

Abstract
Posttranslational histone modifications play an important role in regulating chromatin dynamics and function. One of the modifications, methylation, occurs on both lysine and arginine residues and participates in diverse range of biological processes including heterochromatin formation, X-chromosome inactivation, and transcriptional regulation. While acetylation, phosphorylation, and ubiquitylation are dynamically regulated by enzymes that catalyze the addition and removal of a particular modification, enzymes that are capable of removing methyl groups were not known until recently. Thus far, two families of histone demethylases with distinct cofactor requirements and reaction mechanisms have been identified. One is the FAD (flavin adenine dinucleotide)-dependent amine oxidase family LSD1 (lysine specific demethylase), the other is the Fe(II) and α-KG (α-ketoglutarate)-dependent dioxygenase family JHDM (JmjC domain-containing histone demethylase). Identification and characterization of these histone demethylases is an important step towards understanding both the function and regulation of histone methylation. Here, we describe assays currently used for measuring histone demethylase activity and chromatography strategies used in purifying histone demethylases from HeLa cells.
Keywords: Demethylase, Demethylation, Histone, JmjC, Methylation, Purification
1. Introduction
DNA in a eukaryotic cell is compacted with histones and non-histone proteins in the form of chromatin. Covalent modifications of core histones play an important role in regulating chromatin dynamics and have profound effects on a variety of cellular processes(1). One such modification, methylation, occurs on both lysine and arginine residues. While lysine methylation can take three states (mono-, di-, and tri-), arginine methylation only occurs in the mono- and di(symmetric or asymmetric) state (2). Thus far, 17 lysine residues and 7 arginine residues on histone molecules have been reported to be modified by methylation (3). Enzymes that catalyze histone methylation belong to three distinct protein families including the protein arginine methyltransferase family, the SET domain-containing family, and the Dot1 protein family (2, 4, 5). Studies in the past several years have revealed the involvement of histone methylation in a diverse range of biological processes including heterochromatin formation, X-chromosome inactivation, and transcriptional regulation (4-6). Unlike acetylation and arginine methylation, which generally correlate with transcriptional activation, histone lysine methylation can signal both activation and repression depending on the particular lysine residue that is methylated (2, 5, 7). Even within the same lysine residue, the biological consequence can differ depending on whether it is mono-, di-, or tri-methylated (8, 9).
While histone acetylation, phosphorylation, and ubiquitylation are reversible enzymatic processes, histone methylation was considered to be an irreversible process until recently. This notion was largely based on studies demonstrating that histones and methyl lysine residues within them have similar half-life (10-12), although there was some evidence that active turn over of methyl group does take place at low levels (13, 14). A few models have been proposed to explain the turn over of histone methyl groups. They include clipping of methylated histone tails (15) and replacement of methylated histones with unmethylated variant histones such as H3.3 (16-18). On the other hand, a “binary switch” model, in which the effect of methylation is counteracted by another histone modification, phosphorylation, has been demonstrated for histone H3 lysine 9 (H3K9) (19, 20). However, the most straightforward way to reverse methylation is enzymatic removal of a methyl group. About forty years ago, Paik and colleagues reported an enzymatic activity capable of demethylating free mono- and di-N-methyllysine (21). Subsequently, the same group reported the detection of a histones demethylase activity (22) although this enzymatic activity was not characterized at the molecular level (23). The molecular identities of these putative histone demethylases has remained elusive for the past three decades and consequently histone methylation had been considered as a permanent epigenetic mark.
The first enzyme that has been shown to be capable of turning over a methylated residue is the human peptidyl arginine deiminase 4 (PADI4/PAD4). PADI4/PAD4 antagonizes methylation on arginine residues by converting monomethyl-arginine in histone H3 and H4 to citrulline (24, 25). PADI4/PAD4 is not a bona fide histone demethylase because the enzyme can act on both methylated and non-methylated arginine. In addition, the reaction does not regenerate arginine. These studies nevertheless demonstrats that methyl-arginine is antagonized by an enzymatic process. Shortly after these discoveries, a true histone demethylase LSD1/BHC110 was reported. Using a candidate approach, the Shi group demonstrated that LSD1/BHC110, a protein previously found in several histone deacetylase complexes, can specifically demethylate monoand di-methyl H3K4 (H3K4me1 and H3K4me2) via a FAD-dependent oxidative reaction (26). However, LSD1 cannot demethylate trimethyl-lysine because the reaction requires a protonated nitrogen on the ε-amino group of lysine (26). The lack of apparent LSD1 homolog in budding yeast coupled with the fact that the LSD1 protein family encompasses only about ten related proteins (27) made it unlikely that his family would satisfy the enzymatic requirements to counteract all histone methylation states (28). These observations raised the possibility that additional demethylases that use a different reaction mechanism exist.
