

Abstract
Understanding the molecular mechanisms of transition state regulator proteins is critical, since they play a pivotal role in the ability of bacteria to cope with changing environments. Although much effort has focused on their genetic characterization, little is known about their structural and functional conservation. Here we present the high resolution NMR solution structure of the N-terminal domain of the Bacillus subtilis transition state regulator Abh (AbhN), only the second such structure to date. We then compare AbhN to the N-terminal DNA-binding domain of B. subtilis AbrB (AbrBN). This is the first such comparison between two AbrB-like transition state regulators. AbhN and AbrBN are very similar, suggesting a common structural basis for their DNA binding. However, we also note subtle variances between the AbhN and AbrBN structures, which may play important roles in DNA target specificity. The results of accompanying in vitro DNA-binding studies serve to highlight binding differences between the two proteins.
Transcription factors are central to gene regulation, and elucidating their modes of action is vital for understanding gene expression. Like many bacteria, Bacillus subtilis responds to a multitude of environmental stimuli by using transcription factors to orchestrate gene expression patterns (1–3). The key to bacterial adaptability and survival lies in their capacity to initiate the correct response at the appropriate time under a specific circumstance. Such circumstances include changes in chemical concentration, temperature, osmolarity, viscosity, light, pH, density, and exposure to anti-infectives (2–5). Significant changes in transcriptional regulation occur upon sensing environments that become hostile. A shift from exponential growth phase to a stationary phase prepares a cell for survival by expression of bacterial protection genes. Accordingly, bacterial responses also have numerous forms (e.g. the secretion of toxins, polymer-degrading enzymes, or antibiotics or complete physiological transformations) (6–8). In this final illustration, bacteria can undergo cellular differentiation, leading to the development of highly resistant endospores. Spores are perhaps the ultimate line of bacterial defense, being extremely resistant to heat, sunlight, chemicals, and drugs (9–11).
In B. subtilis, transition state regulators play an essential role in spore formation and survival of the cell (12). Currently, there are several known or projected “AbrB family” transition state regulators found in an array of organisms, including Bacillus, Clostridium, Carboxydothermus, Geobacillus, Listeria, Oceanobacillus, Pyrococcus, Pasteuria, Staphylococcus, Streptococcus, Sulfolobus, and Thermoanaerobacter (12–25). To date, the most widely studied transition state regulator has been the B. subtilis AbrB protein (2, 15, 17–20, 26–31). The NMR solution structure of the DNA-binding N-terminal domain of AbrB from B. subtilis (AbrBN) has been solved independently by Cavanagh and co-workers (Protein Data Bank code 1Z0R) (32) and by Coles and co-workers (Protein Data Bank codes 1YFB and 1YSF) (33). Additionally, there is a great deal of biochemical data in support of its functional role (12, 15, 27–31). However, from a functional standpoint, little data exist for any other transition state regulator, and, to this point, there have been no solution structures determined for any other member of the AbrB family.
Here we present the high resolution NMR solution structure of AbhN, the N-terminal DNA-binding domain of the transition state regulator Abh from B. subtilis (AbhN; residues 1–54) (Protein Data Bank code 2FY9). Abh is a paralogue of AbrB. This represents only the second solution structure of a DNA-binding domain of this class of transition state regulators. Consequently, this affords the first opportunity to directly compare structural similarities and differences within the AbrB family that may influence function.
Based upon significantly different phenotypes of B. subtilis abrB and abh mutant cells,2 the two proteins display (i) different DNA recognition properties, (ii) different regulatory effects, (iii) different temporal effects upon target gene expression, or (iv) a combination of the first three qualities. In this work, we show that several conserved key residues have comparable orientations in both the dimerization and DNA recognition regions of both AbhN and AbrBN. These conserved conformations afford different transition state regulators the ability to bind structurally related subsets of DNA. However, we also note that AbhN and AbrBN display subtle structural and surface differences in some regions that probably contribute to differential DNA binding properties observed for the native proteins in vitro. Abh is capable of binding a high affinity AbrB target (34) but does so with much less affinity and differing DNase I footprinting patterns than AbrB. AbrB and Abh display a pronounced difference in the required optimum pH for binding this target, a fact that may relate to their differing physiological roles. Additionally, Abh seemingly possesses a greater propensity to bind nonspecifically to DNA sequences than does AbrB. This supports a model in which different transition state regulators differentiate between subsets of DNA structures (27).
EXPERIMENTAL PROCEDURES
Cloning
DNA fragments were obtained (via PCR methods) using oligonucleotide primer pairs AbhN-Forward (5′-GCG GGT TCA TAT GAA ATC AAT AGG TGA G-3′) and AbhN-Backward (5′-CGA ATT CCT ATT CTT TTA AAG CGG C-3′).
Primers were purchased from IDT-DNA and designed to incorporate the NdeI and EcoRI restriction sites. These fragments were inserted into the expression vector pET21b from Novagene in order to place the genes under isopropyl 1-thio-β-D-galactopyranoside-inducible transcription. DNA sequencing confirmed that the desired constructions were obtained. The expression plasmids were introduced into Esch-erichia coli cells (Bl(21)DE3 cell line), and transformants were tested for isopropyl 1-thio-β-D-galactopyranoside-induced overexpression of polypeptides of the desired size (~6,150 Da). All transformants tested exhibited induction of protein at about 6–7 kDa.
Expression and Purification of AbhN
DNA was isolated using a Qiagen Miniprep kit. The plasmids were transformed into competent E. coli BL(21)DE3 purchased from Novagene. One liter of LB broth containing 100 μg/ml ampillicin was inoculated and grown at 37 °C at 180 rpm to an A600 of about 0.900. Isopropyl 1-thio-β-D-galactopyranoside was added to 1 mM concentration, temperature was turned down to 30 °C, and the incubation continued for 5–6 h. The cells were pelleted by centrifugation and resuspended with 10 mM Tris-HCl (pH 8.3 at 4 °C or pH 7.9 at room temperature), 1 mM EDTA, 10 mM KCl, 1 mM DTT,3 0.25 μM 4-(2-Aminoethyl)-bezenesulfonylfluoride, and 0.01% Triton X-100. All subsequent steps were performed at 4 °C. The cells were sonicated for 20 cycles of 2-min bursts/3-min rests. The resulting suspension was again centrifuged at 17,000 rpm for 25 min. The supernatant was removed and saved as the crude extract. Solid ammonium sulfate was added slowly to the supernatant to a final concentration of 35%. This was allowed to sit for 30 min and recentrifuged at 17,000 rpm, and the pellet was checked for protein and subsequently discarded. The supernatant was dialyzed into 10 mM Tris-HCl (pH 8.3 at 4 °C or pH 7.9 at room temperature), 1 mM EDTA, 10 mM KCl, and 1 mM DTT. The dialyzed supernatant containing AbhN was purified via column chromatography using Q-Sepharose purchased from Amersham Biosciences. AbhN binds and elutes, using a 0 –500 mM KCl gradient, approximately in the middle of the gradient. The fractions were pooled and dialyzed into the aforementioned buffer. The protein was then loaded onto a heparin-agarose column and eluted using a 0 –200 mM KCl gradient. Fractions containing AbhN were pooled and dialyzed into 10 mM KH2PO4 (pH 5.5), 15 mM KCl, 1 mM EDTA, 1 mM DTT, and 0.02% NaN3 for NMR experiments. For storage, AbhN was dialyzed into 10 mM Tris-HCl (pH 8.3 at 4 °C or pH 7.9 at room temperature), 1 mM EDTA, 10 mM KCl, and 1 mM DTT. Throughout the protocol, the presence of AbhN was monitored by 12% Tricine gel electrophoresis.
Expression and Purification of Abh
The expression and purification is similar to that of AbhN (above). Minor differences were as follows. Solid ammonium sulfate was added slowly to the supernatant to a final concentration of 40%. Fractions containing Abh were pooled and dialyzed into 10 mM Tris-HCl (pH 8.3 at 4 °C or pH 7.9 at room temperature), 1 mM EDTA, 10 mM KCl, and 1 mM DTT.
