing proteins, as well as with results of several reported studies of carbonylation in yeast cells grown on glucose with exogenous oxidants [24,46,47].
As summarized in Table 3, we identified a number of proteins that were carbonylated in the Δidp2Δzwf1 mutant strain but not in the parental strain following 72 h in oleate medium, and that were not modified in the mutant strain shifted to oleate medium in the presence of glutathione and DTT. Although carbonylation and disulfide bond formation measure different types of oxidative effects, approximately half of the carbonylated proteins (Sod1p, Tsa1p, Ahp1p, Pgk1p, Tdh3p, Tpi1p, Adh1p, and Lys9p) identified in the oleate-challenged mutant strain were also identified as disulfide-containing proteins. In some cases, the same apparent isoforms (shown in italics in Table 3) were identified in both screens. Thus, these proteins seem to be particularly redox sensitive under our experimental conditions. Alternatively, these proteins could represent some of the most abundant modified proteins in both screens. However, among carbonylated proteins that did not contain disulfide bonds were heat shock protein Hsp26p, a very abundant protein in stressed yeast cells [48], and Cpr1p, a member of the cyclophilin A family that exhibits peptidyl-prolyl-isomerase activity and is involved as a chaperone in protein localization [49,50]. These proteins, along with glyceraldehyde-3-dehydrogenase isozyme 2 (Tdh2p), mitochondrial alcohol dehydrogenase (Adh3p), and mitochondrial NADH cytochrome b5 reductase (Mcr1p), were not detectably carbonylated in the mutant strain shifted to oleate medium in the presence of external reductants (Table 3). In contrast, enolase isozyme 1 (Eno1p), aldolase (Fba1p), and pyruvate decarboxylase (Pdc1p) were apparently not protected from carbonylation by the presence of glutathione and DTT. Phosphoglucomutase (Gpm1p) exhibited multiple isoforms that demonstrated different patterns of carbonylation; one of these isoforms was uniquely carbonylated in both parental and mutant strains shifted to oleate medium.
Table 3.
Carbonyl-containing proteins
| Parental | Δidp2Δzwf1 | Δidp2Δzwf1+GSH/DTT | |
|---|---|---|---|
| OXIDATIVE STRESS 1 | |||
| Sod1p a, b, c, f 2 | − | + | − |
| d, e | + | + | + |
| Tsa1p a, b | − | + | − |
| Ahp1p b | − | + | − |
| HEAT SHOCK/CHAPERONE | |||
| Hsp26p | − | + | − |
| Cpr1p | − | + | − |
| METABOLIC | |||
| cytosolic | |||
| Tpi1p a, b | − | + | − |
| Pgk1p | − | + | − |
| Tdh2p | − | + | − |
| Tdh3p a, b | − | + | − |
| Eno1p | − | + | + |
| Fba1p | − | + | + |
| Gpm1p a | + | + | − |
| b, c | − | + | − |
| d, e | − | + | + |
| Adh1p a, b | − | + | − |
| Lys9p | − | + | − |
| Pdc1p | − | + | + |
| mitochondrial | |||
| Adh3p | − | + | − |
| Mcr1p a, b | − | + | − |
Different electrophoretic isoforms of identified proteins are indicated by letters and are grouped according to their patterns of disulfide bond content. “+” indicates carbonylation; “−” indicates no detection of carbonylation.
Isoforms listed in italics appear to correspond to those with similar designations in Table II.