Based on the mechanism used by the DNA repair demethylase AlkB (29, 30), we have developed a histone demethylase assay (31). By following the enzymatic activity, we identified a JmjC domain-containing protein JHDM1A which is capable of specifically demethylating H3K36me1 and H3K36me2 via an Fe(II)- and α-KG-dependent oxygenation (hydroxylation) reaction (31). We further demonstrate that the evolutionarily conserved JmjC domain is a signature motif for this family of histone demethylase (31). Consistent with this notion, we and others have recently identified and characterized two additional JmjC domain-containing histone demethylases JHDM2 (32) and JHDM3/JMJD2 (33-35). While JHDM2 specifically demethylates H3K9me1 and H3K9me2, JHDM3 can demethylate both H3K9me3 and H3K36me3. Given that the JmjC domain-containing proteins form a large protein family (36), we anticipate additional members of this protein family will be proved to possess histone demethylase activity.
Identification and characterization of these histone demethylases is an important step towards understanding the regulation and function of histone methylation. In response to the increased interest of the scientific community in these novel enzymes, here we describe several assays for measuring histone demethylase activity and detailed chromatography procedures that allowed us to identify JHDM1A and JHDM2A histone demethylase from HeLa cells.
2.Assays for measuring histone demethylase activity
Based on the proposed mechanism for histone demethylation (Fig. 1), we have devised two different enzymatic reaction conditions. The first reaction condition (type I) is used for analyzing the Fe(II) and α-KG dependent dioxygenase family of histone demethylases, while the second reaction condition (type II) is used for analyzing the FAD-dependent amine oxidase family of histone demethylases. For each assay, we established three different methods for measuring histone demethylase activity. The first method measures release of radioactive formaldehyde, one of the reaction products (Fig. 1), based on a modified NASH method (37) and 3H-labeled methylated histone substrates. The second method measures the change in methylation levels of histone substrates by Western blotting using site-specific methyl-histone antibodies. The third method uses mass spectrometry to detect reduction of histone peptide masses which corresponds to methyl group. The histone demethylation reaction conditions and methods of measuring histone demethylase activity are described below.
Fig. 1.

Two different mechanisms for histone demethylation. (A) Histone demethylation by oxygenation (hydroxylation) catalyzed by JHDM family proteins. JHDM family proteins catalyze addition of hydroxyl group into a methyl group on the methylated lysine using Fe(II) and a-KG as cofactors. As a result, unstable carbinolamine and succinate are generated and formaldehyde is release from carbinolamine. For simplicity, only monomethyl-lysine is illustrated. The same mechanism can be applied for demethylation of di-, or trimethylated lysine residues. (B) Histone demethylation by oxidation catalyzed by LSD1 family proteins. LSD1 catalyzes transfer of two hydrogen atoms from methylated lysine to FAD to form an imine intermediate. The imine intermediate is hydrolyzed via non-enzymatic process to produce an unstable carbinolamine intermediate followed by release of formaldehyde. For simplicity, only monomethyl-lysine is illustrated. The same mechanism can be applied to di-methylated lysine residues. This mechanism cannot be used for demethylation of tri-methylated lysine residues because the reaction requires a protonated nitrogen on the ε-amino group of lysine.
2.1. In vitro histone demethylation reaction
Histone octamers, oligonucleosome (either [3H]-labeled or not), or methylated histone peptides are prepared as described below, and incubated with protein fractions or recombinant enzymes dialyzed against buffer G (40 mM Hepes-KOH pH 7.9, 0.2 mM EDTA, 0.2 mM PMSF, 1 mM DTT, 1 mg/ml each of leupeptin, aprotinin, and pepstatin A, 10% glycerol) with 50 mM KCl (BC50) (without EDTA for the type I reaction) or recombinant enzyme in the type I reaction buffer [50 mM HEPES-KOH (pH 8.0), 7-700 μM Fe(NH4)2(SO4)2. 1 mM α-KG, 2 mM ascorbate] or in the type II reaction buffer [100 mM Glycine (pH 8.0), 50 mM KCl] for 1-3 hr at 37°C before the enzymatic activity is measured by one of the three methods described below.
2.2. Detection of histone demethylase activity by radioactive counting
Substrate preparation
For the preparation of [3H]-labeled methyl-histone octamers or -oligonucleosome substrates, histone methyltransferase reactions are performed. Histone octamers or oligonucleosomes, which are purified from HeLa cells as described previously (38), are incubated with different recombinant histone methyltransferases in the HMT reaction buffer [20 mM Tris-HCl (pH 8.0), 4 mM EDTA, 1 mM PMSF, 0.5 mM DTT, 0.03 mCi/ml S-Adenosyl-L-[methyl-3H] methionine (Perkin Elmer)] for 1-2 hr at 30°C. The reaction mixtures are dialyzed against histone storage buffer [10 mM HEPES-KOH (pH 7.5), 10 mM KCl, 0.2 mM PMSF, and 10% glycerol] in the presence (for type II reaction) or absence (for type I reaction) of 1 mM EDTA overnight to remove unincorporated S-Adenosyl-L-[methyl-3H] methionine. The labeled substrates are then ready for use in the demethylation reaction.
