

Abstract
Stat5 and Stat3, two members of the Stat (signal transducer and activator of transcription) family, are known to play critical roles in mammopoiesis/lactogenesis and involution, respectively, in the mammary gland. Phosphotyrosine phosphatase Shp2 has been shown to dephosphorylate and thus inactivate both Stat5 and Stat3 in vitro. Paradoxically, cell culture studies also suggest a positive role of Shp2 in promoting prolactin-stimulated Stat5 activation. We have shown here that selective deletion of Shp2 in mouse mammary glands suppresses Stat5 activity during pregnancy and lactation, resulting in significant impairment of lobulo-alveolar outgrowth and lactation. In contrast, Stat3 activity was slightly up-regulated shortly before/at involution, leading to normal epithelial cell apoptosis/involution in Shp2-deficient mammary gland. Thus, Shp2 acts to promote Stat5 activation by the JAK2·prolactin receptor complex, while negatively modulating Stat3 activity before the onset of involution. This is the first demonstration that Shp2 manipulates Stat5 and Stat3 activities reciprocally in mammary epithelial cells, providing novel insight into the complex mechanisms for regulation of various Stat family members by a cytoplasmic tyrosine phosphatase.
Unlike most other organs, mammary gland develops primarily in postnatal life, when it undergoes mammogenesis, lactogenesis, and involution. A critical feature of the mammary gland is its ability to produce and secret milk to nourish newborns upon successive cycles of lactation and involution with each pregnancy. During each cycle, epithelial tissue in mammary glands undergoes rapid and drastic changes of regeneration and remodeling. These processes are programmed by a complicated set of mammogenic hormones, including estrogen, progesterone and prolactin (1–3). In particular, prolactin has been shown to play a crucial role in stimulating the mammary epithelium to fully differentiate and lactate (4).
However, intracellular signaling mechanisms directing the developmental program in mammary glands are not fully understood. Members of the Stat (signal transducer and activator of transcription) family mediate cellular responses to cytokines and hormones as well as other extracellular stimuli in a variety of cell types (5, 6). In particular, it has been documented that different Stat family members function at distinct stages during mammary gland development (7–11). Stat5 activity is dramatically increased during pregnancy and lactation but then is rapidly decreased after initiation of involution (8). Consistently, Stat5a knock-out mice exhibit defective lobulo-alveolar outgrowth during pregnancy and fail to lactate after parturition (9). By contrast, Stat3 activity is up-regulated at the initiation of involution, and targeted deletion of Stat3 in the mammary gland suppresses epithelial apoptosis, resulting in delayed involution (7, 10).
Shp2 is a widely expressed protein-tyrosine phosphatase containing two amino-terminal Src homology 2 domains and a single catalytic domain (12, 13). Shp2 has been shown to negatively regulate Stat family proteins, including Stat1, -3, and -5, in different cell types in vitro apparently through direct dephosphorylation of tyrosine-phosphorylated Stats and/or down-regulation of the upstream JAK family kinase activity (14–18). Paradoxically, there are also reports suggesting that Shp2 promotes prolactin-stimulated Stat5 activation as well as up-regulation of Stat5 target gene expression in cell culture (19–21). Thus, it remains to be determined how Shp2 regulates prolactin-stimulated Stat5 activity in mammary gland development and function in vivo. To address this issue, we have created a novel mouse model with Shp2 selectively deleted in the mammary gland. This work not only demonstrates unequivocally a positive role for Shp2 in Stat5 activation by prolactin but also unexpectedly reveals reciprocal action of Shp2 on Stat5 and Stat3 proteins in a single cell type in mammals.
EXPERIMENTAL PROCEDURES
Generation and Phenotypic Analyses of Mutant Mice
A conditional mutant allele of Shp2 (Shp2flox or Shp2F) was generated in mice using the Cre-loxP system by inserting two loxP sites into introns flanking exon 4, as described previously (16). Mammary gland-specific Shp2 knock-out mice were generated by crossing Shp2F/F mice with MMTV-Cre transgenic mice (22). PCR genotyping strategies for the wild-type (Shp2+), Shp2F, and Shp2− alleles as well as the Cre transgene were described previously (16). Mice were housed and bred in compliance with the NIH guidelines and the Burnham Institute Animal Research Committee. Mammary gland tissues were surgically removed, frozen for biochemical analysis, or fixed for histological examination. Adult female mice (6 weeks–6 months) were set up for multiple mating. Litter size from the second time of breeding was counted at weaning, as the mother often ate some pups at the first litter. Only pups from litters of six or more were weighed to assess growth rate.