Enzyme activity assays
To assess any alterations in cellular enzyme activities that might correlate with carbonylation or disulfide bond formation, protein lysates were prepared from the parental strain and the Δidp2Δzwf1 mutant strain before and at 24 h and 72 h following shifts to oleate medium (plus/minus glutathione and DTT for the mutant strain). Glucose-6-phosphate dehydrogenase activity [51] was absent in the mutant strain due to the ZWF1 gene disruption and was unchanged in the parental strain prior to and after the medium shift. NADP+-specific isocitrate dehydrogenase activity [15] was reduced ~four-fold in the mutant strain relative to the parental strain due to loss of Idp2p; these activities did not change as a function of the medium shift. Other assays were conducted to measure activities of aldolase, alcohol dehydrogenase, phosphoglycerokinase, triose phosphate isomerase, and pyruvate decarboxylase [52]; of glyceraldehyde-3-phosphate dehydrogenase [53]; and of saccharopine dehydrogenase [39]. No significant differences were observed for these activities in mutant or parental cell extracts following shifts to oleate medium. We note that multiple cellular isozymes can contribute to measured activities for enolase, glyceraldehyde-3-phosphate dehydrogenase, and alcohol dehydrogenase. However, activities for the other enzymes suggest there is no measurable effect on total cellular activity due to oxidative modifications in this study.
DISCUSSION
In this report, we present a novel comparison of oxidative changes that occur in cellular proteins as a result of challenge due to normal endogenous metabolic pathways or an exogenous oxidant. Our analyses of disulfide bond status are particularly informative about potential consequences of altered cellular levels of NADPH. Moreover, our analyses of changes in yeast cellular proteins following growth in nonfermentable carbon sources offer a unique comparison with published studies of oxidative changes conducted primarily with glucose as the carbon source.
A major conclusion of our study is that oxidative changes in the proteome due to endogenous metabolic sources can be profound and, in many cases, are similar to changes produced by an exogenous oxidant. We also find there are many similar but also many unique changes in specific protein oxidative status as a function of growth on non-fermentable carbon sources versus on glucose.
The fluorescence assay we used to screen cellular proteins provides a snapshot of disulfide bond content and is expected to reflect the flux of reducing equivalents from NADPH. By assaying for disulfide bonds at times prior to a pronounced decrease in viability of the Δidp2Δzwf1 mutant or parental strains, we believe we are primarily observing the more transient protective level of formation of disulfide bonds, i.e. intra- or intermolecular bonds and/or S-thiolation. Our data showing little effect on several enzymatic activities also imply that major irreversible oxidative damage has not occurred. Moreover, our data on carbonylation, a modification to side chains of certain amino acid residues (e.g., methionine, histidine, lysine, tyrosine, and cysteine) that is considered to be irreversible [16], show that the number of proteins that become carbonylated is relatively limited under our experimental conditions.
Our data indicate that labeling with 6-IAF is a sensitive assay for proteins containing disulfide bonds. We detected labeling of both abundant and less abundant cellular proteins. There is, however, an apparent limit to the sensitivity of labeling with 6-IAF, since we did not detect certain low abundance transcription factors (e.g., Yap1p, 54) known to contain redox-sensitive cysteine residues. Also, this method would not detect a constitutive disulfide bond in a protein containing another thiol group that is labeled with 6-IAF. Many of the proteins that were identified in our screen are known to have redox-active cysteine residues involved in detoxification of hydrogen peroxide and/or in catalytic function, as described below. In addition, many of the proteins we identified were present as multiple isoforms with slight differences in apparent pI values and molecular weights. Similar isoform patterns for various proteins were observed in multiple samples and under different conditions, suggesting reproducible and very specific post-translational modifications. These isoforms frequently demonstrated differential labeling with 6-IAF. In terms of sensitivity, Le Moan et al. [55] recently reported a screen for oxidized protein thiols in the yeast proteome using N-[14C]ethylmaleimide for labeling and an affinity column for purification of thiol-containing proteins. They identified ~60 thiol-containing proteins that exhibited various levels of oxidation in parental cells grown on glucose, but they did not distinguish levels of oxidation among different isoforms.
Several proteins with functions related to protection from oxidative stress exhibited the most striking labeling with 6-IAF. In general, labeling was observed in the Δidp2Δzwf1 mutant strain and in the parental strain following shifts to media containing hydrogen peroxide (Table 2). However, in the absence of an exogenous oxidant, several proteins were labeled in the parental strain grown on glucose but not on non-fermentable carbon sources. Le Moan et al. [55] also reported a substantial level of protein-thiol oxidation in glucose-grown yeast cells, and found that the presence of hydrogen peroxide increased the oxidation state of some of these proteins. We previously found that NADPH is required more for biosynthetic reactions than for resistance to oxidative stress in yeast cells growing on glucose [12]. With fermentative growth, macromolecular synthesis is rapid but respiratory rates are relatively slow. Also, Idp2p is not expressed during growth with glucose [15]. Thus, a rapid turnover of reducing equivalents to support rapid growth may result in less availability of NADPH to maintain oxidative stress enzymes in a fully reduced form.