Detection of released radioactive formaldehyde
For detection of [3H]-formaldehyde released from histone demethylation reactions, a modified NASH method (37) is performed. After the demethylation reaction described above, the reaction is stopped by addition of TCA (trichloro acetic acid) to a final concentration of 12.5% to precipitate histone substrate and other proteins present in the reaction mixture. After centrifugation for 10 min, an equal volume of NASH reagent (3.89 M ammonium acetate, 0.1 M acetic acid, 0.2% 2,4-pentandione) is added to the supernatant and the mixture was incubated at 37 for 50 min to convert the formaldehyde to 3, 5-diacethyl-1, 4-dihydrolutidine (DDL). After addition of equal volume of 1-pentanol and vortaxing, the reaction mixture is centrifuged to separate the DDL into 1-pentanol phase. The 1-pentanol-phase is recovered and mixed with liquid scintillation cocktail before being analyzed by scintillation counting. Fig. 2A shows the resulting histone demethylase activity of recombinant JHDM1A towards various methylated histone substrates as measured by this method.
Fig. 2.
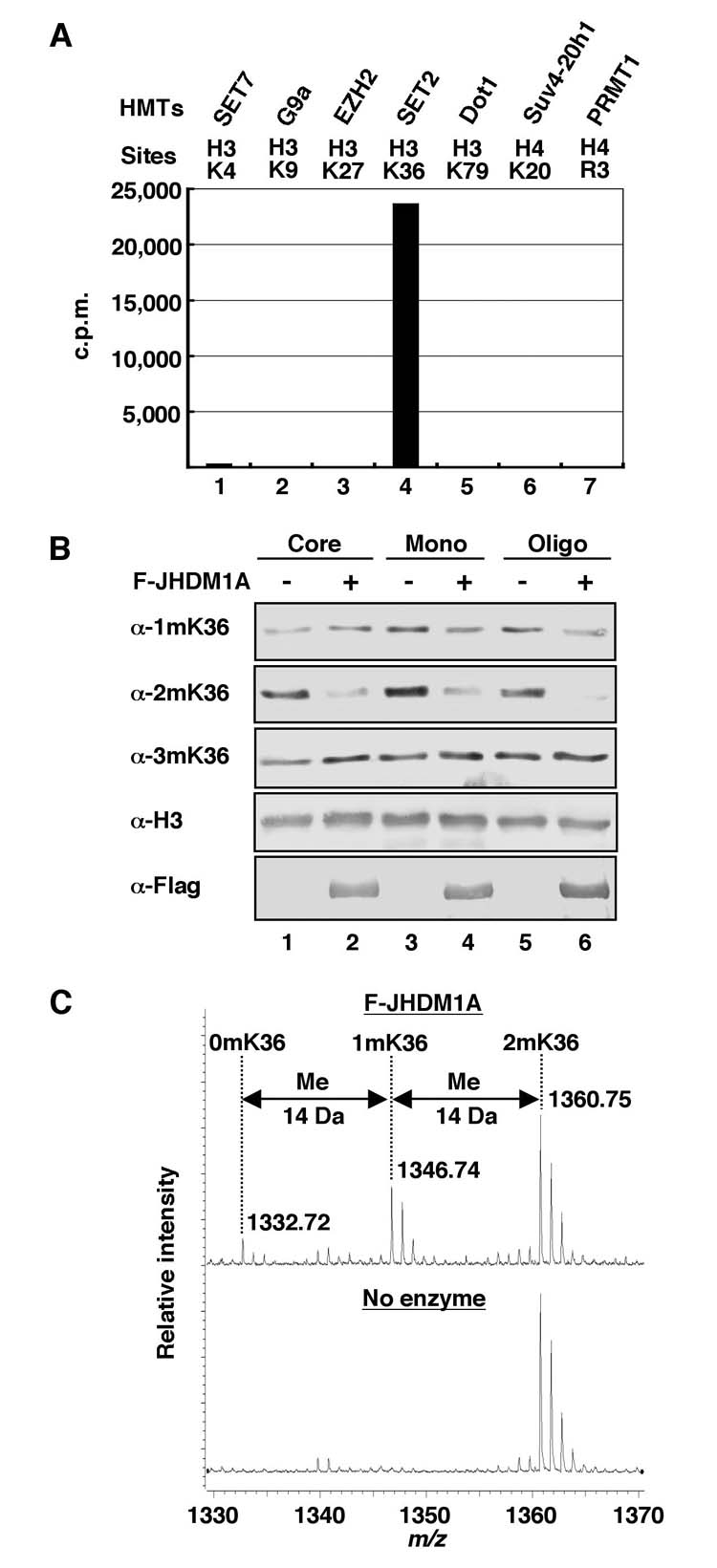
Three methods for measuring histone demethylase activity. (A) Histone demethylase activity of recombinant Flag-JHDM1A towards various methylated histone substrates as measured by release of radioactive formadehyde. The histone methyltransferases (HMTs) and their sites of methylation are indicated on top of the panel. (B) Analysis of histone demethylase activity of recombinant Flag-JHDM1A towards various histone substrates analyzed by Western blotting. Antibodies used are indicated to the left of the panel. (C) Mass spectrometry analysis of demethylation of a H3K36me2 peptide (STGGV2mKKPHRY-C) by recombinant Flag-JHDM1. The enzyme/substrate molar ratio of the reaction is 1:40. Numbers represent the mass-to-charge ratio of the substrate and product peptides.
