

Abstract
Nitros(yl)ation is a widespread protein modification that occurs during many physiological and pathological processes. It can alter both the activity and function of a protein. Nitric oxide (⋅NO) has been implicated in this process, but its mechanism remained uncertain. ⋅NO is unable to react with nucleophiles under oxygen-free conditions, suggesting that its higher oxides, such as N2O3, were actually nitrosylating agents. However, low concentrations and short lifespans of these species in vivo raise the question of how they could efficiently locate target proteins. Here we demonstrate that at physiological concentrations of ⋅NO, N2O3 forms inside protein-hydrophobic cores and causes nitrosylation within the protein interior. This mechanism of protein modification has not been characterized, because all previously described mechanisms (e.g., phosphorylation, acetylation, ADP-ribosylation, etc.) occur via attack on a protein by an external modification agent. Oxidation of ⋅NO to N2O3 is facilitated by micellar catalysis, which is mediated by the hydrophobic phase of proteins. Thus, a target protein seems to be a catalyst of its own nitrosylation. One of the applications of this finding, as we report here, is the design of specific hydrophobic compounds whose cooperation with ⋅NO and O2 allows the rapid inactivation of target enzymes to occur.
The free radical ⋅NO has important biological functions including vasorelaxation, blood clotting, neuronal plasticity, and cytotoxic activity (for reviews, see refs. 1 and 2). In many cases, the physiological effect of ⋅NO can be attributed to the S-nitrosylation of target proteins such as hemoglobin (3), serum albumin (4 and 5), transcription factors (6–9), G proteins (10), ion channels (11), and various enzymes (12–14). Excessive protein nitrosation has been associated also with various pathological situations including myocardial ischemia, atherosclerosis, inflammation, and cancer (for reviews, see refs. 1, 2, 15, and 16). In vivo, nitros(yl)ation can be mediated by dinitrogen trioxide (N2O3; refs. 15 and 17–19 and references therein), ⋅NO carriers such as nitrosothiols [RS(thiols)-NO; refs. 15, 17, and 19–21], NO complexes with transition metals (22, 23), or can result from a direct reaction between ⋅NO and thiols in the presence of electron acceptors (24). It remains uncertain which pathway dominates in vivo and under what conditions it does so. It is also unclear which mechanism is responsible for the high specificity of S-nitrosylation, when only particular nucleophiles are targeted within a protein while others are left unmodified.
⋅NO reacts with O2 according to the following stoichiometry:
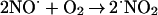 |
1 |
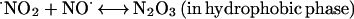 |
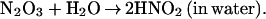 |
Because ⋅NO and O2 are better soluble in hydrophobic solvents than in water, with a partition coefficient (Q) ≫ 1 (25–27), areas of high hydrophobicity can act to increase the local concentration of these molecules by sequestering them from the surrounding aqueous phase. Under aerobic conditions, high local concentrations of ⋅NO and O2 in a hydrophobic phase, such as those within lipid membranes, can significantly accelerate the oxidation of ⋅NO (25–27). These observations led us to ask whether a similar phenomenon of micellar catalysis of ⋅NO oxidation occurs within hydrophobic interiors of proteins. If a considerable portion of the ⋅NO pool were oxidized inside protein globules, N2O3 could form and immediately attack nearby nucleophiles. Such an autocatalytic mechanism exploits protein hydrophobic pockets to target ⋅NO to critical cysteines and thereby may explain the selectivity of S-nitrosylation in regulating the protein function. In this study, we have used previously untried approaches to investigate whether such a pathway is feasible.
Experimental Procedures
⋅NO/H2O or ⋅NO/DMSO solutions were prepared in the airtight device by bubbling ⋅NO gas (Aldrich) that had been purified from higher oxides by passing it through a 1 M solution of KOH into water or DMSO (Aldrich), until the concentration of dissolved ⋅NO reached 1.2 mM. Water (Milli-Q grade) was deaerated by boiling and then cooling under argon (Praxair, Danbury, CT). Immediately before the reaction, the ⋅NO concentration was measured by using an ISO-NO Mark II electrode (WPI Instruments, Waltham, MA). DMSO was dehydrated by distillation in vacuum over CaO.