We also note that many isoforms of the oxidative stress proteins in our screens were found to contain disulfide bonds in the Δidp2Δzwf1 mutant strain under almost all growth conditions (Table 2), i.e., with ‘permissive’ carbon sources glucose or glycerol, and following shifts to non-permissive oleate and acetate media. In a previous study [11], we found that some phenotypes (sensitivity to growth with acetate and mating efficiency) exhibited by this strain become exacerbated just with storage under permissive conditions. The current study suggests that the absence of two major sources of NADPH may result in a relatively oxidative intracellular environment under all growth conditions. This environment may thus produce cumulative effects with time under permissive growth conditions.
Some of the oxidative stress proteins also exhibited interesting patterns of labeling in that specific isoforms appeared to contain constitutive disulfide bonds (i.e., were fluorescently labeled in all cell extracts), whereas other isoforms appeared to be redox sensitive (i.e., were fluorescently labeled primarily in the Δidp2Δzwf1 mutant strain or in the parental strain shifted to media containing hydrogen peroxide). For Sod1p, it is interesting that two isoforms (d and e) exhibit patterns of constitutive disulfide bond content and of carbonylation, whereas three other isoforms (a, b, and c) contain disulfide bonds primarily in the Δidp2Δzwf1 mutant strain and in the exogenously stressed parental strain, and are carbonylated in the mutant strain shifted to oleate medium. This is also the case for isoforms of Tsa1p (a) and of Ahp1p (b), which contain constitutive disulfide bonds and are carbonylated in the oleate-shifted mutant strain. Thus, there is some correspondence between these different indices of oxidative status. Although Le Moan et al. [55] did not distinguish among different electrophoretic isoforms, they also reported that the oxidized cysteine content of Sod1p is high in glucose-grown cells, and that cysteine oxidation in Tsa1p and Ahp1p increases with hydrogen peroxide challenge.
Sod1p has been reported to be carbonylated in wild-type yeast strains grown on glucose following a challenge with hydrogen peroxide [24]. We also observed carbonylation of Sod1p in the Δidp2Δzwf1 mutant shifted to oleate medium. However, we also observed carbonylation of Tsa1p and Ahp1p in this strain; these proteins were not reported to be modified in previous studies of glucose-grown wild-type strains challenged with hydrogen peroxide. This suggests there are some differences as a function of fermentable versus non-fermentable carbon sources.
Interestingly, our results indicate that mitochondrial antioxidant proteins Prx1p and Trx3p contain disulfide bonds only in the hydrogen peroxide-challenged parental cells, suggesting that there is a differential response to exogenous versus endogenous oxidative challenge. In contrast, there was a redox-sensitive pattern for other mitochondrial proteins, Mdh1p and Por1p, i.e., they contain disulfide bonds in both the Δidp2Δzwf1 mutant strain and the challenged parental strain. Le Moan et al. [55] also reported an increase in the oxidative status of Mdh1p following hydrogen peroxide challenge of a parental strain grown on glucose. In addition to Mdh1p and Por1p, the essential cytosolic actin-binding protein, Cof1, which is known to affect mitochondrial morphology and distribution [56], also demonstrates a redox-sensitive labeling pattern. The altered oxidative status for these mitochondrial (and related) proteins is of particular interest due to our observation of a significant increase in petite frequency (i.e., the frequency of generation of colonies containing respiratory defects that are unable to grow on non-fermentable carbon sources) for the Δidp2Δzwf1 mutant strain following shifts to non-permissive media [11, and unpublished results].