2.3. Detection of histone demethylase activity by Western blotting
Substrate preparation
Histone octamers or oligonucleosomes are purified from HeLa cells as described previously (38), and dialyzed against histone storage buffer in the presence (for type II reaction) or absence (for type I reaction) of 1 mM EDTA.
Detection of methylated histones
After the histone demethylase reaction described above, the reaction is stopped by addition of 1/5 volume of 5 × SDS loading buffer [0.25 M Tris-HCl (pH 6.8), 0.5 M DTT, 10% SDS, 0.25% bromophenol blue, 50% glycerol] and incubated at 95°C for 5 min. Histones are separated by 18% SDS-PAGE and transferred onto nitrocellulose membrane. Western blotting with site-specific methylated histone antibodies is performed following standard procedures. Fig. 2B shows histone demethylase activity of recombinant JHDM1A towards various histone substrates as measured by this method.
2.4. Detection of histone demethylase activity by mass spectrometry
Substrate preparation
Methylated histone peptides are synthesized or purchased from commercial companies. The length of peptide may affect recognition of the substrate by different histone demethylases.
Detection of methyl group release
After the histone demethylase reaction, the reaction is stopped by addition of 2 mM EDTA. An aliquot (1/50 volume) of the reaction mixture (50 μl) is diluted 100-fold with 0.1 % formic acid, and loaded onto a 2 μl bed volume of Poros 50 R2 (PerSeptive Biosystems) reversed-phase beads packed in an Eppendorf gel-loading tip. The peptides are eluted with 5 μl of 30% acetonitrile/0.1% formic acid. A fraction (0.5 μl) of this peptide pool is analyzed by MALDI (matrix-assisted laser-desorption/ionization) TOF (time-of-flight) MS (mass spectrometry), using a BRUKER UltraFlex TOF/TOF instrument (Bruker Daltonics; Bremen, Germany), as described (39). Fig. 2C shows the histone demethylase activity of recombinant JHDM1A towards a H3K36me2 peptide substrate as measured by this method.
3. Purification of histone demethylases from HeLa cells
Histone demethylases JHDM1A and JHDM2A were purified from HeLa nuclei by monitoring enzymatic activity using chromatography. The nuclear proteins of cultured HeLa cells can be separated into nuclear extract (NE) and nuclear pellet (NP) fractions based on their differential association with the bulk of chromatin. NE fraction can be further fractionated on ion-exchange phosphocellulose P11 (Sigma) column and NP fraction can be sequentially fractionated on ion-exchange DEAE (diethylaminoethyl) cellulose DE52 (Whatman) and phosphatecellulose P11 columns. Chromatography on these two columns is performed by conventional methods. Chromatography resins are prepared following the manufactures instructions and the appropriate size columns (BioRad). The P11 fractions derived from NE and NP were used as starting materials for purification of histone demethylases.