Whole Mount and Immunohistochemical Staining
The entire inguinal mammary gland (fourth at right side) was excised and fully spread onto glass slides and then fixed in Carnoy’s fixative solution overnight. Tissue preparations were immersed in 70% ethanol for 15 min and changed slowly to distilled water and then stained in carmine alum overnight. Tissue samples were then dehydrated through serial ethanol solutions and cleared in xylene. Finally, the mammary gland was mounted with Permount. For immunohistochemical staining, embedded tissue sections were deparaffinized through serial ethanol solutions and rinsed in distilled water. Sections were washed in phosphate-buffered saline, blocked in 1% goat serum for 30 min, and then incubated at 37 °C for 4 h or 4 °C overnight with primary antibodies at a dilution of 1:600 for Shp2 (C-18, Santa Cruz Biotechnology), 1:300 for β-casein (Santa Cruz Biotechnology), 1:200 for E-cadherin (BD Transduction Laboratories), 1:200 for NPT2b (Alpha Diagnostic), 1:300 for β-actin (Sigma), and 1:500 for phosphorylated Stat5a/b (Cell Signaling Technology). Antibodies for Stat3 and JAK2 were also purchased from Cell Signaling Technology. For apoptosis assay, embedded tissue sections from involuting two-day mammary glands were deparaffinized and analyzed by TUNEL4 assay (ApoAlert DNA fragmentation assay, BD Biosciences) according to the manufacturer’s instructions.
Biochemical Analysis
For in vivo stimulation, mice at pregnancy day 18 were injected intraperitoneally with prolactin from the National Hormone and Peptide Program (Torrance, CA) at 5 μg/g of body weight. Injection of saline solution served as a negative control. After 30 min, inguinal mammary glands were collected for immunoblotting and immunoprecipitation assay. For in vitro stimulation, mammary gland from pregnancy day 18 females were removed and chopped into small pieces in phosphate-buffered saline buffer. Tissues were then starved in F-12 medium (Invitrogen) for 30 min and stimulated with prolactin (100 μg/ml).
RESULTS
Generation of Mice with Selective Deletion of Shp2 in the Mammary Gland
In previous work, we created a conditional Shp2 knock-out allele (Shp2flox or Shp2F) using the Cre-loxP system (16), which allows for selective deletion of Shp2 in a specific cell type or tissue. Mice deficient for Shp2 expression in the mammary gland (Shp2F/F::MMTV-Cre/+ or Shp2M−/−) were generated by breeding Shp2F/F mice with MMTV-Cre transgenic mice (22). As shown in Fig. 1A, PCR analysis using three pairs of primers detected the Cre transgene, the Shp2 floxed (Shp2F), and wild-type (Shp2+) alleles in tail genomic DNA. In homozygous mutant (Shp2M−/−) mice, targeted DNA excision at the Shp2F locus was detected in the mammary gland by allele-specific PCR analysis of genomic DNA isolated from various tissues and organs (Fig. 1B). Consistently, immunoblotting analysis demonstrated decreased Shp2 protein levels selectively in the mammary gland but not in other organs, compared with controls (Fig. 1C). Immunohistochemical analysis revealed that Shp2 was highly expressed in luminal epithelial cells in control animals (Shp2F/F), whereas Shp2 expression levels were dramatically decreased in Shp2M−/− mammary glands (Fig. 1D). The decrease in Shp2 expression occurred progressively with mammogenesis and lactogenesis (Fig. 1E), suggesting that the Cre expression driven by the MMTV-LTRpromoter is up-regulated by mammogenic hormones during pregnancy and lactation, as reported previously (23, 24). Shp2 expression levels were appreciably decreased in the mammary gland of Shp2M−/− mice in virgins and in lactation and involution. There were no general developmental defects and health problems observed in Shp2M−/− mice.
FIGURE 1. Selective deletion of Shp2 in the mammary gland.
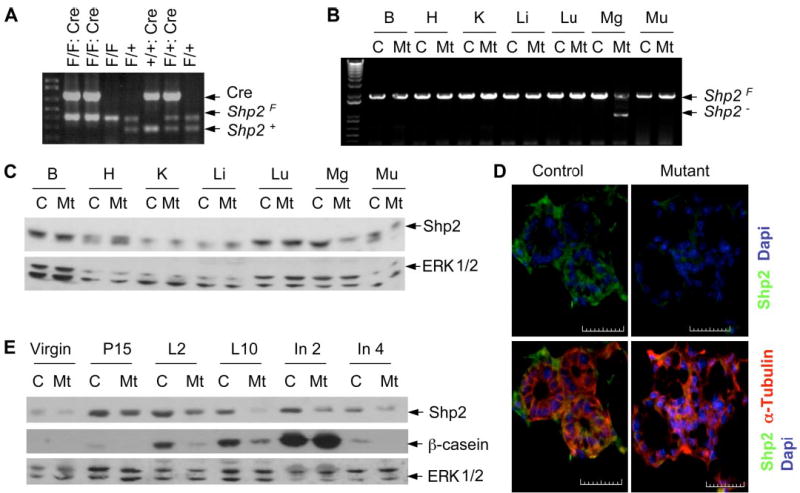
A, genotyping was done by PCR of mouse tail genomic DNA. The predicted PCR products were Cre (650 bp), Shp2F (370 bp), and Shp2+ (250 bp). B, mammary gland-specific deletion of the Shp2F allele was confirmed by PCR of genomic DNA isolated from brain (B), heart (H), kidney (K ), liver (Li), lung (Lu), mammary gland (Mg), and muscle (Mu). C, Shp2 protein levels in various tissue/organs were evaluated by immunoblot using antibodies to Shp2 or Erk1/2 (as the loading control). D, Shp2 expression in control and mutant mammary glands was assessed by immunohistochemical analysis. Mammary tissue was embedded at day 18 of pregnancy and stained with Shp2 (green) and α-tubulin (red). Scale bar, 50 μm. E, immunoblot analysis was done on mammary lysates collected from control and mutant animals at six weeks (Virgin), pregnancy day 15 (P15), lactation days 2 and 10 (L2 and L10), involution days 2 and 4 (In2 and In4). C, control; Mt, mutant. F/F, flox/flex; F/+, flox/+; +, wild-type.