Cytosolic metabolic enzymes showed a variety of patterns of disulfide bond content in our screens. Only Tpi1p appeared to contain constitutive disulfide bonds in three isoforms (a–c). One of these isoforms (b) was carbonylated in the Δidp2Δzwf1 mutant strain shifted to oleate medium. Redox-sensitive disulfide bond content was demonstrated for two isoforms of Adh1p, one of which (b) was also carbonylated in the shifted Δidp2Δzwf1 mutant strain, and for Rki1p, a ribose isomerase that contains a highly conserved cysteine residue near the active site [57].
Other cytosolic enzymes contained disulfide bonds, but without discernible patterns related to carbon source or to chronic oxidative stress in our screens. Le Moan et al. [55] reported that, for many metabolic enzymes in glucose-grown cells, relatively low levels of cysteine oxidation increased upon an acute challenge with hydrogen peroxide or in a mutant lacking enzymes in the thioredoxin antioxidant system. In the latter mutant, Tpi1p and Adh1p were highly oxidized, consistent with our observations of disulfide bonds in isoforms of these enzymes in the Δidp2Δzwf1 mutant strain. There are also some interesting correlations between results of these proteomic screens for cysteine oxidation [55, current study] with those obtained by Shenton and Grant [17] who examined yeast proteins that were S-thiolated, i.e., that formed mixed disulfide bonds with compounds like glutathione, in response to an acute challenge with hydrogen peroxide. Commonly labeled proteins included the glycolytic enzymes Tpi1p, Tdh3p, Eno2p, and Adh1p. A protein translation factor, Tef2p, which demonstrated disulfide bond content in some of our cellular extracts, was also identified as an S-thiolated protein. Shenton and Grant [17] found that S-thiolation correlated with a reduction in activities of several glycolytic enzymes and in rates of protein synthesis. These effects could be reversed by removal of hydrogen peroxide, suggesting that S-thiolation functions to redirect metabolic flux through the pentose phosphate pathway to increase production of NADPH during transient exogenous oxidative challenge. We think it is likely that the disulfide bonds in many of the proteins in our study are a consequence of S-thiolation, and some of the variability in patterns in different conditions may be due the relatively mild challenges that we have employed that do not produce obvious changes in enzymatic activities. However, it is also possible that some of the cysteine side chains have been oxidized to forms that would not be detected in our screen. What is quite clear in a comparison of the current and other global analyses of yeast proteins [17,55] is that there are only a limited number of cellular proteins that contain reactive cysteine side chains, and most of these proteins are cytosolic. Also, the oxidative status of the cysteine residues in these proteins appears to be a good index for cellular redox status.
Finally, there is significant overlap in proteins identified as carbonylated in the Δidp2Δzwf1 mutant strain following a shift to oleate medium and proteins previously reported to be carbonylated in glucose-grown yeast cells in response to acute challenge with hydrogen peroxide. In particular, all of the glycolytic and cytosolic metabolic enzymes listed in Table 3, with the exception of Lys9p, were identified in both types of studies [24,46,47]. In other functional categories, Sod1p and cyclophilin Cpr1p were similarly identified as carbonylated in the Δidp2Δzwf1 mutant shifted to oleate medium, and in wild-type glucose-grown hydrogen peroxide-challenged cells [24]. Other carbonylated proteins, including Tsa1p, Ahp1p, Hsp26p, Adh3p, and Mcr1p, were uniquely identified in the Δidp2Δzwf1 mutant. Interestingly, several of the proteins (including Eno1p, Eno2p, Fbpa1p, Adh1p, Tdh, and other heat shock proteins) identified in this and related studies were also found to be carbonylated in response to replicative and/or chronological aging [26]. Because of these similarities in patterns of carbonylation, examination of the state of cysteine side-chain oxidation and of levels of NADP(H) as a function of yeast cellular aging would clearly be of interest for future studies.
Acknowledgments
This work was supported by a grant (AG017477) from the National Institutes of Health (to L.M-H.) and by a grant (7P30CA54174) from the San Antonio Cancer Institute (to S.T.W.). We thank Dr. Asish R. Chaudhuri for providing detailed protocols for labeling with 6-IAF, and Kevin Hakala for assistance with some mass spectrometry analyses. The latter analyses were conducted in the Institutional Mass Spectrometry Laboratory at the UTHSCSA.