3.1. Preparation of NE and NP
HeLa nuclear proteins are extracted and separated into NE and NP fractions essentially as previously described (38). All of the procedures described below are performed at 4°C or on ice. Briefly, after collection and washing with PBS, cultured HeLa cells are swelled in 5 pellet-volumes of buffer A (10 mM Tris-HCl pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2 mM PMSF, 0.5 mM DTT) for 10 min. Cells are then collected by centrifugation and homogenized using a type B (loose) pestle in 2 pellet-volume of buffer A. After collection by centrifuging at 2,500 rpm (Sorvall RTH-750) for 10 min, nuclei are resuspended in 3 ml buffer C (20 mM Tris-HCl pH 7.9, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM PMSF, 0.5 mM DTT, 25% glycerol) per 109 cells and homogenized again using a type B pestle. The suspension is then stirred gently using a magnetic stirring bar for 30 min and cleared by centrifugation at 15,000 rpm in a Sorvall SS-34 rotor for 30 min. The resulting supernatant is NE and the pellet is NP. The supernatant is dialyzed against buffrer D (20 mM Tris-HCl pH 7.9, 0.1 M KCl, 0.2 mM EDTA, 0.2 mM PMSF, 10 mM β-mercaptoethanol, 20% glycerol) for further fractionation or stored as the NE fraction. To extract the proteins from the pellet, the pellet is resuspended and homogenized in equal volume of buffer E (50 mM Tris-HCl pH 7.9, 5 mM MgCl2, 0.5 mM EDTA, 0.2 mM PMSF, 5 mM DTT, 25% glycerol). After adding 1/10 volume of 3 M (NH4)2SO4, the suspension is mixed immediately and DNA is sheared by sonication until the solution is no longer viscous anymore. After centrifugation at 25,000 rpm in a KAL-40.100 rotor (KOMPSPIN) for 1 hr to remove debris, the supernatant is diluted by addition of 2-volumes of buffer E and cleared of debris again by centrifuging at 25,000 rpm for 1 hr. Proteins are precipitated by addition of 0.42 g (NH4)2SO4 per ml supernatant and centrifuge at 30,000 rpm for 1 hr. The pellet is then resupended in appropriate volume of buffer F (50 mM Tris-HCl pH 7.9, 0.1 mM EDTA, 0.2 mM PMSF, 2 mM DTT, 25% glycerol) and centrifuge again at 12,000 rpm for 20 min. The ammonium sulfate concentration of the supernatant is adjusted to 20 mM for further fractionation.
3.2. Distribution of histone demethylase activities in NE and NP fractions from HeLa cells
Proteins in the NE fractions (6 g) were dialyzed against buffer G (40 mM Hepes-KOH pH 7.9, 0.2 mM EDTA, 0.2 mM PMSF, 1 mM DTT, 1 mg/ml each of leupeptin, aprotinin, and pepstatin A, 10% glycerol) containing 0.1 mM KCl (BC100) and further fractionated on a 700 ml P11 column equilibrated with BC100. Proteins that bound to the column were step eluted with buffer G containing 0.3 (BC300), 0.5 (BC500), and 1.0 M KCl (BC1000), respectively. Proteins in the NP fraction (7 g), were fractionated on a 800 ml DE52 column equilibrated with buffer G containing 20 mM (NH4)2SO4 (BD20). Proteins bound to the column were step eluted with buffer G containing 0.35 (BD350) and 0.5 M (NH4)2SO4 (BD500). The 0.35 M fraction was then dialyzes against BC100 and loaded onto a 600 ml P11 column equilibrated with BC100. Proteins bound to the column were step eluted with BC300, BC500, and BC1000, respectively. Small aliquots from every P11 fractions were dialyzed against BC50 without EDTA and then assayed for histone demethylase activity using the released radioactive formadehyde detection method described above. The distribution of histone demethylase activity towards various histone substrates in the different P11 fractions derived from HeLa nuclear proteins is shown in Fig. 3.
Fig. 3.

Distribution of histone demethylase activity towards various histone substrates in different P11 fractions derived from HeLa nuclear proteins. The numbers above the top panel represent the molar concentration of KCl in the elution buffers. Histone demethylase activity towards methylated H3K36 in type I reaction buffer (top panel), H3K9 with type I reaction buffer (middle panel), and H3K4 with type II reaction buffer (bottom panel) were measured by release of radioactive formadehyde using SET2-methylated oligonucleosomes (top panel), G9a-methylated histone octamers (middle panel), and SET7-methylated histone octamers (bottom panel) as substrates.
Using the P11 fractions as starting material, we have purified two histone demethylases JHDM1A and JHDM2A. We describe below how these two histone demethylases were purified using the AKTA FPLC (Fast Protein Liquid Chromatography) system (Amersham Biosciences). In each chromatographic step, every second to forth fraction (or portion of a fraction) was dialyzed against BC50 without EDTA before histone demethylase activity was assayed by the radioactive formaldehyde detection method. In parallel, the same fractions were also analyzed by silver staining on a 6.5-15% linear gradient SDS-PAGE to evaluate purity of enzymes.