BSA, CM-BSA, glutathione (GSH), Trp, Cys (Sigma), and the Trp peptide (USBiological, San Antonio, TX) were dissolved in water (1 mM stock solutions). The nitrosation reaction was carried out at room temperature in the 1-ml quartz cuvette. The blank probe contained 0.5 ml of K2HPO4/KH2PO4 buffer (25 mM; pH 7.0) and 0.4 ml each of the tested reagent. An aqueous ⋅NO solution (0.4 ml) was added for 5, 10, 15, 20, 25, or 30 min, and then 0.1 ml of 0.1% ammonium sulfamate in 0.4 M HCl was added for 1 min to remove HNO2 from the sample. The UV-visible spectra were recorded by using an Ultrospec 3000 spectrophotometer (Amersham Pharmacia). Spectra were digitized and analyzed by win dig and origin 6.0 software (Microcal Software, Northampton, MA).
ANSA (5-aminonaphthaline-sulfonamides) were synthesized from 5-nitronaphthalinsulphoacids and 5-aminonaphthalinsulphoacids (Reahim, Budapest, Hungary) as described (28). TLC of ANSA and their derivatives was carried out on silica gel 60 plates (Merck) in the hexane/ethyl acetate system. Gel filtration was performed by using the Sephadex G25 M size-exclusion column (Amersham Pharmacia). Fluorescence of ANSA was observed and measured under 365 nm UV light by using the Bio-Rad Fluor-S MultiImager system.
Transcription reactions (total volume = 20 μl) were performed in a buffer containing 20 mM Tris⋅HCl (pH 7.5) 10 mM MgCl2, 50 mM KCl; 8 μM ATP, GTP, UTP; 1.8 μM CTP; and 0.15 μM [α-32P] CTP (3,000 Ci/mmol) for 5 min at 37°C. The RNA polymerase (RNAP) was purified as described (28). The DNA template containing the A1 promoter of phage T7 was obtained by PCR from the plasmid pENtR2 (29).
Results
Stimulating Effect of the Protein Environment on S- and N-Nitrosation.
We first tested model protein BSA for its ability to serve as a ⋅NO concentrator and a catalyst of ⋅NO oxidation. Albumin, the most abundant transport and depot protein in the vasculature, was chosen because previous studies have shown that its specific cysteine residue (Cys-34) is a target for nitrosylation, resulting in modification of its biological activity (4 and 5). Our hypothesis of autocatalytic protein S-nitrosylation predicts that at physiological concentrations of ⋅NO and O2, the rate of Cys-34 modification in BSA should be considerably higher than that of free Cys, or when it was in the context of a short peptide.
Fig. 1 demonstrates the time-dependent formation of nitroso-Cys in two representative experiments in which the initial concentration of ⋅NO was 100 or 10 μM. Aqueous solutions of BSA, natural peptide–GSH, or free Cys were tested in equal molar concentrations. In each case, six UV-visible absorption spectra were collected at 5-min intervals under aerobic conditions after the addition of a freshly prepared, oxygen-free, aqueous solution of ⋅NO. The overlapping nitrosation spectra of nitroso-Trp were subtracted from that of nitroso-Cys. The rate and efficiency of nitroso-Cys formation in BSA was considerably higher than that of low-molecular-weight thiols. The most significant difference in the nitrosation rates was observed during the first few minutes of the reaction (Fig. 1C). Nitrosation profiles of free Cys and GSH were almost identical.
Figure 1.

Comparative analysis of Cys nitrosation. (A and B) UV-visible absorption spectra of newly generated chromophore during the reaction of BSA, GSH, or free Cys with the aqueous solution of ⋅NO. After addition of 100 μM (A) or 10 μM (B) ⋅NO to the aqueous solution of either reagent, spectra were recorded immediately and repeated every 5 min for a total of six cycles. Recording was conducted against a blank sample containing BSA, GSH, or free Cys. Overlapping spectra corresponding to nitroso-Trp were subtracted. The final concentration of each of the reagents was 0.4 mM in both A and B. (C) Graphic representation of the results of A. (D) Difference between the initial nitrosation rate of BSA (RBSA) and that of free Cys (RCys) as a function of ⋅NO concentration. Experiments similar to those shown in A and B have been done by using eight other ⋅NO concentrations: 30, 60, 120, 200, 300, 500 μM, and 1 mM. In each case, the experiment was repeated four times, and the averages of RBSA and RCys were calculated. Then the ratio between RBSA and RCys was plotted as a function of ⋅NO concentration. ΔD346, absorbance (OD units) with the maximum at λ 346 nm.