References
- 1.Costa V, Moradas-Ferreira P. Oxidative stress and signal transduction in Saccharomyces cerevisiae: insights into aging, apoptosis and diseases. Mol Aspects Med. 2001;22:217–246. doi: 10.1016/s0098-2997(01)00012-7. [DOI] [PubMed] [Google Scholar]
- 2.Jamieson DJ. Oxidative stress responses of the yeast Saccharomyces cerevisiae. Yeast. 1998;14:1511–1527. doi: 10.1002/(SICI)1097-0061(199812)14:16<1511::AID-YEA356>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 3.Park SG, Cha M-K, Jeong W, Kim I-H. Distinct physiological functions of the thiol peroxidase isoenzymes in Saccharomyces cerevisiae. J Biol Chem. 2000;275:5723–5732. doi: 10.1074/jbc.275.8.5723. [DOI] [PubMed] [Google Scholar]
- 4.Inoue Y, Matsuda T, Sugiyama K, Izawa S, Kimura A. Genetic analysis of glutathione peroxidase in oxidative stress response of Saccharomyces cerevisiae. J Biol Chem. 1999;274:27002–27009. doi: 10.1074/jbc.274.38.27002. [DOI] [PubMed] [Google Scholar]
- 5.Pedrajas JR, Kosmidou E, Miranda-Vizuete A, Gustafsson JA, Wright AP, Spyrou G. Identification and functional characterization of a novel mitochondrial thioredoxin system in Saccharomyces cerevisiae. J Biol Chem. 1999;274:6366–6273. doi: 10.1074/jbc.274.10.6366. [DOI] [PubMed] [Google Scholar]
- 6.Collinson LP, Dawes IW. Isolation, characterization and overexpression of yeast gene, GLR1, encoding glutathione reductase. Gene. 1995;156:123–127. doi: 10.1016/0378-1119(95)00026-3. [DOI] [PubMed] [Google Scholar]
- 7.Trotter EW, Grant CM. Non-reciprocal regulation of the redox state of the gluthione-glutaredoxin and thioredoxin systems. EMBO Rep. 2003;4:184–188. doi: 10.1038/sj.embor.embor729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trotter EW, Grant CM. Overlapping roles of the cytoplasmic and mitochondrial redox regulatory systems in the yeast Saccharomyes cerevisiae. Eukaryot Cell. 2005;4:392–400. doi: 10.1128/EC.4.2.392-400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minard KI, Jennings GT, Loftus TM, Xuan D, McAlister-Henn L. Sources of NADPH and expression of mammalian NADP+-specific isocitrate dehydrogenase in Saccharomyces cerevisiae. J Biol Chem. 1998;273:31486–31493. doi: 10.1074/jbc.273.47.31486. [DOI] [PubMed] [Google Scholar]
- 10.Minard KI, McAlister-Henn L. Dependence of peroxisomal beta-oxidation on cytosolic sources of NADPH. J Biol Chem. 1999;274:3402–3406. doi: 10.1074/jbc.274.6.3402. [DOI] [PubMed] [Google Scholar]
- 11.Minard KI, McAlister-Henn L. Antioxidant function of cytosolic sources of NADPH in yeast. Free Rad Biol Med. 2001;31:832–843. doi: 10.1016/s0891-5849(01)00666-9. [DOI] [PubMed] [Google Scholar]
- 12.Minard KI, McAlister-Henn L. Sources of NADPH vary with carbon source. J Biol Chem. 2005;280:39890–39896. doi: 10.1074/jbc.M509461200. [DOI] [PubMed] [Google Scholar]
- 13.Thomas D, Cherest H, Surdin-Kerjan Y. Identification of the structural gene for glucose-6-phosphate dehydrogenase in yeast. Inactivation leads to a nutritional requirement for organic sulfur. EMBO J. 1991;10:547–553. doi: 10.1002/j.1460-2075.1991.tb07981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagae I, Johnson M. Isolation and characterization of the ZWF1 gene of Saccharomyces cerevisiae, encoding glucose-6-phosphate dehydrogenase. Gene. 1990;96:161–166. doi: 10.1016/0378-1119(90)90248-p. [DOI] [PubMed] [Google Scholar]