3.3. Purification of the H3K36-specific histone demethylase JHDM1A
To identify the protein or protein complex responsible for H3K36 demethylation, we used the 0.3 M P11 fraction derived from NP as a starting material (Fig. 3). The 0.3 M P11 fraction was dialyzed against BD50 and loaded onto a 45 ml DE5PW column (TosoHaas) equilibrated with BD50. The bound proteins were eluted with 12 column-volume (cv) liner gradient from BD50 to BD500. Every forth fraction was dialyzed against BC50 without EDTA and analyzed for histone demethylase activity by measuring release of radioactive formadehyde using SET2-methylated [3H]-methyl oligonucleosome substrates. The protein purity assay by silver staining was also performed. The fractions containing the demethylase activity, which eluted between 140-185 mM ammonium sulfate, were combined and adjusted to 700 mM ammonium sulfate by addition of 3 M (NH4)2SO4 drop by drop before loading onto a 22 ml Phenyl Sepharose column (Amersham Biosciences) equilibrated with BD700. The bound proteins were eluted with 8 cv linear gradient from BD700 to BD50. Every third fraction was dialyzed against BC50 without EDTA and analyzed for histone demethylase activity and protein purity. The active fractions, which eluted between 450-360 mM ammonium sulfate, were pooled before being concentrated to 0.5 ml by centrigugation using Amicon (Millipore). The concentrated sample was loaded onto a 24 ml Superose 6 gel filtration column (Amersham Biosciences). The Superose 6 column was eluted with BC400. Every third fraction was dialyzed against BC50 without EDTA and the histone demethylase activity and protein purity were analyzed. The active fractions, which eluted between 240-320 kDa, were then combined and adjusted to 200 mM KCl by addition of BC50 dropwise before being loaded to a 0.1 ml MonoQ column (Amersham Biosciences). This chromatographic step was performed on an AKTA Purifier system using a 0.1 ml MonoQ column (Amersham Biosciences). The bound proteins were eluted with 20 cv linear gradient from BC200 to BC500. Every second fraction was analyzed for histone demethylase activity and protein purity. Two polypeptides (130 kDa and 18 kDa) correlated with the histone demethylase activity (Fig. 4B). The proteins in the active fractions that eluted from the column between 315-345 mM KCl were combined and resolved in a 6.5-15% gradient SDS-PAGE. After Coomassie staining, candidate polypeptides were excised for protein identification. Mass spectrometric analysis of the 130 kDa protein identified the protein as the human protein FBXL11 (Fig. 4C). Because histone demethylase activity is the first function attributed to FBXL11 and FBXL11 is the first JmjC domain-containing protein shown to possess histone demethylase activity, we have named the protein JHDM1A (JmjC domain-containing histone demethylase 1A).
Fig. 4.
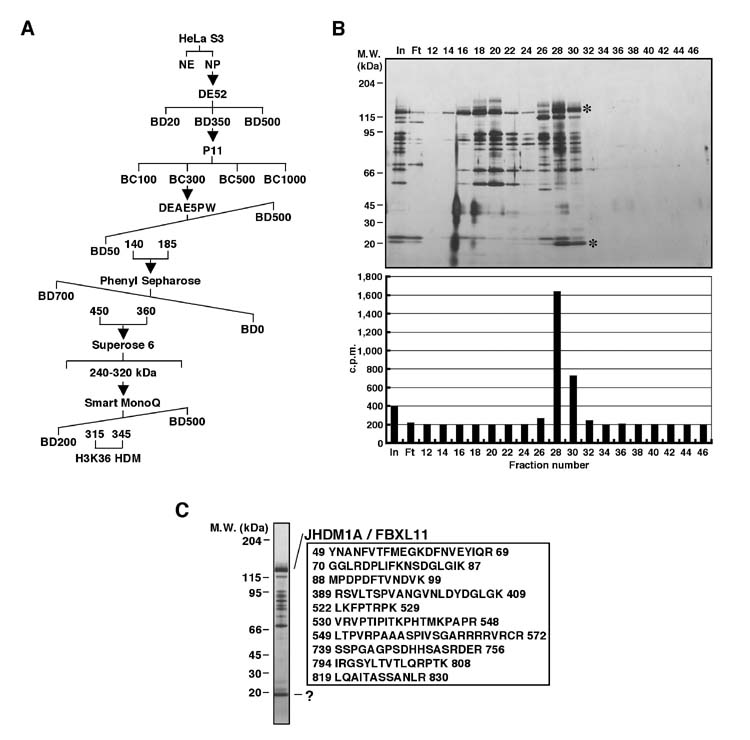
Purification of a H3K36-specific histone demethylase JHDM1A. (A) Schematic representation of the steps used in purifying the demethylase activity. Numbers represent the salt concentrations (mM) at which the histone demethylase activity elutes from the column. (B) A silver stained protein gel (top panel) and histone demethylase activities (bottom panel) of the protein fractions derived from a 0.1 ml MonoQ column. The candidate proteins that co-fractionated with the demethylase activity are indicated by *. The positions of the protein size markers on SDS-PAGE are indicated to the left of the panel. (C) A silver stained protein gel containing the samples for protein identification. The candidate protein band was identified by mass spectrometry. A total of 34 peptides covering 30% of JHDM1A/FBXL11 (NP_036440) from the top protein band were identified. Selected peptides identified from mass spectrometry are listed. The numbers correspond to the amino acid numbers in the JHDM1A protein. The “?” represents an unidentified JHDM1A associated protein.