Impaired Lobulo-alveolar Outgrowth in Shp2-deficient Mammary Gland
Whole mount and hematoxylin and eosin (H&E) staining were performed to analyze the role of Shp2 in mammary development (Fig. 2, A and B). There were no significantly defective structures seen at early stages of mammary gland development in virgin animals. Both control and mutant female mammary glands reached puberty around 4–6 weeks and remained mature but inactive until pregnancy. Mammary glands in Shp2M−/− mice displayed normal branched ductal structures and terminal end buds. During pregnancy, the mammary gland epithelium undergoes rapid morphogenesis. In response to various hormones, terminal end buds fully differentiate to form lobulo-alveolar structures where milk is synthesized and secreted. Compared with control littermates, Shp2M−/− mice exhibited significantly delayed and impaired morphogenesis and remodeling during pregnancy and lactation (Fig. 2C). The lobulo-alveolar structures in Shp2-deficient mammary glands were incompletely developed, resulting in highly decreased numbers of luminal cells during lactation. Whole mount staining further demonstrated that, in mutant females, some undifferentiated end bud-like structures persisted at lactation day 2 (Fig. 2D).
FIGURE 2. Impaired mammary gland development.
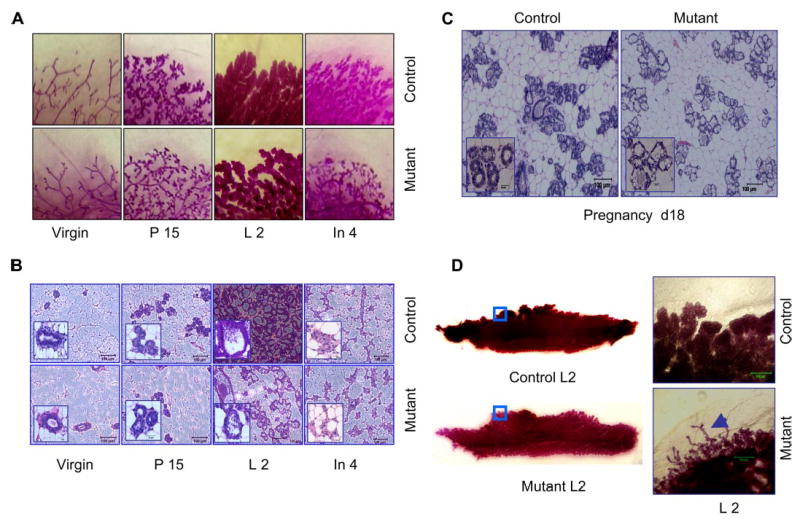
A, whole-mount staining with carmine was done for inguinal mammary gland at virgin (6 weeks), pregnancy day 15, lactation day 2, and involution day 4. B, hematoxylin and eosin staining was done on paraffin-embedded gland tissue sections at different stages. Scale bar, 100 μm (inset, high power magnification; scale bar, 20 μm). C, impaired development of lobular-alveolar structures in Shp2M−/− mammary gland at a late stage of pregnancy day 18. Scale bar, 100 μm (inset, high power magnification; scale bar, 20 μm). D, undifferentiated end bud-like structures in Shp2M−/− mammary gland at lactation day 2. Inguinal mammary glands were stained with carmine, and high power magnification was used. Arrowhead in mutant mammary gland indicates immature lobular-alveoli. Scale bar, 500 μm.
Reduced Milk Production
Toward the end of pregnancy, lobule-alveoli differentiated from end buds are fully expanded and predominate throughout the whole gland. Following parturition, this functional architecture becomes highly active and manufactures a large volume of milk. Notably, milk secretion was significantly decreased in Shp2M−/− females during lactation, consistent with impaired mammopoiesis during pregnancy. β- and γ-casein are unique lactic proteins that constitute 80% of the total milk proteins. Paired comparison of casein expression between control and mutant animals was performed for mammary tissue extracts at virgin, pregnancy, lactation, and involution stages. As shown in Fig. 1E, expression levels of β-casein were much lower in mutants than in controls during late pregnancy and lactation. Consistently, comparative analysis of several pairs of control and mutant littermates indicated that the amounts of secreted milk in mutant mice were decreased to variable extents (Fig. 3A). β-casein expression levels in mutants were highly correlated with the extent of structural defects in lobule-alveoli as revealed by H&E staining (Fig. 3B). With the unique morphological changes, the ratio of luminal cells to adipocytes changes dramatically in lactating mammary glands, which correlates well with milk production capacity. As shown in Fig. 3C, consistent with impaired milk secretion and defective morphological architecture, significant reduction in the number of luminal cells was observed in Shp2M−/− mammary glands at lactation day 2 (controls, 136.6 ± 9.64 cells/mm2; mutants, 63.4 ± 12.09; p < 0.01; n = 15–39). Conversely, an increased number of adipocytes was observed in mutant mammary gland tissue compared with controls (mutants, 189.7 ± 10.9; controls, 82.3 ± 5.15). Reduced milk secretion in Shp2-deficient mammary glands was also confirmed by immunohistochemical staining for NaPi-type IIb cotransporter (NPT2b) and a marker of lactation, casein (Fig. 3D). NPT2b is a secretion-associated apical protein produced at high levels during late pregnancy and lactation but undetectable in virgins and at involution (9). NPT2b was not expressed in ductal cells, although its expression correlates with epithelial differentiation to a secretory function, which correlates with defective milk secretion. Notably, impaired milk production in lactating Shp2M−/− females decreased the growth rate and viability of their pups (Fig. 3, E and F). Litter size or pup viability from mutant females appeared slightly reduced at both day 0 and weaning day (controls, 98.2 ± 1.02%; mutants, 88.1 ± 3.97%; p = 0.09; n = 12–16). To evaluate growth rate, newborn animals were weighed at postnatal days 2, 6, and 10 and at 4 weeks for 5–8 independent litters, and significant differences were observed during early developmental stages (Fig. 3F).