- 15.Loftus TM, Hall LV, Anderson SL, McAlister-Henn L. Isolation, characterization, and disruption of the yeast gene encoding cytosolic NADP-specific dehydrogenase. Biochemistry. 1994;33:9661–9667. doi: 10.1021/bi00198a035. [DOI] [PubMed] [Google Scholar]
- 16.Giustarini D, Rossi R, Milzani A, Colombo R, Dalle-Donne I. S-Glutathionylation: from redox regulation of protein functions to human diseases. J Cell Mol Med. 2004;8:201–212. doi: 10.1111/j.1582-4934.2004.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shenton D, Grant CM. Protein S-thiolation targets glycolysis and protein synthesis in response to oxidative stress in the yeast Saccharomyces cerevisiae. Biochem J. 2003;374:513–519. doi: 10.1042/BJ20030414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaudhuri AR, Khan IA, Luduena RF. Detection of disulfide bonds in bovine brain tubulin and their role in protein folding on and microtubule assembly in vitro: a novel disulfide detection approach. Biochemistry. 2001;40:8834–8841. doi: 10.1021/bi0101603. [DOI] [PubMed] [Google Scholar]
- 19.Botstein D, Falco SC, Stewart SE, Brennan M, Scherer S, Stinchcomb DT, Struhl K, Davis RW. Sterile host yeast (SHY); a eukaryotic system of biological containment for recombinant DNA experiments. Gene. 1979;8:17–24. doi: 10.1016/0378-1119(79)90004-0. [DOI] [PubMed] [Google Scholar]
- 20.Thompson LM, McAlister-Henn L. Dispensable presequence for cellular localization and function of mitochondrial malate dehydrogenase from Saccharomyces cerevisiae. J Biol Chem. 1989;264:12091–12096. [PubMed] [Google Scholar]
- 21.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez-Manzaneque MT, Ros J, Cabiscol E, Sorribas A, Herrero E. Grx5 glutaredoxin plays a central role in protection against protein oxidative damage in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:8180–8190. doi: 10.1128/mcb.19.12.8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 24.Costa VMV, Amorim MA, Quintanilha A, Moradas-Ferreira P. Hydrogen peroxide-induced carbonylation of key metabolic enzymes in Saccharomyces cerevisiae: the involvement of the oxidative stress response regulators Yap1 and Skn7. Free Rad Med Biol. 2002;33:1507–1515. doi: 10.1016/s0891-5849(02)01086-9. [DOI] [PubMed] [Google Scholar]
- 25.O’Brien KM, Dirmeier R, Engle M, Poyton RO. Mitochondrial protein oxidation in yeast mutants lacking manganese-(MnSOD) or copper-and zinc-containing superoxide dismutase (CuZnSOD): evidence that MnSOD and CuZnSod have both unique and overlapping functions in protecting mitochondrial proteins from oxidative damage. J Biol Chem. 2004;279:51817–51827. doi: 10.1074/jbc.M405958200. [DOI] [PubMed] [Google Scholar]
- 26.Reverter-Branchat G, Cabiscol E, Tamarit J, Ros J. Oxidative damage to specific proteins in replicative and chronological-aged Saccharomyces cerevisiae: common targets and prevention by calorie restriction. J Biol Chem. 2004;279:31983–31989. doi: 10.1074/jbc.M404849200. [DOI] [PubMed] [Google Scholar]
- 27.Sickmann A, Reinders J, Wagner Y, Joppich C, Zahedi R, Meyer HE, Schonfisch B, Perschil I, Chacinska A, Guiard B, Rehling P, Pfanner N, Meisinger C. The proteome of Saccharomyces cerevisiae mitochondria. Proc Natl Acad Sci USA. 2003;100:13207–13212. doi: 10.1073/pnas.2135385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sumner ER, Shanmuganathan A, Sideri TC, Willetts SA, Houghton JE, Avery SV. Oxidative protein damage causes chromium toxicity in yeast. Microbiology. 2005;151:1939–1948. doi: 10.1099/mic.0.27945-0. [DOI] [PubMed] [Google Scholar]