The difference between the initial S-nitrosation rate of BSA (RBSA) and that of free Cys (RCys) increased sharply with decreasing ⋅NO concentration (Fig. 1D). The lowest concentration that allowed consistent spectroscopic measurements was 10 μM ⋅NO. At this concentration, RBSA was more than 30 times greater than RCys. The curve (Fig. 1D) was constructed based on eight statistically significant points. Extrapolation of this curve suggests that, at the physiological concentrations of ⋅NO (10 nM–1 μM), the difference between RBSA and -RCys should be at least two orders of magnitude.
Besides the proposed mechanism of micellar catalysis, the relatively high efficiency of BSA S-nitrosylation can be explained whether the microenvironment of Cys-34 significantly affects its pKa. To examine this possibility, we did similar experiments to compare the rate of nitrosation of the only tryptophan residue (Trp-214) in BSA with that of the randomly chosen Trp-containing peptide, FPRAWTHTGFI, and free Trp (Fig. 2). Trp-214 is located far (≈33Å) from Cys-34 in the folded albumin polypeptide (30) and thus represents a convenient, independent “marker” of nitrosation. To monitor N-nitrosation by spectroscopy unambiguously, we used CM-BSA—a modified version of BSA where Cys-34 is blocked covalently (4). The results indicate that, similar to what was observed in the case of S-nitrosation, N-nitrosation of BSA occurs much more rapidly and efficiently than that of either free Trp or Trp peptide (Fig. 2 A and B). Again, the difference between the initial N-nitrosation rate of CM-BSA (RCM-BSA) and that of free Trp (RTrp) increased rapidly with decreasing ⋅NO concentration (Fig. 2D). At 10 μM ⋅NO, RCM-BSA was more than 40 times greater than RTrp.
Figure 2.

Comparative analysis of Trp nitrosation. UV-visible absorption spectra of newly generated chromophore during the reaction of CM-BSA, Trp-containing peptide, or free Trp with an aqueous solution of ⋅NO. After the addition of 100 μM (A) or 10 μM (B) of ⋅NO to the solution of one of the reagents, spectra were recorded immediately and repeated every 5 min for a total of eight cycles. Recording was conducted against a blank sample containing CM-BSA, the peptide, or free Trp. The final concentration of each of the reagents was 0.4 mM in both A and B. (C) Graphic representation of the experiment analogous to the one in A but with the addition of a situation when 0.4 mM Trp was dissolved in DMSO and then treated with ⋅NO. (D) Difference between the initial nitrosation rate of CM-BSA (RCM-BSA) and that of free Trp (RTrp) as a function of ⋅NO concentration. Experiments similar to those shown in A and B have been done by using eight other ⋅NO concentrations: 30, 60, 120, 200, 300, 500 μM, and 1 mM. In each case, the experiment was repeated four times, and the averages of RCM-BSA and RTrp were calculated. Then the ratio between RCM-BSA and RTrp was plotted as a function of ⋅NO concentration. ΔD330, absorbance (OD units) with the maximum at λ 330 nm.
To imply further a nonpolar protein environment in the stimulation of nitrosation, we used water-free DMSO/⋅NO solution. In DMSO, RTrp was greater than in water, and after the first 5 min of reaction, an accumulation of nitroso-Trp continued almost linearly during the time of the experiment (Fig. 2C). The diffusion of O2 in DMSO is similar to that in water and cannot explain the overall stimulating effect of DMSO on nitrosation. The major difference, however, can be attributed to the fact that in DMSO, the majority of newly formed N2O3 molecules interacted with Trp rather than with water. A similar mechanism can explain the high efficiency of Trp nitrosation in CM-BSA; the protein hydrophobic core should sequester ⋅NO from the water solution. Noticeably, during the first few minutes, RCM-BSA in water was considerably higher than RTrp in DMSO, suggesting that the hydrophobic core of BSA not only protects N2O3 from water molecules but also potentiates its formation by concentrating ⋅NO and O2 in a small volume.
Protein Hydrophobic Core Is a Catalyst of Nitrosylation.