Construction and Purification of the AbrB(E30A) Mutant Protein
A gene encoding the mutant AbrB protein in which an alanine residue replaces the glutamate residue at position 30 was constructed using the material and protocols in the QuikChange ® II site-directed mutagenesis kit (Stratagene). Mutagenic oligonucleotides were obtained from the Biopolymer core facility (University of Maryland, Baltimore), and sequence confirmation of the desired clones was performed at the aforementioned facility. A fragment containing the mutagenized gene under translational control of its native ribosome binding site was inserted in the pKQV4 expression vector (35) and transformed into E. coli DH5 α competent cells (Invitrogen). The resultant expression strain was grown at 37 °C in LB medium containing 50 μg/ml ampicillin to an A600 of 0.600. Isopropyl 1-thio-β-D-galactopyranoside was added to 1 mM , and incubation continued for 2 h. The cells were harvested by centrifugation, washed, and cracked open as has been described (35). Throughout the procedure, the presence of the AbrB(E30A) protein was monitored by SDS-polyacrylamide gel fractionation (4% stacking/18% separating, Laemmli buffer system). All of the following steps were performed at 4 °C. The buffer (B*) used to prepare the crude extract and in the following steps was 10 mM Tris, pH 8, 10 mM KCl, 0.1 mM MgCl2, 1 mM Na2 EDTA, 10 mM β-mercaptoethanol, 40 μg/ml phenyl-methylsulfonyl fluoride. This crude extract was subjected to streptomycin sulfate and ammonium sulfate precipitation steps as has been described for the wild type AbrB protein (35), except that the ammonium sulfate cut used for subsequent steps was 50–70%. After dialysis versus buffer B*, the (NH4)2SO4 cut was applied to a DEAE-trisacryl (BioSepra) column, washed with B*, and eluted with a 10–200 mM gradient of KCl in B*. Fractions containing the AbrB(E30A) protein were pooled, dialyzed versus buffer B*, and applied to a heparin-agarose (Sigma) column equilibrated with the same buffer. The column was washed with B* and eluted with a 10 –300 mM KCl gradient in B*. Fractions containing the AbrB(E30A) protein were pooled and concentrated to ~0.5 mg/ml using Centriplus YM3 and Centricon YM3 devices (Amicon). Glycerol was added to ~25% (final concentration) for storage at −80 °C. Protein concentrations were determined using the Bio-Rad (Bradford) assay reagent with bovine serum albumin as the standard. Final protein purity was judged to be greater than 95% by SDS-PAGE. Wild-type B. subtilis AbrB was expressed in E. coli and purified as has been described (35) with the following modifications. Buffer B* replaced buffer A of the original purification scheme, and the desalting step prior to fractionation on DEAE-trisacryl column was accomplished by dialysis rather than use of a desalting column.
Size Exclusion Liquid Chromatography
Size exclusion liquid chromatography (SELC) was performed on AbhN, with thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobin (17.5 kDa), and vitamin B12 (1,350 Da) as sizing standards. Analysis was performed at room temperature with a TOSOH SuperSW2000 gel filtration column on a Waters Breeze system with a flow rate of 0.3 ml/min and monitored at 280 nm. SELC protein samples (1–2 mM ) were exchanged into 10 mM KH2PO4 (pH 5.5), 15 mM KCl, 1 mM EDTA, 1 mM DTT, and 0.02% NaN3.
In Vitro DNA Binding Assays
The DNA binding targets designated BS18 and C47 are aptomer sequences originally selected in vitro as displaying high affinity binding to AbrB (34). For binding studies, 124-bp EcoRI-HindIII fragments, containing the target sequences in the middle, were used. In the case of BS18, AbrB has been shown to protect from DNase I cleavage a 25-bp sequence (AAATTGGAAAACATTGCCAGTAGAA); in the case of C47, a 45-bp sequence (TAAACAGGAA-GGTATTTCCATTTTTGGGGGTATAAGGATCCTGAC) is protected (34). The Z81 target, present in a 414-bp EcoRI-BamHI fragment, contains the sequence from 26 bp upstream to 96 bp downstream (404 bp total) of the B. subtilis abrB coding frame. Wild-type AbrB protein does not bind the Z81 target in vitro, and it contains no known in vivo AbrB-dependent regulatory sites. For footprinting and gel mobility shift assays (see below), the target DNA fragments were singly end-labeled using [α-32P]dATP (Amersham Biosciences) and the Klenow enzyme. Fragments were purified using polyacrylamide electrophoresis (1 × TBE buffer) and electroelution of the DNA from excised gel slices. DNA fragment concentrations were determined using scintillation counting.
Binding buffers (1 × concentration) consisted of 50 mM buffering agent, 100 mM KCl, 10 mM MgCl2, 0.1 mM Na2EDTA, 0.04 mM DTT, and 0.1 mg/ml bovine serum albumin. Sodium phosphate buffers were used for pH values 6.0–7.0, Tris buffers were used for pH 7.0–9.4, and sodium carbonate buffers were used for pH >9.4. Gel mobility shift assays were performed at 22 °C (utilizing 10% polyacrylamide gels) essentially as has been described (35). Apparent dissociation constants (Kd) were derived using data of phosphorimaging quantification of dried gels obtained with a Typhoon 9400 apparatus (Amersham Biosciences) equipped with ImageQuant TL software. DNase I footprinting assays were performed at 22 °C as has been described (35).
Nuclear Magnetic Resonance Spectroscopy
All NMR experiments were performed at 305 K on a Varian INOVA 600. 1.0–2.0 mM protein samples in the following buffer were used: 90:10% or 1:99% H2O/D2O, 15 mM KH2PO4 (pH 5.5), 10 mM KCl, 1 mM EDTA, 0.02% NaN3, and 1 mM DTT. Sequential assignments were made from HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO, and HN(CA)CO experiments (36–40). Side chains were assigned from H(CCO)NH, (H)C(CO)NH, and HCCH-TOCSY experiments (36, 38, 39). Exchange-protected amides were monitored by sequentially recording 100 12-min two-dimensional 1H-15N HSQC experiments over a 24-h period. HNHA, CSI, and TALOS experiments were used to determine coupling constants for assigning backbone ψ and ϕ angles (41). NOEs were obtained from three-dimensional 15N NOESY-HSQC experiments with 120- and 150-ms mixing times and three-dimensional 13C NOESY-HSQC experiments with 120- and 150-ms mixing times. Structures were calculated with NOEs, hydrogen bond restraints (inter- and intramolecular) (CSI predictions and amide exchange experiments), and ψ and ϕ angles (TALOS predictions). Intermolecular NOEs were assigned as previously described (29, 32). ARIA (version 1.2) and CNS (version 1.1) programs were used to compute the solution structure starting from an extended structure with random side chain conformations (42, 43). The CNS protocols used simulated annealing with torsion angle and Cartesian space dynamics using the default parameters. Manually assigned inter- and intramolecular NOEs were input to ARIA as assigned and uncalibrated with respect to distance. The total number of ambiguous NOE restraints allowed for each peak in the NOESY spectra was set to 20. Distance restraints, derived from the manually assigned NOEs, were set to 1.8–6.0 Å. The dihedral angle restraints were taken to be ±2 S.D. values or at least ±20 from the average values predicted by TALOS (41). In this study, the dihedral angles were restrained to ϕ = − 70 ± 50° and ϕ = − 50 ± 50° for the helical regions. The noncrystallographic symmetry energy term was used to keep the Cα atoms of the monomers superimposable, and distance symmetry potential was used to ensure that the relative orientations of all of the Cα atoms of the monomers were symmetric (44). The spectra were processed with NMRPIPE and analyzed with NMRVIEW on LINUX workstations running Fedora Core 1 (45, 46). Molecules were visualized and aligned with MOLMOL (47). The 10 lowest energy structures were further water-refined with ARIA. Analysis of the Ramachandran plot, from the robust structure analysis and validation program MolProbity (48), showed that 98.88% of modeled residues were in allowed or favored regions, indicative of a well comprised solution structure.
RESULTS
N-terminal Domain of Abh Is Dimeric
The N-terminal domain of Abh (AbhN) comprises amino acid residues 1–54 of the full protein, and the N-terminal domain of AbrB (AbrBN) comprises residues 1–53 of the full protein (31, 49). Fig. 1A shows the sequence alignment of the two domains. Fig. 1B shows the results of SELC for AbhN, AbrBN, thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobin (17 kDa), and vitamin B12 (1,350 Da). The elution profiles of AbhN and AbrBN are similar, with both showing a solution size of ~12 kDa. This indicates that the stable macromolecular state of AbhN is that of a dimer, corroborating previous AbrBN data (26, 27).