- 29.Furukawa Y, Torres AS, O’Halloran TV. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 2004;23:2872–2881. doi: 10.1038/sj.emboj.7600276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown NM, Torres AS, Doan PE, O’Halloran TV. Oxygen and the copper chaperone CCS regulate posttranslational activation of Cu, Zn superoxide dismutase. Proc Natl Acad Sci USA. 2004;101:5518–5523. doi: 10.1073/pnas.0401175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Rad Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 32.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 33.Trivelli X, Krimm I, Ebel C, Verdoucq LI, Prouzet-Mauleon V, Chartier Y, Tsan P, Lauquin G, Meyer Y, Lancelin JM. Characterization of the yeast peroxiredoxin Ahp1 in its reduced active and overoxidized inactive forms using NMR. Biochemistry. 2003;42:14139–14149. doi: 10.1021/bi035551r. [DOI] [PubMed] [Google Scholar]
- 34.Prouzet-Mauleon V, Monribot-Espagne C, Boucherie H, Lagniel G, Lopez S, Labarre J, Garin J, Lauquin GJ. Identification in Saccharomyces cerevisiae of a new stable variant of alkyl hydroperoxidase reductase 1 (Ahp1) induced by oxidative stress. J Biol Chem. 2002;277:4823–4830. doi: 10.1074/jbc.M109614200. [DOI] [PubMed] [Google Scholar]
- 35.Rabilloud T, Heller M, Gasnier F, Luche S, Rey C, Aebersold R, Benahmed M, Louisot P, Lunardi J. Proteomics analysis of cellular response to oxidative stress. Evidence for an in vivo overoxidation of peroxiredoxins at their active site. J Biol Chem. 2002;277:19396–19401. doi: 10.1074/jbc.M106585200. [DOI] [PubMed] [Google Scholar]
- 36.Chae HZ, Kim IH, Kim K, Rhee SG. Cloning, sequencing, and mutation of thiol-specific antioxidant gene of Saccharomyces cerevisiae. J Biol Chem. 1993;268:16815–16821. [PubMed] [Google Scholar]
- 37.Lee SM, Park JW. Theromosensitive phenotype of yeast mutant lacking thioredoxin peroxidase. Arch Biochem Biophys. 1998;359:99–106. doi: 10.1006/abbi.1998.0896. [DOI] [PubMed] [Google Scholar]
- 38.Amutha B, Pain D. Nucleoside diphosphate kinase of Saccharomyces cerevisiae, Ynk1p: localization to the mitochondrial intermembrane space. Biochem J. 2003;370:805–815. doi: 10.1042/BJ20021415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Storts DR, Bhattacharjee JK. Purification and properties of saccharopine dehydrogenase (glutamate forming) in the Saccharomyces cerevisiae lysine biosynthetic pathway. J Bacteriol. 1987;169:416–418. doi: 10.1128/jb.169.1.416-418.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iida K, Moriyama K, Matsumoto S, Kawasaki H, Nishida E, Yahara I. Isolation of a yeast essential gene, COF1, that encodes a homologue of mammalian cofilin, a low-M(r) actin-binding and depolymerizing protein. Gene. 1993;124:115–120. doi: 10.1016/0378-1119(93)90770-4. [DOI] [PubMed] [Google Scholar]
- 41.Mihara K, Sato R. Molecular cloning and sequencing of cDNA for yeast porin, an outer mitochondrial membrane protein: a search for targeting signal in the primary structure. EMBO J. 1985;4:769–774. doi: 10.1002/j.1460-2075.1985.tb03695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Bras M, Clement MV, Pervaiz S, Brenner C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol Histopathol. 2005;20:205–219. doi: 10.14670/HH-20.205. [DOI] [PubMed] [Google Scholar]