3.4. Purification of H3K9-specific histone demethylase JHDM2A
To purify the protein or protein complex responsible for H3K9 demethylation, we started the purification using the 0.3 M P11 fraction derived from NE (Fig. 3). The 0.3 M P11 fraction was dialyzed against BD50 and loaded to a 45ml DE5PW column (TosoHaas) equilibrated with BD50. The bound proteins were eluted with 12 cv linear gradient from BD50 to BD450. Every forth fraction was dialyzed against BC50 without EDTA and analyzed for histone demethylase activity by measuring release of radioactive formadehyde using G9a-methylated [3H]-methyl core histone substrates. The protein purity assay by silver staining was also performed. The flow-through, which contained histone demethylase activity, was adjusted to 700mM ammonium sulfate by addition of 3 M (NH4)2SO4 dropwise before loading onto a 22 ml Phenyl Sepharose column (Amersham Biosciences). The bound proteins were eluted with 10 cv linear gradient from BD700 to BD0. Every forth fraction was dialyzed against BC50 without EDTA and analyzed for histone demethylase activity and protein purity. The fractions eluted from the column between 150-50 mM ammonium sulfate were pooled based on histone demethylase activity and protein purity, and concentrated to 5ml before being loaded onto a 120ml Sephacryl S–300 gel filtration column (Amersham Biosciences). The Sephacryl S–300 gel filtration column was eluted with BC400. Every third fraction was dialyzed against BC50 without EDTA and analyzed for histone demethylase activity and protein purity. The active fractions, which eluted between 150-443 kDa, were then combined and dialyzed against BC50 without EDTA before being loaded onto a 1ml Mono S column (Amersham Biosciences) equilibrated with BC50. Bound proteins were eluted with 20 cv linear gradient from BC50 to BC400. Every third fraction was analyzed for histone demethylase activity and protein purity. The active fractions were eluted from BC100 to BC150. Silver staining analysis allowed us to correlate the enzymatic activity with a 150 kDa polypeptide (Fig. 5B). To identify the proteins responsible for enzymatic activity, we concentrated two fractions (17 and 20) and compared their protein composition by 6.5-15% gradient SDS-PAGE with silver staining. Because there is a dramatic difference in the enzymatic activities between these two fractions (Fig. 5B), we were able to correlate the activity to some protein bands. After Coomassie staining performed in parallel with silver staining, candidate polypeptides that were unique to fraction 20, which also include the 150 kDa polypeptide, were excised for protein identification. Mass spectrometric analysis of the 150 kDa protein identified the human protein JMJD1A (jumonji domain-containing 1A) or TSGA (testis-specific gene A) (Fig. 5C). Because this is the second JmjC domain-containing histone demethylase, we have named the protein JHDM2A.
Fig. 5.

Purification of an H3K9-specific histone demethylase JHDM2A. (A) Schematic representation of the steps used in purifying the demethylase activity. Numbers represent the salt concentrations (mM) at which the histone demethylase activity elutes from the column. (B) A silver stained protein gel (top panel) and histone demethylase activities (bottom panel) of the protein fractions derived from a MonoS column. The candidate proteins that co-fractionated with the demethylase activity are indicated by *. The positions of the protein size markers on SDS-PAGE are indicated to the left of the panel. (C) A silver stained protein gel comparing the composition of the histone demethylase positive fraction 20 and the adjacent histone demethylase negative fraction 17. The candidate protein band was identified by mass spectrometry. A total of 63 peptides covering 53% of the JHDM2A/JMJD1A (NP_060903) were identified. Selected peptides identified from mass spectrometry are listed. The numbers correspond to the amino acid numbers in the JHDM2A protein.
4. Concluding remarks
One of the reasons that histone demethylases have been elusive until recently was the lack of a sensitive assay to detect their activity. We have developed a very sensitive assay which measures release of radioactive formadehyde. Using this assay, we have successfully purified two histone demethylases and have defined the JmjC domain as a histone demethylase signature motif. The methods described here can be applied to the study of any demethylase that uses the same reaction mechanism for demethylation of any protein or nucleic acids. Thus, we believe that these methods will be helpful in our understanding of biological events relevant to protein methylation.
Acknowledgements
We thank Robert Klose for critical reading of the manuscript. Research in the lab of Y. Z. is supported by a NIH grant (GM68804) and the Howard Hughes Medical Institute.