FIGURE 1. Multimerization state and sequence alignment of AbrBN and AbhN.
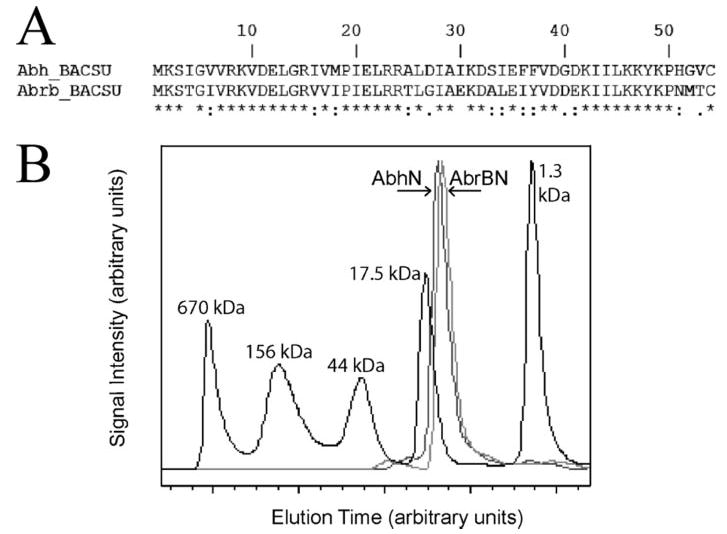
A, N-terminal sequence alignment of Abh and AbrB transition state regulators from B. subtilis. Identical residues are marked with an asterisk, strongly similar are marked with a colon, somewhat similar are marked with a dot, and dissimilar residues are not marked. The N-terminal domains are ~ 70% identical between AbrB and Abh from B. subtilis. B, size exclusion chromatography of AbhN, AbrBN, thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobin (17 kDa), and vitamin B12 (1,350 Da). Signal intensity and elution time points are in arbitrary units; higher molecular weight species elute at lower times points.
NMR Structural Determination of Dimeric AbhN
Heteronuclear multidimensional NMR methods were used to assign 1H, 13C, and 15N backbone and side chain resonances, provide sequential connectivities, and define distance relationships between protons for the development of a high resolution solution structure of AbhN (50). The 1H-15N HSQC spectrum of AbhN shows good 1H-15N peak dispersion, and 50 such peaks are fully resolved. All NH resonances were assigned, with the exception of residues 1 and 2, whereas nearly all side chain resonances for residue 2 were assigned from H(CCO)NH- TOCSY and (H)C(CO)NH-TOCSY spectra. Side chain assignments for residues 9, 13, 22, 26, 28, 31, 34, 42, 45–47, and 49 were either weak or not observed, suggesting some conformational flexibility. The majority of the aforementioned residues are located within loops or loosely associated β-strands 1 and 3. Ultimately, 95% of the backbone resonance assignments (HN, N, Cα, C′) were made. Chemical shifts of all assigned resonances are deposited in the BioMagResBank, code 6826 (available on the World Wide Web at www.bmrb.wisc.edu).
Hydrogen exchange experiments were performed by running 100 sequential 12-min 1H-15N HSQCs in deuterated NMR buffer to determine the existence of hydrogen bonds. In particular, slow exchange was observed for residues 16, 34 –38, and 42– 46. Medium exchange was observed for residues 4 – 8 and 20 –23. Other residues were in fast exchange. CSI values and TALOS predictions are consistent with the hydrogen exchange experiments in terms of secondary structure.
The solution structures of AbhN are shown in Fig. 2, A and B. These structures were generated using the combined ARIA/CNS protocol described under “Experimental Procedures.” The restraint statistics for the 10 lowest energy structures, generated by ARIA in the final iteration, are shown in Table 1. These structures were determined with 1,426 unambiguous restraints (765 ambiguous): 109 intraresidue, 572 sequential, 356 medium range, and 389 long range. Manual checks of NOEs assigned by ARIA were performed to assess the accuracy of the assignments and were found to be consistent with the structures. TALOS/CSI predictions agree well with the average energy-minimized structure containing one α-helix (residues 20–26) and four β-strands (residues 4–9, 15–17, 34–39, and 42–46). Regions of secondary structure defined by NOEs present in each monomer are classified as random coil (residues 1–3 and 47–53); loops 1, 2, and 3 (residues 11–14, 28–32, and 40–41, respectively); α-helix (residues 20–26); and β-sheet (β-strand 1 residues 4–10, β-strand 2 residues 15–17, β-strand 3 residues 33–39, and β-strand 4 residues 41–46). Since AbhN and AbrBN are homodimers, primes will be used hereafter to differentiate residues and structural elements from each monomeric unit of the dimer. The β-sheet presents an extensive scaffold between β-strands 3, 4, 3′, and 4′. The structure is well defined, as reflected by the low r.m.s. deviation values and is of good quality as indicated by the energies and Ramachandran statistics (Table 1). The ensemble of the 10 lowest energy structures for residues 1–108 (two 54 residue monomeric subunits of the homodimer) exhibits a backbone r.m.s. deviation of 0.19 ± 0.02 Å for the secondary structure regions and mean backbone and heavy atom r.m.s. deviations of 0.69 ± 0.09 and 1.01 ± 0.06 Å, respectively (Table 1 and Fig. 2, A and B). Reflecting their positions at the termini, residues 1–3 and 49–54 show higher r.m.s. deviation values and are less well defined. The reported Ramachandran errors from loop 1 and loop 2 are slight, with both being on the edge of accepted values as determined via MolProbity.
FIGURE 2. Stereo view of the ensemble of the 10 lowest energy structures for AbhN from B. subtilis and overlay of AbhN and AbrBN.
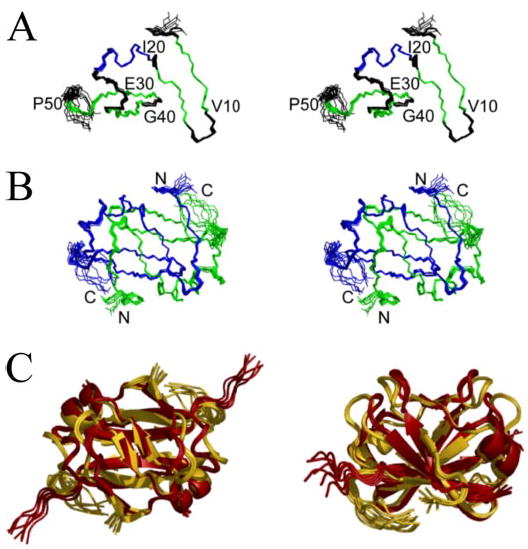
A, AbhN monomer colored by secondary structure; strands are colored green, whereas the helix is shown in blue. B, AbhN dimer colored by monomer. The superimposition is over residues 1–54 of each monomer, resulting in an r.m.s. deviation of 0.19 Å for backbone atoms. C, ribbon structure overlay of AbhN (gold ) and AbrBN (red ), highlighting the similarities and differences in the structures. Images were generated with Pymol (62).
TABLE 1.
NMR and refinement statistics
| NMR distance and dihedral constraints | AbhN dimer |
|---|---|
| Distance constraints | |
| Total NOE | 2,191 |
| Intramolecular | 1,678 (~76%)a |
| Intermolecular | 513 (~24%)a |
| Unambiguous | 1,426 |
| Intraresidue | 109 |
| Sequential (|i − j| = 1) | 572 |
| Medium range (|i − j| <4) | 356 |
| Long range (|i −j| >;5) | 389 |
| Ambiguous | 765 |
| Hydrogen bonds | 70 |
| Total dihedral angle restraints | |
| φ | 48 |
| ψ | 48 |
| Structure statistics | |
| Energies (kcal mol− 1) | |
| van der Waals | −1,059 ± −72 |
| Electrostatic | −4,182 ± −136 |
| Average violations per structure | |
| NOEs and/or hydrogen bonds | 0.1b |
| Dihedrals | 0 |
| Violations (mean and S.D.) | |
| Distance constraints (Å) | 0.032 ± 0.007 |
| Hydrogen bonds (Å) | 0.050 ± 0.002 |
| Dihedral angle constraints (degrees) | 0.445 ± 0.008 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.00447 ± 0.00013 |
| Bond angles (degrees) | 0.641 ± 0.019 |
| Impropers (degrees) | 1.67 ± 0.06 |
| Average pairwise r.m.s.c (Å) | |
| Secondary structure (backbone) | 0.19 ± 0.02 |
| Secondary structure (heavy) | 0.56 ± 0.07 |
| Backbone | 0.69 ± 0.09 |
| Heavy | 1.01 ± 0.06 |
| Ramachandran analysis | |
| Allowed or better | 98.88% |
Percentage of total NOEs.