- 43.Schirmaier F, Phillippsen P. Identification of two genes coding for the translation elongation factor EF-1 alpha of S. cerevisiae. EMBO J. 1984;3:3311–3315. doi: 10.1002/j.1460-2075.1984.tb02295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cottrelle P, Cool M, Thuriaux P, Price VL, Thiele D, Buhler JM, Fromageot P. Either one of the two yeast EF-1 alpha genes is required for cell viability. Curr Genet. 1985;9:693–697. doi: 10.1007/BF00449823. [DOI] [PubMed] [Google Scholar]
- 45.Heinemeyer W, Trondle N, Albrecht G, Wolf DH. PRE5 and PRE6, the last missing genes encoding 20S proteasome subunits from yeast? Indication for a set of 14 different subunits in the eukaryotic proteasome core. Biochemistry. 1994;33:12229–12237. doi: 10.1021/bi00206a028. [DOI] [PubMed] [Google Scholar]
- 46.Cabiscol E, Piulats E, Echave P, Herrero E, Ros J. Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J Biol Chem. 2000;275:27393–27398. doi: 10.1074/jbc.M003140200. [DOI] [PubMed] [Google Scholar]
- 47.Yoo BS, Regnier FE. Proteomic analysis of carbonylated proteins in two-dimensional gel electrophoresis using avidin-fluorescein affinity staining. Electrophoresis. 2004;25:1334–1341. doi: 10.1002/elps.200405890. [DOI] [PubMed] [Google Scholar]
- 48.Susek RE, Lindquist S. Transcriptional derepression of the Saccharomyces cerevisiae HSP26 gene during heat shock. Mol Cell Biol. 1990;10:6362–6373. doi: 10.1128/mcb.10.12.6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown CR, Cui DY, Hung GG, Chiang HL. Cyclophilin A mediates Vid22p function in the import of fructose-1,6-bisphosphatase into Vid vesicles. J Biol Chem. 2001;276:48017–48026. doi: 10.1074/jbc.M109222200. [DOI] [PubMed] [Google Scholar]
- 50.Ansari H, Greco G, Luban J. Cyclophilin A peptidyl-prolyl isomerase activity promotes ZPR1 nuclear import. Mol Cell Biol. 2002;22:6993–7003. doi: 10.1128/MCB.22.20.6993-7003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuby SA, Noltman EA. Glucose 6-phosphate dehydrogenase (crystalline) from brewer’s yeast. Meth Enzymol. 1966;9:116–125. [Google Scholar]
- 52.Maitra PK, Lobo Z. A kinetic study of glycolytic enzyme synthesis in yeast. J Biol Chem. 1971;246:475–488. [PubMed] [Google Scholar]
- 53.McAlister L, Holland M. Isolation and characterization of yeast strains carrying mutations in the glyceraldehyde-3-phosphate dehydrogenase genes. J Biol Chem. 1985;260:15013–15018. [PubMed] [Google Scholar]
- 54.Gulshan K, Rovinsky SA, Coleman ST, Moye-Rowley WS. Oxidant-specific folding of Yap1p regulates both transcriptional activation and nuclear localization. J Biol Chem. 2005;280:40524–40533. doi: 10.1074/jbc.M504716200. [DOI] [PubMed] [Google Scholar]
- 55.Le Moan N, Clement G, Le Maout S, Tacnet F, Toledano MB. The S. cerevisiae proteome of oxidized proteinthiols: Contrasted functions for the thioredoxin and GSH pathways. J Biol Chem. 2006;281:10420–10430. doi: 10.1074/jbc.M513346200. [DOI] [PubMed] [Google Scholar]
- 56.Altmann K, Westermann B. Role of essential genes in mitochondrial morphogenesis in Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:5410–5417. doi: 10.1091/mbc.E05-07-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Graille M, Meyer P, Leulliot N, Sorel I, Janin J, Van Tilbeurgh H, Quevillon-Cheruel S. Crystal structure of the S. cerevisiae D-ribose-5-phosphate isomerase: comparison with the archaeal and bacterial enzymes. Biochimie. 2005;87:763–769. doi: 10.1016/j.biochi.2005.03.001. [DOI] [PubMed] [Google Scholar]