FIGURE 3. Decreased lactation in Shp2-deficient mammary gland.
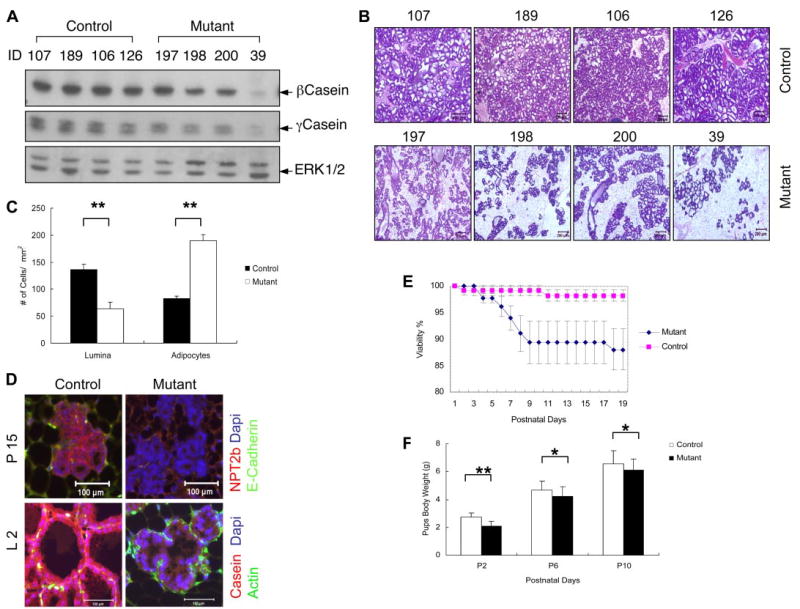
A, control and mutant animals (4/group) were analyzed for expression levels of β- and γ-casein in mammary glands at lactation day 2 with anti-Erk staining as loading control. B, histological analysis of H&E-stained sections of mammary glands from the same two groups of mice. C, the numbers of luminal cells and adipocytes/mm of mammary gland tissue were counted. D, immunohistochemical analysis was done for Npt2b and β-casein at day 15 of pregnancy and day 2 of lactation. Scale bar, 100 μm. E, litter size from control and mutant females was determined on the delivery and weaning days. Shown are: mutants (8.8 ± 0.4/cage at day 0 and 7.7 ± 0.5 at weaning day (n = 12)) and controls (8.56 ± 0.35 at day 0 and 8.44 ± 0.37 at weaning day (n = 16)). Shown for pup viability are: control (98.2 ± 1.02% and mutant (88.1% ± 3.97; p = 0.09 (n = 12–16)). F, growth rate of pups. Newborns were weighed at days 2, 6, and 10. *, p < 0.05; **, p < 0.01 (n = 5– 8). ID, mouse tag ID number.
Normal Involution and Epithelial Apoptosis in Shp2M−/− Mice
To determine the effect of Shp2 deletion on involution and epithelial cell apoptosis, pups were removed from their mothers to forcibly terminate lactation at day 10 of lactation (also designated day 0 of involution). Milk accumulated at similar high levels in both control and mutant animals at day 2 of forced involution and then was abruptly reduced at day 4, as assessed by the β-casein levels (Fig. 1E). This result suggests that the Shp2-deficient mammary gland can effectively regress to a resting state through alveolar cell apoptosis. The normal remodeling process in Shp2M−/− mice was also witnessed by whole mount and H&E staining (Fig. 2, A and B). Consistently, there was no significant difference in cell apoptosis rates between control and mutant mammary glands measured by TUNEL assay on days 2 and 4 of involution (Fig. 4).
FIGURE 4. Normal involution and apoptosis in Shp2M−/− mice.