One NOE violation for the ensemble of 10 lowest energy structures.
Pairwise r.m.s. deviation was calculated among the 10 lowest energy refined structures for residues 1–54.
Comparative DNA Binding of Abh and AbrB with High Affinity AbrB Targets
Fig. 2C shows a ribbon structure overlay of AbhN and AbrBN. Given the significant degree of structural similarities in their DNA-binding N-terminal domains, we performed a set of comparative DNA binding studies of full-length Abh and AbrB. We first examined the ability of purified, intact Abh to bind known, high affinity AbrB targets (BS18, C47) (34). Gel mobility shift assays show that Abh binds to both targets with significantly reduced affinity compared with AbrB under conditions (pH 8, 100 mM KCl) previously shown to be optimal for AbrB binding (e.g. the Kd of Abh binding to BS18 is ~0.7 μM , whereas the Kd of AbrB binding to BS18 is ~0.007 μM ). A semi- quantitative examination of the effects of salt concentration and pH upon Abh binding to these targets revealed that although KCl concentrations between 50 and 150 mM had little effect (data not shown), Abh binds with significantly higher affinity at pH 7 than at pH 6 or pH 8 (see below).
Fig. 3 shows the results of DNase I footprinting assays of Abh and AbrB binding to the BS18 target. The vertical lines in Fig. 3A highlight regions where Abh binding is seen to induce hypersensitivity to DNase I cleavage. Compared with the well delineated region of protection afforded by AbrB binding (Fig. 3B), it is clear that, at both pH 7 and pH 8, Abh binds the target in a significantly different manner (Fig. 3A). Abh binding does not protect the entire region protected by AbrB. Most noticeably, Abh binding results in enhanced sensitivity to DNase I cleavage in regions adjacent to the region of protection.
FIGURE 3. DNase I footprinting analysis of Abh and AbrB binding to the BS18 target.
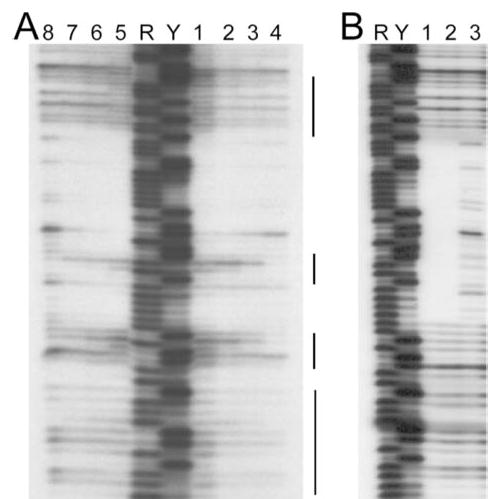
Shown are the results using 124-bp EcoRI-HindIII fragments containing the BS18 target sequence (34) labeled at the EcoRI end. A, Abh binding reactions performed at pH 7 (lanes 1– 4) versus pH 8 (lanes 5– 8). Lanes 1 and 5, 30 μM Abh; lanes 2 and 6, 10 μM Abh; lanes 3 and 7, 3 μM Abh; lanes 4 and 8, no Abh. B, AbrB binding reactions performed at pH 8. Lane 1, 10 μM AbrB; lane 2, 2 μM AbrB; lane 3, no AbrB. Maxam-Gilbert purine (R) and pyrimidine (Y) sequencing ladders are shown for reference. The vertical lines marked indicate regions showing significant enhancement of DNase I cleavage due to the presence of bound Abh.
Fig. 4 shows the binding propensities of AbrB and Abh for a DNA fragment containing the C47 target (see “Experimental Procedures”). Fig. 4A shows the DNase I footprinting profile of Abh and AbrB binding at pH 7. At pH 7, Abh and AbrB show quite similar binding specificity, with both protecting approximately the same region from DNase I attack. However, as in the case of BS18 binding (Fig. 3), subtle differences are evident. Abh binding leads to an enhancement of DNase I cleavage in positions adjacent to the major protection region (see region denoted with a bar in Fig. 4A). Because of their similar recognition profiles toward C47, pH-dependent profiles of Abh and AbrB binding affinity to the C47 target were examined. These results are depicted in Fig. 4B. Neither protein displays a very strong binding affinity for C47 at pH values of 6.8 or lower. This may be attributed to the fact that measured pI values of both Abh and AbrB are between 6.0 and 6.5. Abh is seen to bind C47 within a very narrow range, centered about pH 7. In contrast, strong AbrB-C47 binding occurs across a broad range of pH values. At pH 7, the affinity of AbrB for C47 was only slightly less than that of Abh. However, Abh affinity for C47 begins to plummet at values above pH 7 and is very weak at pH 8 and above. In stark contrast to this profile, AbrB-C47 binding reaches a peak at pH ~8 and maintains this plateau for at least 2 pH units.
FIGURE 4. Abh and AbrB binding to the C47 target.
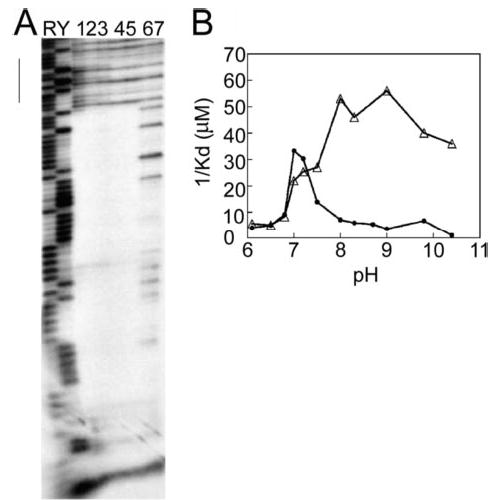
Shown are results using 124-bp EcoRI-HindIII fragments containing the C47 target sequence (34) labeled at the EcoRI end. A, DNase I footprinting analysis performed at pH 7. Lanes 1–3, 30 μM , 10 μM , and 3 μM Abh, respectively; lanes 4 and 5, 10 and 2 μM AbrB, respectively; lanes 6 and 7, no protein. The vertical line indicates a region showing significant enhancement of DNase I cleavage due to the presence of bound Abh. B, pH dependence of Abh (filled circles) and AbrB (triangles) binding to C47. Apparent Kd values were determined using a gel mobility shift assay as described under “Experimental Procedures.” For this presentation, the reciprocals of the empirically determined Kd values are plotted as a function of binding reaction pH, so that higher affinity interactions have larger values on the ordinate axis. Maxam-Gilbert purine (R) and pyrimidine (Y) sequence ladders are used for reference.
Comparison of Nonspecific Binding Propensities of Abh and AbrB
Fig. 5 shows footprinting studies of Abh, wild-type AbrB, and the E30A AbrB mutant binding to the DNA target Z81 (from the coding region of the abrB gene). As shown in Fig. 5A, Abh interacts extensively with the DNA target fragment Z81 at both pH 7 (lanes 1–5) and pH 8 (lanes 6–9). Binding occurs in a concentration-dependent manner, producing numerous regions displaying heightened sensitivity to DNase I cleavage with the occasional appearance of interspersed sites (one or two cleavage bands) showing a slight degree of protection (e.g. positions indicated as a and b in Fig. 5A). This behavior is in contrast to that observed when wild-type AbrB protein is mixed with the Z81 fragment. In this case, there is very little, if any, evidence of significant AbrB binding to Z81 (Fig. 5B, lanes 1 and 2). We suspected that an E30A substitution in AbrB might partially mimic the loop 2 region (see “Discussion”) of Abh, which has an isoleucine residue at position 30 rather than a glutamate. Lanes 3 and 4 in Fig. 5B show that E30A AbrB interacts extensively with the Z81 sequence. In contrast to the Abh interaction, the E30A AbrB interaction promotes DNase I cleavage enhancement rather than protection. Nevertheless, both observations can be classified as resulting from relatively nonspecific binding recognition, and these data show that changing residue 30 in AbrB from charged (Glu) to neutral (Ala) removes its ability to discriminate against Z81. In other words, a charged residue at position 30 contributes to AbrB specificity. It is important to note that position 30 in Abh is a neutral residue (isoleucine) and that wild-type Abh, like E30A AbrB, interacts with Z81.