The observed phenomenon of BSA-mediated catalysis of its own nitrosylation can be explained by the principle of micellar catalysis: the hydrophobic core of BSA accelerates formation of N2O3 by concentrating ⋅NO and O2 from the water solution. To test this hypothesis directly, we synthesized fluorescent versions of Griess indicators of nitrosation, ANSA. ANSA are aromatic amines that contain a sulfonamide group with one or two aliphatic substitutions (R1 and R2; ref. 28; Fig. 3A). They have a relatively low fluorescence yield in polar solvents, which is greatly enhanced in the nonpolar environment. Differences in the fluorescence activity can be readily visualized by the naked eye. Nitrosation of the NH2 group converts the ANSA molecule to an aryldiazonium cation, which cross-links with another ANSA molecule (azocoupling reaction) to generate nonfluorescent, deeply colored azoderivatives (Fig. 3A). The origin of each azoderivative dye can be defined by TLC. Additionally, the hydrophobicity of ANSA can be changed by varying the identity of R1 and R2 groups.
Figure 3.

BSA-mediated catalysis of ANSA nitrosation. (A) Chemical structure and properties of ANSA. Nitrosation of the NH2 group converts ANSA into the aryldiazonium cation that cross-links to another ANSA molecule in solution or within the interior of a protein. Coupling of two or more ANSA molecules produces nonfluorescent dyes. R1, R2, and R3 stand for the variable substitutions. When R3 was substituted, ANSA was unable to perform azocoupling reactions. (B) ⋅NO extinguishes fluorescence (F) of ANSA in the presence (lanes 3 and 4) or absence (lanes 6 and 7) of 5 μM BSA. The picture of the multiwell plate was taken under 365-nm UV light. The content of each well is indicated on top of the panel. The final concentration of ANSA I and ANSA II was 2 μM each. An aqueous solution of ⋅NO was added to a final concentration of 10 μM (lanes 3 and 6) or 50 μM (lanes 4 and 7), and incubation was carried out for 5 min. (C) Effect of BSA on azoderivative dyes formation. ANSA and their azoderivatives were extracted by CHCl3 from the reaction mixtures of B, as well as other mixtures where proportions of ANSA I and ANSA II were not equal, and loaded onto a TLC plate. The picture of the plate was taken under UV light (lane 1) or daylight (other lanes). Without BSA, 50 μM ⋅NO converts ANSA I and ANSA II (lane 1) into dye I and dye II, respectively (lanes 2–6). In the presence of BSA, only ANSA I forms dyes on incubation with 50 μM ⋅NO (lane 8). No detectable dyes were produced with 10 μM ⋅NO (lane 7). Minor bands represent dyes that originated from more than one azocoupling reaction.
We first synthesized two ANSA with opposite hydrophobic properties (Fig. 3B). In the low-hydrophobic ANSA molecule (ANSA I), the R1 group is -CH3 and the R2 group is -H. The highly hydrophobic ANSA (ANSA II) has C3H7 for both R1 and R2. Experiments were done by mixing an aqueous solution of ANSA with the test protein and then initiating the nitrosation reaction by the aerobic addition of an aqueous solution of ⋅NO. Two initial concentrations of ⋅NO were used at a 25-molar excess over ANSA (high dose) and a 5-molar excess (low dose). In the absence of BSA, treatment of the ANSA I + ANSA II mixture with the high dose of ⋅NO almost completely extinguished the fluorescence (Fig. 3B, lane 7). It produced a set of dyes that accumulated proportionally and originated from both ANSA I and ANSA II (Fig. 3C, lane 4), indicating that both types of ANSA were nitrosated at the same rate. Under the same conditions, the low dose of ⋅NO had little nitrosating effect, because the fluorescence was decreased only slightly (Fig. 3B, lane 6). We did not observe any nitrosation of ANSA under oxygen-free argon conditions, even when the high dose of ⋅NO was applied (data not shown), indicating that the nitrosating agents must be intermediates of ⋅NO oxidation. Because the pH of the reaction buffer was 7.5, it is likely that N2O3 rather than HNO2 was the major nitrosating agent.