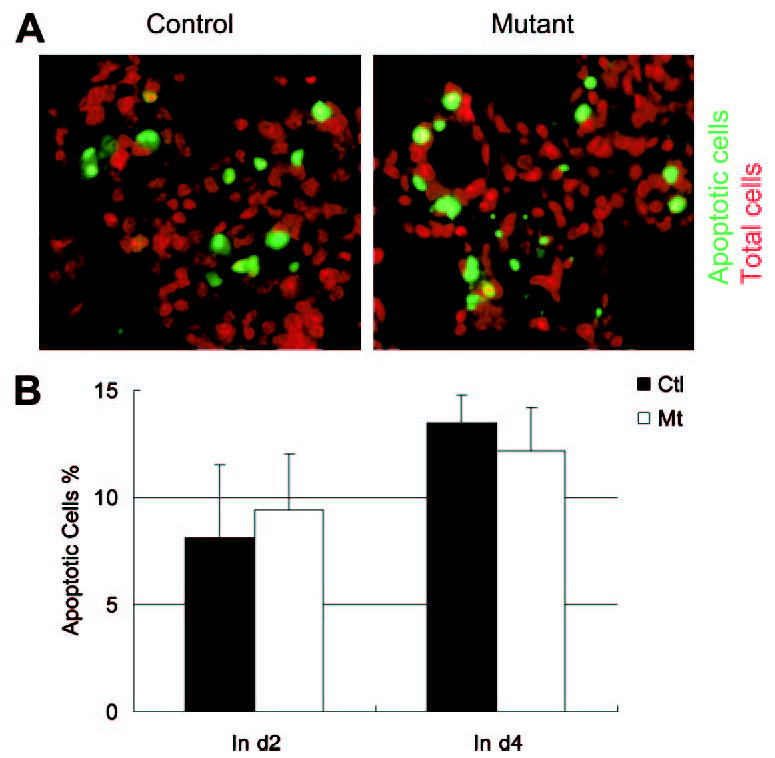
Paraffin-embedded mammary gland sections at involution day 2 were analyzed by TUNEL assay. Apoptotic luminal cells were visualized using fluorescent-conjugated DNA fragment; total cells were indicated by propidium iodide staining. A, a representative TUNEL assay on day 2 (d2) of involution. B, quantitative analysis of the data collected at days 2 and 4 of involution. No significant differences were found in apoptotic cells between controls (Ctl ) and mutants (Mt).
Bidirectional Regulation of Stat5 and Stat3 Activities by Shp2
To dissect the mechanism of Shp2 function in mammary glands, we evaluated the activation status of Stat5 and Stat3, which are strongly implicated in mammopoiesis/lactation and involution, respectively. Phospho-Stat5 levels were low in virgin mammary glands, rose dramatically during lactation, and rapidly declined following involution in control animals (Fig. 5A). Notably, both Stat5 protein contents and phospho-Stat5 levels were significantly lower in Shp2M−/− mice than in control animals during lactation, consistent with impaired lactogenesis illustrated in Fig. 3. When normalized against Stat5 protein levels, phospho-Stat5 signals were decreased during lactation in mutants compared with controls (Fig. 5B). Thus, Shp2 is positively required for Stat5 activation during pregnancy and lactation.
FIGURE 5. Bidirectional regulation of Stat5 and Stat3 activities by Shp2.
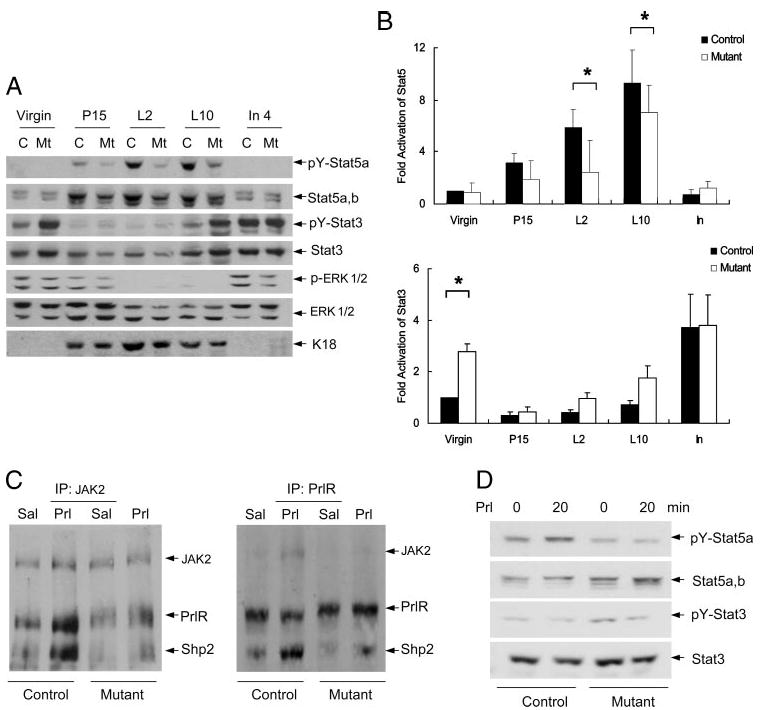
A, immunoblot analysis of phospho-Stat5, phospho-Stat3, and phospho-ERK levels was done on mammary glands at virgin (6 weeks), pregnancy day 15 (P15), lactation days 2 and 10 (L2, L10), and involution day 4 (In4). B, signals for phospho-Stat5 and phospho-Stat3 were densitometrically quantitated and normalized against the protein amounts using NIH Image J software by setting the value at virgin as 1 unit. For Stat5 (n = 5), Shp2-deficienct mammary gland exhibited significantly reduced p-Stat5 contents at lactation days 2 and 10 (p = 0.034 at lactation day 2; p = 0.022 at lactation day 10), slightly decreased at pregnancy day 15 (p = 0.056), and no difference at virgin and involution. For Stat3 (n = 3), statistical analysis indicated only a significant difference at virgin (p = 0.032), no difference at pregnancy day 15 (p = 0.115), lactation day 2 (p = 0.073), lactation day 10 (p = 0.082), and involution day 2 (p = 0.507). *, P < 0.05. C, late pregnant mice were injected intraperitoneally (IP) with prolactin or saline, and inguinal mammary glands were dissected and lysates were prepared. Lysates were immunoprecipitated with antibodies against JAK2 or PrlR. The resulting immunoprecipitates were blotted with antibodies against PrlR, JAK2, and Shp2 on the same membrane. D, activation of Stat5 and Stat3 was analyzed following in vitro stimulation of isolated mammary gland tissue with prolactin. Prl, prolactin, Sal, saline.