FIGURE 5. DNase I footprinting analysis of Abh, AbrB, and AbrB(E30A) binding to the Z81 target fragment (coding region of abrB gene).
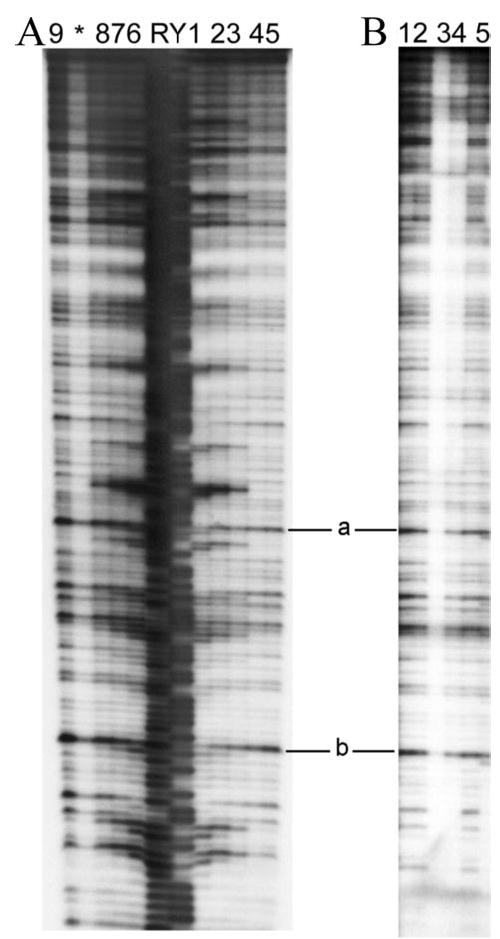
The 414-bp EcoRI-HindIII Z81 fragments containing the abrB coding region were labeled at their EcoRI ends. A, Abh binding reactions performed at pH 7 (lanes 1–5) versus pH 8 (lanes 6 –9). Lanes 1 and 6, 30 μM Abh; lanes 2 and 7, 10 μM Abh; lanes 3 and 8, 3 μM Abh; lanes 4, 5, and 9, no Abh. B, wild-type AbrB and AbrB(E30A) binding to Z81 at pH 8. Lane 1, 20 μM wild type; lane 2, 2 μM wild type; lane 3, 7 μM E30A; lane 4, 0.7 μM E30A; lane 5, no protein. Two positions showing limited Abh-afforded protection are marked a and b (see “Results”). Maxam-Gilbert purine (R) and pyrimidine (Y) sequencing ladders are shown for reference. *, artifactual lane between lanes 8 and 9.
DISCUSSION
AbrB interacts with numerous DNA targets present on the B. subtilis chromosome that show little or no obvious consensus base sequence (12, 35, 51–53). However, AbrB does not bind to just any or every nucleotide sequence; rather it displays a “limited promiscuity” in its binding preferences. In addition, AbrB shows explicit high affinity binding specificity to a finite subset of DNA targets (27, 34, 53). The DNA-binding nature of AbrB is 3-fold: (i) nonspecific interactions due to recognition of general DNA features; (ii) limited promiscuity contacts, which allow interactions with a subset of further structurally related DNA structures; and (iii) high affinity interactions at specific sites where more extensive thermodynamically favorable contacts can be made (54, 55). AbrB achieves this DNA-binding nature through an orchestrated mechanism of dimerization of the N-terminal domain and the proper orientation of specific arginine residues. Understanding the balance between general non-specific binding, limited promiscuity binding, and high affinity specific binding can be achieved only by comparative structural and functional studies. Such studies must be carried out using structurally homologous proteins (or mutant variants) that display both differential binding properties while retaining general recognition characteristics. With this in mind, we have elucidated the high resolution NMR solution structure of the DNA-binding domain of Abh and compared it with the high resolution NMR solution structure of the DNA-binding domain of its paralogue AbrB. Additionally, we have provided the first comparison between the DNA binding proclivities of AbrB and Abh.
The similarities between the DNA-binding domains AbhN and AbrBN are abundant. SELC shows that both AbhN and AbrBN are ~12-kDa dimers (Fig. 1B) (26, 27). Comparison of hydrogen exchange data between the two proteins reveals similar protection of the backbone amides. In particular, analogous hydrogen exchange protection is observed for residues 4–8, 16, 20–26, 34–38, and 42–46. Overall, the solution structure of AbhN (Fig. 2, A and B) is extremely similar to the solution structure of AbrBN (Protein Data Bank codes 1Z0R, 1YFB, and 1YSF), affording an r.m.s. deviation of 1.73 Å (for residues 4–47 from 1Z0R) (29, 32, 33). Structural comparisons between AbhN (Protein Data Bank code 2FY9) and AbrBN (Protein Data Bank code 1Z0R) are made here, since both of these structures were determined in this laboratory under similar conditions and employing identical protocols.
Fig. 2C shows the structure overlays of AbhN and AbrBN in two positions. Both proteins are composed of four antiparallel β-strands and one α-helix. Both structures contain a domain swap fold, which is defined as a set of β-strands (3 and 4) from monomeric unit A interacting with β-strands 1 and 2 from monomeric unit B. A four-stranded β-sheet scaffold consisting of β-strands 3, 4, 3′, and 4′ is seen in both AbhN and AbrBN. In each case, strong interactions between β-strand 4 (residues 41–46) and β-strand 4′ (residues 41′ -46′) contribute significantly to the dimerization interface. Particularly important are the hydrogen bonding interactions between Ile-43 (Ile-43′) and Leu-45′ (Ile-45). This interface is supplemented by a weaker interaction between β-strand 2 and β-strand 2′ using Ile-16 (Ile-16′) and Met-18 (Met-18′). Ile-43 and Leu-45 (β-strand 4) also show intense NOEs to Ile-16′ and Met-18′ (β-strand 2), further defining the extent of this dimer interface. The presence of backbone amide proton resonances in a 1H-15N HSQC spectrum after 24 h in deuterated NMR buffer defines residues 43 and 45′(43′ and 45) and 16 and 16′(16′ and 16) as intermolecular hydrogen-bonded partners at the dimer interface. Slow exchange is not seen for residue 14 or residue 18, suggesting that only one pair of strong hydrogen bonds between β-strand 2 and β-strand 2′. Ile-44 (Ile-44′) marks the center of symmetry between the two monomers. The extensive dimerization interface is therefore conserved between AbhN and AbrBN (29, 32, 33).
Also displaying significant similarities are the α-helices, which contain essential residues for DNA binding (29, 30). In both structures, the α-helices are of similar length, residues 20–26. The angle of the α-helix with respect to β-strand 2 within each structure differs by only 2.9°. The pitch of the α-helices with respect to one another is nearly identical. The general dynamic nature of proteins suggests that the 2.9° difference in the angle of the α-helix with respect to β-strand 2 may not be significant. The pitch similarity between the α-helices in AbhN and AbrBN is probably functionally relevant in terms of general target recognition. Each α-helix is dominated by intramolecular contacts and hydrogen bonds (slow to medium amide exchange for residues 20–23) that define the secondary structure of the α-helices. In addition, both α-helices contain intermolecular contacts to both loop 1 and loop 3. This underscores the extensive multimerization domain throughout the whole of the protein (Fig. 2, B and C).
Mutagenesis data define Arg-8, Arg-15, Arg-23, and Arg-24 as key residues in the ability for AbrB to bind DNA (12, 29, 30). In the sequence and structure alignment of AbhN and AbrBN, these residues occupy identical positions. In AbhN and AbrBN, Arg-23 and Arg-24 are contained in the α-helix. This helix has been hypothesized to contact moieties in the minor groove of double-stranded DNA (32). The orientations of Arg-23 and Arg-24 within those helices are conserved. This is shown in the top left panel of Fig. 6. The orientation of Arg-8 within β-strand 1 is also conserved. This is shown in the bottom left panel of Fig. 6. AbrB mutagenesis studies identify Arg-15 as being important for in vivo regulatory activity (29, 30). In both the AbhN and AbrBN structures, Arg-15 is located within the β-strand 2 dimerization interface and is conserved in sequence and orientation (Fig. 6, middle left panel). This suggests a dual function for this region: dimerization and DNA binding. In particular, Arg-15 has significant intermolecular NOE contacts to Val-17′(Val-17 is conserved in both Abh and AbrB). These contacts serve to hold the dimerization interface together by providing an abundance of constraints for this region and may serve to restrict Arg 15, in both Abh and AbrB, to an appropriate subset of functional conformations for correct interactions with DNA.