References
- 1.Strahl BD, Allis CD. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Reinberg D. Genes Dev. 2001;15:2343–60. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 3.Bannister AJ, Kouzarides T. Nature. 2005;436:1103–6. doi: 10.1038/nature04048. [DOI] [PubMed] [Google Scholar]
- 4.Lachner M, O'Sullivan RJ, Jenuwein T. J Cell Sci. 2003;116:2117–24. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- 5.Martin C, Zhang Y. Nat Rev Mol Cell Biol. 2005;6:838–49. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 6.Margueron R, Trojer P, Reinberg D. Curr Opin Genet Dev. 2005;15:163–76. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Kouzarides T. Curr Opin Genet Dev. 2002;12:198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 8.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Nature. 2002;419:407–11. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, An W, Cao R, Xia L, Erdjument-Bromage H, Chatton B, Tempst P, Roeder RG, Zhang Y. Mol Cell. 2003;12:475–87. doi: 10.1016/j.molcel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Byvoet P, Shepherd GR, Hardin JM, Noland BJ. Arch Biochem Biophys. 1972;148:558–67. doi: 10.1016/0003-9861(72)90174-9. [DOI] [PubMed] [Google Scholar]
- 11.Duerre JA, Lee CT. J Neurochem. 1974;23:541–7. doi: 10.1111/j.1471-4159.1974.tb06057.x. [DOI] [PubMed] [Google Scholar]
- 12.Thomas G, Lange HW, Hempel K. Hoppe Seylers Z Physiol Chem. 1972;353:1423–8. [PubMed] [Google Scholar]
- 13.Annunziato AT, Eason MB, Perry CA. Biochemistry. 1995;34:2916–24. doi: 10.1021/bi00009a023. [DOI] [PubMed] [Google Scholar]
- 14.Borun TW, Pearson D, Paik WK. J Biol Chem. 1972;247:4288–98. [PubMed] [Google Scholar]
- 15.Allis CD, Bowen JK, Abraham GN, Glover CV, Gorovsky MA. Cell. 1980;20:55–64. doi: 10.1016/0092-8674(80)90234-2. [DOI] [PubMed] [Google Scholar]
- 16.Ahmad K, Henikoff S. Mol Cell. 2002;9:1191–200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- 17.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, Spector DL. Cell. 2004;116:683–98. doi: 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson K, Pflugh DL, Yu D, Hesslein DG, Lin KI, Bothwell AL, Thomas-Tikhonenko A, Schatz DG, Calame K. Nat Immunol. 2004;5:853–61. doi: 10.1038/ni1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. Nature. 2005;438:1116–22. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 20.Fischle W, Wang Y, Allis CD. Nature. 2003;425:475–9. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 21.Kim S, Benoiton L, Paik WK. J Biol Chem. 1964;239:3790–6. [PubMed] [Google Scholar]
- 22.Paik WK, Kim S. Biochem Biophys Res Commun. 1973;51:781–8. doi: 10.1016/0006-291x(73)91383-1. [DOI] [PubMed] [Google Scholar]
- 23.Paik WK, Kim S. Arch Biochem Biophys. 1974;165:369–78. doi: 10.1016/0003-9861(74)90175-1. [DOI] [PubMed] [Google Scholar]
- 24.Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, Kouzarides T. Cell. 2004;118:545–53. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, Roeder RG, Clarke S, Stallcup MR, Allis CD, Coonrod SA. Science. 2004;306:279–83. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- 26.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Cell. 2004;119:941–53. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Kubicek S, Jenuwein T. Cell. 2004;119:903–6. doi: 10.1016/j.cell.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 28.Wysocka J, Milne TA, Allis CD. Cell. 2005;122:654–8. doi: 10.1016/j.cell.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 29.Falnes PO, Johansen RF, Seeberg E. Nature. 2002;419:178–82. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 30.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Nature. 2002;419:174–8. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 31.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Nature. 2006;439:811–6. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 32.Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. Cell. 2006;125:483–95. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 33.Robert J,K, Kenichi Y, Yangjin B, Dianzheng Z, Hediye E-B, Paul T, Jiemin W, Yi Z. Nature. 2006 doi:10.1038/nature04837. [Google Scholar]
- 34.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Cell. 2006;125:467–81. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 35.Cloos PAC, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. Nature. 2006 doi: 10.1038/nature04837. doi:10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi T, Watanabe Y, Takano-Shimizu T, Kondo S. Dev Dyn. 2006 doi: 10.1002/dvdy.20851. [DOI] [PubMed] [Google Scholar]
- 37.Kleeberg U, Klinger W. J Pharmacol Methods. 1982;8:19–31. doi: 10.1016/0160-5402(82)90004-3. [DOI] [PubMed] [Google Scholar]
- 38.Fang J, Wang H, Zhang Y. Methods Enzymol. 2004;377:213–26. doi: 10.1016/S0076-6879(03)77012-8. [DOI] [PubMed] [Google Scholar]
- 39.Erdjument-Bromage H, Lui M, Lacomis L, Grewal A, Annan RS, McNulty DE, Carr SA, Tempst P. J Chromatogr A. 1998;826:167–81. doi: 10.1016/s0021-9673(98)00705-5. [DOI] [PubMed] [Google Scholar]