The results of nitrosation were completely different in the presence of BSA. ANSA II, but not ANSA I, was able to form a stable complex with BSA as evidenced by gel filtration analysis (not shown). Addition of BSA to the mixture of ANSA I + ANSA II increased the fluorescence of the solution significantly (Fig. 3B, lane 2), indicating that ANSA II was solubilized by the hydrophobic core of BSA. The low dose of ⋅NO was enough to decrease the fluorescence of the ANSA I + ANSA II + BSA mixture to approximately half that of the intact ANSA I + ANSA II mixture (Fig. 3B, compare lanes 3 and 5), indicating that approximately half the ANSA molecules became nitrosated. Because we did not observe any dye formation with BSA and the low dose of ⋅NO (Fig. 3C, lane 7), we conclude that only BSA-bound ANSA II, not free ANSA I, was nitrosated. We confirmed this conclusion by isolating intact ANSA I from the mixture by gel filtration and CHCl3 extraction. The absence of ANSA II-generated dyes can be explained only if azoderivatives of ANSA II were cross-linked to amino acid residues of BSA and not to other ANSA molecules. Correspondingly, when the high dose of ⋅NO was applied, we observed only dyes that originated from ANSA I (Fig. 3C, lane 8). These results indicated that BSA acted as a catalyst of nitrosation of the bound hydrophobic compounds (ANSA II), i.e., the nitrosating activity is greater in the hydrophobic interior of BSA than in solution.
Hydrophobic Amines (ANSA) as ⋅NO-Dependent Enzyme Inhibitors.
To investigate whether this mechanism might be functioning in proteins other than BSA, we examined the effect of nitrosation on a completely distinct protein—RNA polymerase (RNAP) from Escherichia coli. The experiments were done by exposing RNAP to a low dose of ⋅NO and then adding the DNA template and ribonucleotide substrates to initiate the transcription reaction. For a control, we substituted a solution of NO2− for the ⋅NO, by oxidizing ⋅NO before the experiment. As Fig. 4 shows, ⋅NO partially inhibited transcription (lane 2), whereas NO2− had no effect (lane 3). The ⋅NO-dependent inhibition of RNAP was complex. Nitrosation of the RNAP decreased the overall amount of transcripts, suggesting that the early steps of the transcription cycle were suppressed (e.g., DNA binding and open promoter complex formation). Also, the addition of ⋅NO decreased significantly the accumulation of the full-length, 49-nt transcript. Shorter transcripts represent paused RNAP molecules resulting from the limited amount of CTP in the reaction. This redistribution of RNA transcripts on nitrosation suggests that the catalytic parameters of RNAP (Km for ribonucleotides and/or Vmax) have been changed. The complex inhibitory effect of nitrosation on RNAP is not surprising, because many potential nitrosation targets are distributed in structurally and functionally distinct regions of the enzyme (31).
Figure 4.

Inactivation of E. coli RNAP by ⋅NO and ANSA derivatives. (A) The autoradiogram shows RNA products from transcription reactions performed in the presence of 10 pmol RNAP; 30 pmol DNA; 8 μM each ATP, GTP, and UTP; 1.8 μM CTP; and [32P]CTP. Before transcription initiation, RNAP (with or without ANSA) was treated with the 120 μM aqueous solution of ⋅NO (even lanes) or NO2− (odd lanes) for 15 min. Numbers on the left indicate the lengths of the transcripts. Roman numerals stand for ANSA species: (I) 5-amino-1-(N-methyl)-ANSA; (II) 5-amino-1-(N,N-dipropyl)-ANSA; (III) 5-amino-1-(N,N-dimethyl)-ANSA; (IV) 5-amino-1-[N,N-(3′-oxapentamethylene)]-ANSA; (V) 5-amino-1-(N-pentyl)-ANSA; (VI) 5-methylamino-1-(N,N-dimethyl)-ANSA; (VII) 5-peptidylamino-1-(N-pentyl)-ANSA. All ANSA have been used as aqueous solutions at a final concentration 10 μM. In each case, the relative inhibition of transcription (i) was calculated by dividing the amount of [32P]RNA obtained with RNAP that had been challenged with ⋅NO (even lanes) to that with NO2− (odd lanes) and multiplying by 100. (B) Summary table. For each ANSA used in the experiment, the chemical structures of substitution groups and properties important for their ⋅NO-dependent inhibitory activity are indicated. GGRR stands for the peptidyl (R3) substitution where “G” represents glycine and “R” represents arginine.
The experiments with BSA demonstrated that on nitrosation, ANSA II could cross-link to its “host” protein molecule. To determine whether this process could also occur with RNAP, we treated the mixture of RNAP + ANSA II with ⋅NO (Fig. 4A, lane 4). In the presence of ANSA II, the same low dose of ⋅NO used in lane 2 (Fig. 4B) completely inactivated RNAP. ANSA II alone did not affect transcription (Fig. 4A, lane 5).