FIGURE 6. Arginine residues involved in AbhN/AbrBN DNA binding.
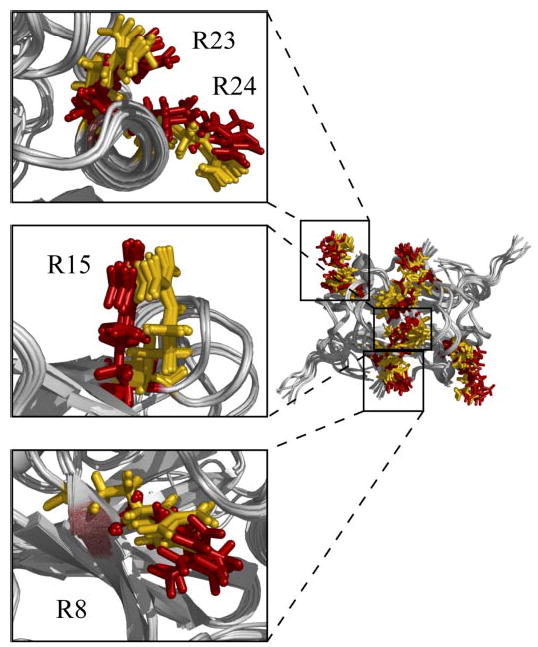
Backbone overlay of the ensemble solution structures of AbhN and AbrBN showing the orientations of the side chain residues of important DNA-binding arginine residues, AbrBN (red) and AbhN (gold). The insets are shown with the five lowest energy average side chain positions for visual clarity. Images were generated with Pymol (62).
Fig. 7, A and B, shows the electrostatic potential of the putative DNA-binding surfaces of AbhN and AbrBN, respectively. This orientation is the same as that shown in the left panel of Fig. 2C. These plots have many similarities. Both surfaces have considerable positive electrostatic character, dominated by the conserved arginine residues (Arg-8, Arg-15, Arg-23, and Arg-24) previously identified as essential for either in vivo AbrB-de pendent regulatory properties, in vitro AbrB-DNA binding, or both (29, 30). In one model for binding DNA, residues in the center of this site, particularly Arg-8 and Arg-15, are responsible for recognizing different nucleotide sequences within the major groove (32). In both AbrBN and AbhN, Arg-23 and Arg-24 display similar electrostatic patterns. Arg-8, Arg-15, Arg-23, and Arg-24 have similar orientations and may hydrogen-bond to the acceptor groups of all base pair types, preferably with guanine and thymine bases (Fig. 6). In both AbhN and AbrBN, loop 1 possesses identical residues, providing a negative electrostatic potential (Figs. 1A and 7, A and B), particularly Asp-11 and Glu-12. These residues may restrict movement of Arg-15 (Arg-15′) to a suitable subset of positions for optimum interaction with DNA. The observations noted above highlight many similarities between AbhN and AbrBN and may play a significant role in conserving the functionality of the transition state regulator/DNA interaction. We suggest that these structural features contribute to a general mechanism of DNA binding employed by transition state regulators as a family.
FIGURE 7. Comparison of the electrostatic surface potential and the orientation of loops 1 and 2 between AbhN and AbrBN.
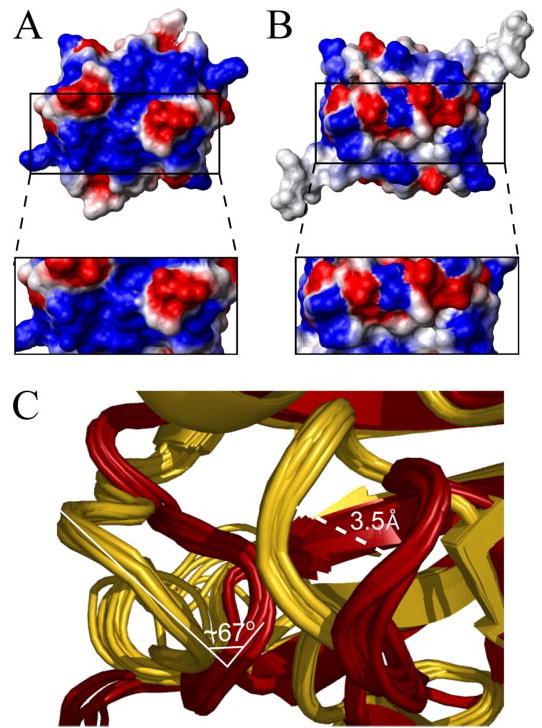
A, electrostatic surface plot of AbhN. B, electrostatic surface plot of AbrBN. Blue, positive charge; red, negative charge. In each case, the protein is oriented as depicted in Fig. 2C. The box inset is a “zoomed-in” region of loop 2 and strand 1 containing Arg-8. This illustrates the extent and similarity and difference of the charged DNA interaction surface on AbhN with respect to AbrBN. Images were generated with MOLMOL (64). C, overlay of the ensemble solution structures of AbhN (gold) and AbrBN (dark red), highlighting the similarities and differences in the loop 1 and 2 regions. The shift between loops 1 of AbhN and AbrBN is ~3.5 Å, whereas the angle between loop 2 from AbrBN and AbhN shows a rotation of ~67° centered on residue Lys-31.
On the other hand, AbhN and AbrBN exhibit structural and/or electrostatic differences in three regions: loop 1, loop 2, and the area immediately surrounding Arg-8 (Fig. 7). In loop 1, although all residues are conserved in sequence, there is an ~3.5-Å shift between the position of AbhN loop 1 and AbrBN loop 1 (Fig. 7C). This shift serves to slightly redistribute the electrostatic surface profile between the two proteins. Differences in AbhN loop 2 and AbrBN loop 2 come from both nonconservation of residue 30 and a reorientation of the loop. In AbrBN, residue 30 is a glutamate, whereas in AbhN, residue 30 is an isoleucine. The change in charge between an Ile and Glu at position 30 can modify interactions with the adjacent residue Lys-31. Not surprisingly, AbrBN shows a more negative electrostatic surface characteristic in loop 2 than does AbhN. In addition, there is a notable difference in the orientation of loop 2, between AbhN and AbrBN. A contributing factor to this feature may be the change from a Gly (AbhN) to an Asp (AbrBN) at position 27 (Figs. 1 and 7C). There is an approximately 67° rotation between AbhN loop 2 and AbrBN loop 2 (centered on Lys-31), with the AbhN loop 2 pointing more toward the α-helix than loop 2 in AbrBN (Fig. 7C). The resulting difference in charge and orientation in this region due to these effects probably plays a role in DNA sequence specificity. Consistent with this proposition is the observed loss of binding specificity of the E30A AbrB mutant relative to wild-type AbrB (Fig. 5B). The biggest structural difference in the two structures is where there is the least sequence conservation (Fig. 1A). Also contributing to differences in the overall electrostatic surface is the area surrounding Arg-8. All arginines in AbrBN and AbhN are solvent-exposed, contribute significantly to the general positive electrostatic surface, and are involved in DNA binding. However, there is a disparity in the amount of positive electrostatic surface charge around Arg-8 when comparing AbhN with AbrBN. Taken together, these differences can be expected to play a role in the ability of transition state regulators to (i) bind different DNA sequences or (ii) bind similar DNA sequences with different affinities. We suggest that these structural features may contribute to the specific DNA binding properties of individual transition state regulator proteins.