In agreement with the literature (25, 26), phospho-Stat3 levels were high in the resting mammary gland (virgin), decreased during pregnancy/lactation, and returned to high levels before/at the onset of involution (Fig. 5A). In contrast to the phospho-Stat5 signal change, phospho-Stat3 levels were elevated in Shp2-deficient mammary glands compared with controls in virgin animals and at the late stage of lactation or before the onset of involution. Strikingly, targeted deletion of Shp2 in the mammary gland resulted in opposite effects on Stat5 and Stat3 activities, when examined on lactation day 10. As compared with the control, the phospho-Stat5 signal was decreased, while the phospho-Stat3 level was increased in the mutant (Fig. 5A).
We also evaluated the activation status of ERK by monitoring the phospho-ERK1/2 levels. Compared with controls, phospho-ERK levels were modestly decreased in mutants during virgin and involution states, and phospho-ERK signals were undetectable during lactation (Fig. 5A), consistent with observations in other cell types that Shp2 acts to promote signaling through the ERK pathway (12, 13).
Previous experiments have shown that prolactin expression rises during late pregnancy and throughout lactation and falls rapidly during involution and that prolactin promotes maturation of epithelial lumen and stimulates milk secretion by activating the Stat5 pathway (2). To define a physiological role for Shp2 in prolactin action, late pregnant mice were injected intraperitoneally with prolactin and mammary lysates prepared. Co-immunoprecipitation assay with anti-prolactin receptor (PrlR) and anti-JAK2 antibodies showed that, upon prolactin treatment, the PrlR rapidly recruited JAK2 and Shp2 to form a PrlR·JAK2·Shp2 complex (Fig. 5C). Notably, in Shp2-deficient mammary tissue lysates, prolactin-stimulated association of JAK2 with PrlR was impaired, suggesting a role for Shp2 in promoting assembly of a JAK2·PrlR complex essential for Stat5 activation. Consistently, previous in vitro experiments demonstrated that formation of a trimetric complex is indispensable for transmission of lactogenic signals (20). To test this hypothesis further, we treated the isolated mammary tissues with prolactin or saline in vitro for 20 min and examined phospho-Stat5 and -Stat3 levels (Fig. 5D). Compared with controls, Shp2-deficient mammary tissue exhibited appreciably lower phospho-Stat5 signals following prolactin stimulation in vitro. Together, our physiological and biochemical data in vivo and in vitro argue strongly that Shp2 is positively required for prolactin-stimulated Stat5 activity in mammopoiesis and lactogenesis.
DISCUSSION
We have demonstrated here a positive role of Shp2 in mammopoiesis and lactogenesis but not in involution through generating a mutant mouse model with targeted deletion of Shp2 in the mammary gland. More importantly, we have shown that Shp2 has opposite roles in modulating the activities of two Stat family members, Stat5 and Stat3, which function at different developmental stages of the mammary gland. Shp2 has a positive role in promoting prolactin-stimulated Stat5 activation during pregnancy/lactation, while acting as a negative regulator of Stat3. Thus, this work reveals that Shp2 acts not only as a negative regulator of signaling pathways mediated by various Stats, as generally accepted but also functions as a positive one in some cases. Notably, the increase in Stat5 expression during mammopoiesis was also down-regulated in Shp2M−/− mice, which could be due to a positive role of Shp2 in inducing Stat5 expression or alternatively could be secondary to the impaired development of the lobulo-alveolar outgrowth and therefore the epithelial tissue mass in the architecture. Defective lactation in Shp2M−/− mice is likely caused by the combined direct effect of Shp2 deletion on Stat5 activation by prolactin and impaired lobulo-alveolar structures.