The difference in pH-dependent binding profiles of AbrB and Abh toward a common target (C47 in Fig. 4) can be straightforwardly explained by consideration of the sequences and structures of AbhN and AbrBN (Figs. 1, 6, and 7). The side chains of Cys, Tyr, His, Arg, Lys, Asp, and Glu are sensitive to pH changes. The structures of AbrBN and AbhN show that Lys-2, Cys-54, His-51 (Abh), Tyr-37 (AbrB), and Tyr-48 are on the opposite face of each protein from the DNA binding surface. These residues do not contribute to the DNA binding event. This leaves Arg, Lys, Asp, and Glu residues as potentially responsible for the differing pH curves in Fig. 4b. We note that in both proteins, strand 1-loop 1-strand 2 are identical. Additionally, the helices in both proteins are identical. This eliminates Arg-8, -15, -23, and -24, Lys-9, and Asp-11 as being responsible for the pH binding differences. The only part of the DNA binding surface to show notable differences is loop 2. Loop 2 in AbrB is Ile-Ala-Glu-Lys; loop 2 in Abh is Ile-Ala-Ile-Lys. The only change is a Glu-30 (AbrBN) to Ile-30 (AbhN) modification. Changing the protonation state of Glu-30 (AbrBN) will certainly affect those residues close by, including Lys-31 (and vice versa). This cannot happen in AbhN (Ile-30). Lys-31 is conserved in both sequences. Consequently, the difference in the pH profile must be due to the surface region presented by loop 2 alone.
The data describing the in vitro binding of AbrB and Abh to the C47, BS18, and Z81 DNA targets, along with the structural comparison between AbhN and AbrBN, are all consistent with a model in which transition state regulators have different, but partially overlapping, binding specificities toward DNA sequence-dependent structures. Based upon the phenotypic difference of mutant strains, AbrB and Abh must play different in vivo physiological roles, although other evidence indicates that their regulons overlap at least partially.2 The evidence also indicates that Abh can interact with some, but not all, promoter regions that are targets of AbrB binding.
The enhanced cleavage regions seen in Fig. 3, comparing AbrB and Abh binding to BS18, can be interpreted as being due to induced structural changes in the DNA regions adjacent to the site of Abh interaction that result in increased sensitivity to DNase I cleavage (56, 57). Such regions of significant cleavage enhancement are not seen adjacent to the AbrB protection region on BS18 or in any of the over 100 different sites of AbrB binding that we have previously examined (for representative examples, see Refs. 34, 35 and 52, 53). The notable difference in pH-dependent binding profiles of AbrB and Abh toward a common target (e.g. C47; Fig. 4A) suggests a possible physiological role played by Abh. During sporulation, it has been observed that the pH of the developing forespore drops from pH 8 to pH 7 by stage IV of the process (58). During the initial stages of sporulation, the intracellular concentration of AbrB drops below effective regulatory levels due to Spo0A~P-mediated repression of abrB transcription (59, 60). This results in lifting AbrB-dependent repressive effects on gene expression that had been exerted during active growth. The drop in AbrB concentration, coupled with the drop in pH to suboptimal levels for AbrB-DNA interactions, means that AbrB-dependent regulatory effects will become negligible in the developing spore. It is possible that one important role played by Abh is in the resumption of repressive effects at some AbrB-dependent promoters whose expression is no longer needed after a given point in the sporulation process.
During the course of investigating potential in vitro binding of Abh to promoter regions from putative in vivo regulatory targets, we observed indications that Abh might have a significantly greater propensity than AbrB for forming relatively stable interactions with DNA in a generalized, nonspecific fashion (data not shown). To determine whether this behavior might only be correlated with interactions to promoter-derived sequences, Abh binding to an ~400-bp promoterless fragment of B. subtilis DNA designated Z81 was examined via DNase I footprinting (Fig. 5). When mixed with the Z81 fragment, wild-type AbrB showed essentially no binding interactions. Conversely, Abh interacts extensively with the fragment at both pH 7 and pH 8. This is the first example of an in vitro protein-DNA interaction showing definite binding specificity differences between extremely similar members of the AbrB family of transition state regulators.
Recent results (61) have shown that a chimeric protein composed of the N-domain of AbrB (residues 1–50) fused to the C-domain of Abh (residues 54–92) is capable of regulating AbrB-dependent promoters in vivo and indicate that the C-domains of AbrB and Abh are independent multimerization modules with little, if any, role in conferring DNA binding specificity. Such observations argue that the observed differences in binding properties of AbrB and Abh must lie in the limited differences in amino acid sequence of their N-domains and the subtle structural alterations caused by these differences. Further comparison of AbrB and Abh presents an ideal system for detailed functional and biophysical studies aimed at dissecting the effects of individual residues and localized structural variations upon DNA-binding mechanisms and specificity. Preliminary evidence in both an in vitro system and a yeast cell two-hybrid system suggests that Abh and AbrB may be able to form heterotetramers by subunit mixing and that these entities may have DNA binding properties different from the homotetramers. We are exploring these initial findings more thoroughly.4
Elucidating the mechanisms by which transition state regulator proteins interact with their DNA targets is of great importance, since these proteins allow bacteria, including pathogenic species, to adapt to changing environments by forming spores and expression of toxins (15–17). Here we have solved the NMR solution structure of the DNA-binding domain of AbhN, to our knowledge only the second solution structure of a transition state regulator domain. We have compared this structure with that of AbrBN to highlight similarities and differences that may contribute to DNA binding promiscuity and specificity. We are currently pursuing extensive mutational studies and the identification of more DNA binding targets for transition state regulators in order to further refine these binding and recognition models. This work solves the NMR solution structure of AbhN and represents the first step in identifying the structural contributions that help balance general and specific DNA binding traits in transition state regulators.
Acknowledgments
We thank Daniel M. Sullivan for preparation of AbhN protein for the SELC work, Katherine Zoller for constructing the AbrB(E30A) mutant, Austin Rowshan for assistance in purification of AbrB and AbrB(E30A) proteins, and Erin J. Regel and Daniel M. Sullivan for helpful discussions.
Footnotes
This work was supported in part by National Institutes of Health (NIH) Grants GM55769 (to J. C.) and GM46700 (to M. A. S.), a grant from the Kenan Institute for Engineering, Technology and Science (to J. C.) and the Intramural Research Program of NIEHS, NIH.
M. A. Strauch, unpublished observations.
The abbreviations used are: DTT, dithiothreitol; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine; SELC, size exclusion liquid chromatography; r.m.s., root mean square; NOE, nuclear Overhauser effect; NOESY, NOE spectroscopy.
M. A. Strauch, submitted for publication.
The atomic coordinates and structure factors (code 2FY9) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The AbhN atomic coordinates have been deposited in the BioMagResBank (6826).