Several groups have shown previously that Shp2 dephosphorylates and therefore negatively regulates Stat1, -2, -3, and -5 activities in vitro (14, 17, 18, 27). Shp2-deficient mouse fibroblast cells are more sensitive to the anti-proliferative/pro-apoptotic effects of interferons than wild-type cells due to increased activation of Stat1 and -2 (14). Shp2 (but not its close relative Shp1) directly dephosphorylates tyrosine-phosphorylated Stat5 in vitro (17, 18). Shp2-deficient mouse embryonic stem cells displayed defective differentiation and more efficient self-renewal in vitro at least in part due to increased Stat3 activity induced by leukemia inhibitory factor (27, 28), suggesting a negative role of Shp2 in modulating Stat3 activation. Watson and colleagues (29) have demonstrated a non-redundant role of leukemia inhibitory factor in inducing Stat3 activity essential for epithelial apoptosis during involution in mammary gland. Consistently, we found that, in Shp2-deficient mammary gland, Stat3 activity was up-regulated shortly before the initiation of involution as compared with controls.
In contrast, we observed that deletion of Shp2 resulted in down-regulation of prolactin-induced Stat5 activation during mammopoiesis and lactation, which argues for a positive role of Shp2 in modulating Stat5 activity. This observation seems at odds with the dephosphorylation effect of Shp2 on Stat5 observed in vitro by several groups including us (17, 18). However, this in vivo data is consistent with previous experiments showing that Shp2 promotes prolactin-stimulated Stat5 activity in a reporter assay in cell culture (20). Shp2 appears to regulate the stability of JAK2 association with PrlR through modulating JAK2 interaction with Socs1 (17, 19–21, 30, 31). JAK2 is associated with PrlR and is activated upon prolactin stimulation, which in turn phosphorylates Stat5, leading to its activation and translocation into the nucleus, where Stat5 acts as a transcription activator. Recent in vitro experimental results suggest that Shp2 can dephosphorylate a phosphotyrosyl site (Tyr-1007) on JAK2, thereby preventing JAK2 association with Socs1, a negative regulator of the prolactin response during lactation (30, 32). By binding to JAK2 via p-Tyr-1007, Socs1 targets JAK2 to an ubiquitin-dependent degradation pathway, thus regulating protein stability (30, 33). Consistent with a previous in vitro observation, our experiments showed that in Shp2-deficient mammary lysates, the amount of JAK2 associated with PrlR was decreased following prolactin stimulation, leading to impaired Stat5 activation by prolactin. To our knowledge, this is the first physiological data showing a positive role of Shp2 in regulating the activity of a Stat family member.
It is important to note that the data shown here does not rule out the possibility that Shp2 subsequently has a negative effect in dephosphorylating Stat5, leading to down-regulation of the active Stat5 signal. It is likely that Shp2 has dual functions in regulating Stat5 activity in the mammary gland, first acting as a positive regulator for the activation of Stat5 by prolactin and then as a negative effector in dephosphorylation/down-regulation of the activated Stat5 pathway. As the positive role of Shp2 precedes its negative effect on Stat5 activity, we observed defective mammopoiesis/lactation in Shp2M−/− mice. A similar positive and then negative effect of CD45 tyrosine phosphatase in regulation of lymphocyte activation has been documented (34). Furthermore, this work does not rule out the possibility that Shp2 acts primarily as a negative regulator of Stat5 activity in other cell types. In this regard, the cell type-specific or inducible gene deletion of Shp2 in mice will prove to be a powerful experimental approach to elucidating the mechanisms underlying the control of signaling specificity in vivo.
Acknowledgments
We thank our laboratory members for helpful discussion and critical reading of the manuscript.
Footnotes
This work was supported by National Institutes of Health Grants CA078606 and CA102583.
The abbreviations used are: TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling; H&E, hematoxylin and eosin.
References
- 1.Hennighausen L, Robinson GW. Genes Dev. 1998;12:449–455. doi: 10.1101/gad.12.4.449. [DOI] [PubMed] [Google Scholar]
- 2.Hennighausen L, Robinson GW. Dev Cell. 2001;1:467–475. doi: 10.1016/s1534-5807(01)00064-8. [DOI] [PubMed] [Google Scholar]
- 3.Radisky DC, Hirai Y, Bissell MJ. Trends Cell Biol. 2003;13:426–434. doi: 10.1016/s0962-8924(03)00146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hennighausen L, Robinson GW, Wagner KU, Liu W. J Biol Chem. 1997;272:7567–7569. doi: 10.1074/jbc.272.12.7567. [DOI] [PubMed] [Google Scholar]