References
- 1.Dubnau D, Hahn J, Roggiani M, Piazza F, Weinrauch Y. Res Microbiol. 1994;145:403–411. doi: 10.1016/0923-2508(94)90088-4. [DOI] [PubMed] [Google Scholar]
- 2.Driks A. Cell Mol Life Sci. 2002;59:389–391. doi: 10.1007/s00018-002-8430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonenshein AL, Hoch JA, Losick R. Bacillus subtilis and Its Closest Relatives: From Genes to Cells. American Society for Microbiology Press; Washington, D. C: 2002. [Google Scholar]
- 4.Shafikhani SH, Partovi AA, Leighton T. Curr Microbiol. 2003;47:300–308. doi: 10.1007/s00284-002-4012-2. [DOI] [PubMed] [Google Scholar]
- 5.Persuh M, Mandic-Mulec I, Dubnau D. J Bacteriol. 2002;184:2310–2313. doi: 10.1128/JB.184.8.2310-2313.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujita M, Gonzalez-Pastor JE, Losick R. J Bacteriol. 2005;187:1357–1368. doi: 10.1128/JB.187.4.1357-1368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soriano M, Diaz P, Pastor FI. Curr Microbiol. 2005;50:114–118. doi: 10.1007/s00284-004-4382-8. [DOI] [PubMed] [Google Scholar]
- 8.Rowbury RJ. Sci Prog. 2004;87:193–225. doi: 10.3184/003685004783238508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furukawa S, Narisawa N, Watanabe T, Kawarai T, Myozen K, Okazaki S, Ogihara H, Yamasaki M. Int J Food Microbiol. 2005;102:107–111. doi: 10.1016/j.ijfoodmicro.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Nicholson WL, Munakata N, Horneck G, Melosh HJ, Setlow P. Microbiol Mol Biol Rev. 2000;64:548–572. doi: 10.1128/mmbr.64.3.548-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Setlow P. Annu Rev Microbiol. 1995;49:29–54. doi: 10.1146/annurev.mi.49.100195.000333. [DOI] [PubMed] [Google Scholar]
- 12.Strauch MA, Hoch JA. Mol Microbiol. 1993;7:337–342. doi: 10.1111/j.1365-2958.1993.tb01125.x. [DOI] [PubMed] [Google Scholar]
- 13.Huang X, Helmann JD. J Mol Biol. 1998;279:165–173. doi: 10.1006/jmbi.1998.1765. [DOI] [PubMed] [Google Scholar]
- 14.Dong TC, Cutting SM, Lewis RJ. FEMS Microbiol Lett. 2004;233:247–256. doi: 10.1016/j.femsle.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Strauch MA, Ballar P, Rowshan AJ, Zoller KL. Microbiology. 2005;151:1751–1759. doi: 10.1099/mic.0.27803-0. [DOI] [PubMed] [Google Scholar]
- 16.Koehler TM. Curr Top Microbiol Immunol. 2002;271:143–164. doi: 10.1007/978-3-662-05767-4_7. [DOI] [PubMed] [Google Scholar]
- 17.Saile E, Koehler TM. J Bacteriol. 2002;184:370–380. doi: 10.1128/JB.184.2.370-380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baillie L, Moir A, Manchee R. J Appl Microbiol. 1998;84:741–746. doi: 10.1046/j.1365-2672.1998.00405.x. [DOI] [PubMed] [Google Scholar]
- 19.Shcheptov M, Chyu G, Bagyan I, Cutting S. Gene (Amst) 1997;184:133–140. doi: 10.1016/s0378-1119(96)00603-8. [DOI] [PubMed] [Google Scholar]
- 20.Bagyan I, Hobot J, Cutting S. J Bacteriol. 1996;178:4500–4507. doi: 10.1128/jb.178.15.4500-4507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomas CA, Alsaker KV, Bonarius HP, Hendriksen WT, Yang H, Beamish JA, Paredes CJ, Papoutsakis ET. J Bacteriol. 2003;185:4539–4547. doi: 10.1128/JB.185.15.4539-4547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alsaker KV, Spitzer TR, Papoutsakis ET. J Bacteriol. 2004;186:1959–1971. doi: 10.1128/JB.186.7.1959-1971.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grandvalet C, Gominet M, Lereclus D. Microbiology. 2001;147:1805–1813. doi: 10.1099/00221287-147-7-1805. [DOI] [PubMed] [Google Scholar]
- 24.Vary P. Biotechnology. 1992;22:251–310. [PubMed] [Google Scholar]
- 25.Dubnau EJ, Cabane K, Smith I. J Bacteriol. 1987;169:1182–1191. doi: 10.1128/jb.169.3.1182-1191.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benson LM, Vaughn JL, Strauch MA, Bobay BG, Thompson R, Naylor S, Cavanagh J. Anal Biochem. 2002;306:222–227. doi: 10.1006/abio.2002.5704. [DOI] [PubMed] [Google Scholar]
- 27.Bobay BG, Benson L, Naylor S, Feeney B, Clark AC, Goshe MB, Strauch MA, Thompson R, Cavanagh J. Biochemistry. 2004;43:16106–16118. doi: 10.1021/bi048399h. [DOI] [PubMed] [Google Scholar]
- 28.Phillips ZE, Strauch MA. FEMS Microbiol Lett. 2001;203:207–210. doi: 10.1111/j.1574-6968.2001.tb10842.x. [DOI] [PubMed] [Google Scholar]
- 29.Vaughn JL, Feher V, Naylor S, Strauch MA, Cavanagh J. Nat Stuct Mol Biol. 2005;12:380. doi: 10.1038/81999. [DOI] [PubMed] [Google Scholar]
- 30.Xu K, Clark D, Strauch MA. J Biol Chem. 1996;271:2621–2626. doi: 10.1074/jbc.271.5.2621. [DOI] [PubMed] [Google Scholar]
- 31.Xu K, Strauch MA. J Bacteriol. 2001;183:4094–4098. doi: 10.1128/JB.183.13.4094-4098.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bobay BG, Andreeva A, Mueller GA, Cavanagh J, Murzin AG. FEBS Lett. 2005;579:5669–5674. doi: 10.1016/j.febslet.2005.09.045. [DOI] [PubMed] [Google Scholar]
- 33.Coles M, Djuranovic S, Soding J, Frickey T, Koretke K, Truffault V, Martin J, Lupas AN. Structure (Camb) 2005;13:919–928. doi: 10.1016/j.str.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 34.Xu K, Strauch MA. Mol Microbiol. 1996;19:145–158. doi: 10.1046/j.1365-2958.1996.358882.x. [DOI] [PubMed] [Google Scholar]
- 35.Strauch MA, Spiegelman GB, Perego M, Johnson WC, Burbulys D, Hoch JA. EMBO J. 1989;8:1615–1621. doi: 10.1002/j.1460-2075.1989.tb03546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grzesiek S, Bax A. J Biomol NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 37.Ikura M, Kay LE, Bax A. Biochemistry. 1990;29:4659–4667. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- 38.Logan TM, Olejniczak ET, Xu RX, Fesik SW. FEBS Lett. 1992;314:413–418. doi: 10.1016/0014-5793(92)81517-p. [DOI] [PubMed] [Google Scholar]
- 39.Logan TM, Olejniczak ET, Xu RX, Fesik SW. J Biomol NMR. 1993;3:225–231. doi: 10.1007/BF00178264. [DOI] [PubMed] [Google Scholar]
- 40.Montelione GT, Emerson SD, Lyons BA. Biopolymers. 1992;32:327–334. doi: 10.1002/bip.360320406. [DOI] [PubMed] [Google Scholar]
- 41.Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 42.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 43.Linge JP, O’Donoghue SI, Nilges M. Methods Enzymol. 2001;339:71–90. doi: 10.1016/s0076-6879(01)39310-2. [DOI] [PubMed] [Google Scholar]
- 44.Junius FK, O’Donoghue SI, Nilges M, Weiss AS, King GF. J Biol Chem. 1996;271:13663–13667. doi: 10.1074/jbc.271.23.13663. [DOI] [PubMed] [Google Scholar]
- 45.Johnson BA. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 46.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 47.Koradi R, Billeter M, Wuthrich K. J Mol Graph. 1996;14:29–32. 51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 48.Davis IW, Murray LW, Richardson JS, Richardson DC. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaughn JL, Feher V, Naylor S, Strauch MA, Cavanagh J. Nat Struct Biol. 2000;7:1139–1146. doi: 10.1038/81999. [DOI] [PubMed] [Google Scholar]
- 50.Cavanagh J, Fairbrother WJ, Palmer AG, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Academic Press, Inc; San Diego: 1996. [Google Scholar]
- 51.Strauch MA. Mol Gen Genet. 1996;250:742–749. doi: 10.1007/BF02172986. [DOI] [PubMed] [Google Scholar]
- 52.Strauch MA. J Bacteriol. 1995;177:6999–7002. doi: 10.1128/jb.177.23.6999-7002.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strauch MA. J Bacteriol. 1995;177:4532–4536. doi: 10.1128/jb.177.15.4532-4536.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lilley DMJ. DNA-Protein: Structural Interactions. Oxford University Press; Oxford: 1995. [Google Scholar]
- 55.Travers AA. DNA-Protein Interactions. 1st Ed. Chapman & Hall; London: 1993. [Google Scholar]
- 56.Drew HR. J Mol Biol. 1984;176:535–557. doi: 10.1016/0022-2836(84)90176-1. [DOI] [PubMed] [Google Scholar]
- 57.Suck D. J Mol Recognit. 1994;7:65–70. doi: 10.1002/jmr.300070203. [DOI] [PubMed] [Google Scholar]
- 58.Magill NG, Cowan AE, Koppel DE, Setlow P. J Bacteriol. 1994;176:2252–2258. doi: 10.1128/jb.176.8.2252-2258.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strauch M, Webb V, Spiegelman G, Hoch JA. Proc Natl Acad Sci U S A. 1990;87:1801–1805. doi: 10.1073/pnas.87.5.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Phillips ZE, Strauch MA. Cell Mol Life Sci. 2002;59:392–402. doi: 10.1007/s00018-002-8431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao F, Strauch MA. J Bacteriol. 2005;187:6354–6362. doi: 10.1128/JB.187.18.6354-6362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DeLano WL. Pymol. DeLano Scientific; South San Francisco, CA: 2002. [Google Scholar]