- 5.Darnell JE, Jr, Kerr IM, Stark GR. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 6.Ihle JN. Curr Opin Cell Biol. 2001;13:211–217. doi: 10.1016/s0955-0674(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 7.Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K, Akira S, Clarke AR, Watson CJ. Genes Dev. 1999;13:2604–2616. doi: 10.1101/gad.13.19.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groner B, Hennighausen L. Breast Cancer Res. 2000;2:149–153. doi: 10.1186/bcr47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyoshi K, Shillingford JM, Smith GH, Grimm SL, Wagner KU, Oka T, Rosen JM, Robinson GW, Hennighausen L. J Cell Biol. 2001;155:531–542. doi: 10.1083/jcb.200107065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humphreys RC, Bierie B, Zhao L, Raz R, Levy D, Hennighausen L. Endocrinology. 2002;143:3641–3650. doi: 10.1210/en.2002-220224. [DOI] [PubMed] [Google Scholar]
- 11.Iavnilovitch E, Groner B, Barash I. Mol Cancer Res. 2002;1:32–47. [PubMed] [Google Scholar]
- 12.Lai LA, Zhao C, Zhang EE, Feng GS. In: Protein Phosphatases. Arino J, Alexander D, editors. Springer-Verlag; Berlin Heidelberg, Germany: 2003. pp. 275–299. [Google Scholar]
- 13.Neel BG, Gu H, Pao L. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 14.You M, Yu DH, Feng GS. Mol Cell Biol. 1999;19:2416–2424. doi: 10.1128/mcb.19.3.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu TR, Hong YK, Wang XD, Ling MY, Dragoi AM, Chung AS, Campbell AG, Han ZY, Feng GS, Chin YE. J Biol Chem. 2002;277:47572–47580. doi: 10.1074/jbc.M207536200. [DOI] [PubMed] [Google Scholar]
- 16.Zhang EE, Chapeau E, Hagihara K, Feng GS. Proc Natl Acad Sci U S A. 2004;101:16064–16069. doi: 10.1073/pnas.0405041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Wen R, Yang S, Schuman J, Zhang EE, Yi T, Feng GS, Wang D. J Biol Chem. 2003;278:16520–16527. doi: 10.1074/jbc.M210572200. [DOI] [PubMed] [Google Scholar]
- 18.Yu CL, Jin YJ, Burakoff SJ. J Biol Chem. 2000;275:599–604. doi: 10.1074/jbc.275.1.599. [DOI] [PubMed] [Google Scholar]
- 19.Berchtold S, Volarevic S, Moriggl R, Mercep M, Groner B. Mol Endocrinol. 1998;12:556–567. doi: 10.1210/mend.12.4.0086. [DOI] [PubMed] [Google Scholar]
- 20.Ali S, Chen Z, Lebrun JJ, Vogel W, Kharitonenkov A, Kelly PA, Ullrich A. EMBO J. 1996;15:135–142. [PMC free article] [PubMed] [Google Scholar]
- 21.Chughtai N, Schimchowitsch S, Lebrun JJ, Ali S. J Biol Chem. 2002;277:31107–31114. doi: 10.1074/jbc.M200156200. [DOI] [PubMed] [Google Scholar]
- 22.Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ. Proc Natl Acad Sci U S A. 2000;97:3444–3449. doi: 10.1073/pnas.050408497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner KU, McAllister K, Ward T, Davis B, Wiseman R, Hennighausen L. Transgenic Res. 2001;10:545–553. doi: 10.1023/a:1013063514007. [DOI] [PubMed] [Google Scholar]
- 24.Li G, Robinson GW, Lesche R, Martinez-Diaz H, Jiang Z, Rozengurt N, Wagner KU, Wu DC, Lane TF, Liu X, Hennighausen L, Wu H. Development. 2002;129:4159–4170. doi: 10.1242/dev.129.17.4159. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Robinson GW, Hennighausen L. Mol Endocrinol. 1996;10:1496–1506. doi: 10.1210/mend.10.12.8961260. [DOI] [PubMed] [Google Scholar]
- 26.Philp JA, Burdon TG, Watson CJ. FEBS Lett. 1996;396:77–80. doi: 10.1016/0014-5793(96)01069-1. [DOI] [PubMed] [Google Scholar]
- 27.Chan RJ, Johnson SA, Li Y, Yoder MC, Feng GS. Blood. 2003;102:2074–2080. doi: 10.1182/blood-2003-04-1171. [DOI] [PubMed] [Google Scholar]
- 28.Qu CK, Feng GS. Oncogene. 1998;17:433–440. doi: 10.1038/sj.onc.1201920. [DOI] [PubMed] [Google Scholar]
- 29.Kritikou EA, Sharkey A, Abell K, Came PJ, Anderson E, Clarkson RW, Watson CJ. Development. 2003;130:3459–3468. doi: 10.1242/dev.00578. [DOI] [PubMed] [Google Scholar]
- 30.Ali S, Nouhi Z, Chughtai N. J Biol Chem. 2003;278:52021–52031. doi: 10.1074/jbc.M306758200. [DOI] [PubMed] [Google Scholar]
- 31.Yamada S, Shiono S, Joo A, Yoshimura A. FEBS Lett. 2003;534:190–196. doi: 10.1016/s0014-5793(02)03842-5. [DOI] [PubMed] [Google Scholar]
- 32.Lindeman GJ, Wittlin S, Lada H, Naylor MJ, Santamaria M, Zhang JG, Starr R, Hilton DJ, Alexander WS, Ormandy CJ, Visvader J. Genes Dev. 2001;15:1631–1636. doi: 10.1101/gad.880801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ungureanu D, Saharinen P, Junttila I, Hilton DJ, Silvennoinen O. Mol Cell Biol. 2002;22:3316–3326. doi: 10.1128/MCB.22.10.3316-3326.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huntington ND, Tarlinton DM. Immunol Lett. 2004;94:167–174. doi: 10.1016/j.imlet.2004.05.011. [DOI] [PubMed] [Google Scholar]