To test further the autocatalytic mechanism of enzyme inactivation and the key role of nitrosation-azocoupling in this process, we synthesized additional ANSA and tested them in the ⋅NO-dependent RNAP inactivation assay (Fig. 4B). Fig. 4B shows ANSA that were designed to have different degrees of hydrophobicity (I < III ≈ IV < VI < V ≈ VII < II). We found that the hydrophobicity of ANSA directly correlated with its ability to stimulate the ⋅NO-dependent inactivation of the enzyme. The least hydrophobic ANSA I had no effect on transcription (Fig. 4A, lane 10); ANSA III and ANSA IV exhibited a partial negative effect (Fig. 4A, lanes 6 and 14), and ANSA V completely inactivated RNAP (Fig. 4A, lane 12). ANSA VI and ANSA VII, with methyl and peptidyl substitutions in the NH2 group, were incapable of forming aryldiazonium cation on nitrosation. Although ANSA VI and ANSA VII were more hydrophobic than ANSA III and ANSA IV, they were unable to enhance inactivation of RNAP (Fig. 4A, lanes 8 and 16). This experiment raises the possibility that hydrophobic amines could be used as efficient tools for the ⋅NO-dependent inactivation of enzymes.
Our results with ANSA can be described by the following scheme (Fig. 5B): First, a protein solubilizes hydrophobic ANSA. Then, the hydrophobic phase formed by nonpolar protein residues acts as an efficient micellar catalyst of ⋅NO oxidation and formation of the nitrosating agent. Finally, if an NH2 group of ANSA is free, the nitrosation reaction converts it into a highly reactive aryldiazonium cation that immediately cross-links to the protein interior and suppresses the enzymatic activity.
Figure 5.

Autocatalytic mechanism of protein nitrosation and its implication for NO-donors formation and also target enzyme inactivation. (A) Micellar catalysis of ⋅NO oxidation and nitrosation. The hydrophobic core of the protein globule (shaded in dark gray) concentrates ⋅NO and O2 from solution in a small volume thus accelerating N2O3 formation. 1–3SHs, cysteines; W, tryptophan; RS, small-molecular-weight thiols. The lipophilic N2O3 molecules can attack nucleophiles directly in the hydrophobic core (e.g., cysteine 1SH). At the same time, NO+ can reach other nucleophiles in the hydrophilic phase (e.g., W and 2SH) and also RS and H2O outside the protein through the intermolecular transport and transnitrosation (arc-shaped arrows). 3SH cannot be nitrosated because of constraints imposed by the protein structure. (B) The mechanism of ANSA-dependent protein inactivation. The hydrophobic ANSA molecules with intact NH2 group (ANH2) accumulate inside the protein-hydrophobic core and form reactive diazonium cations (AN2+) on nitrosation. AN2+ cross-links with nearby amino acids and inactivates the protein.
Discussion
Our data suggest a mechanism in which hydrophobic compartments of proteins concentrate ⋅NO and O2 and thus catalyze formation of N2O3, the primary nitrosating agent (Fig. 5B). As soon as N2O3 is formed within the protein interior, it attacks nearby nucleophilic amino acid groups. Short-lived intermediates of this reaction transfer the nitroso-group further to less nucleophilic competitors and finally to molecules of water or thiols in the surrounding media. This model, by which nitrosating agents are created in regions of hydrophobicity, accounts for the high selectivity of S-nitrosylation protein modifications (19). In this case, the overall structure of the protein (the size and geometry of the hydrophobic core and distribution of nucleophiles) determines its ability to generate N2O3 and also transfer NO+ to particular Cys targets.
According to our model, N2O3 is synthesized not only during nitrosative stress, i.e., when the concentration of ⋅NO donors in solution rises substantially (refs. 15, 17, and 19 and references therein), but constitutively in the hydrophobic protein interior where the local concentration of ⋅NO and O2 is much higher than in solution. Thus, the concentration of N2O3 depends not only on the initial concentrations of ⋅NO and O2 in the whole system but also on the size and geometry of the hydrophobic phase as well as the distribution of available targets in the protein molecule. Such a mechanism further suggests that both S-nitrosylation and N-nitrosation can be controlled, not only by the activity of NO synthases and O2 concentrations, but also by conformational transitions in the protein molecule that change its hydrophobic properties. Additionally, nitros(yl)ation itself may be able to induce further conformational transitions, which may be favorable or unfavorable to continue the process. Further, solubilization of hydrocarbons and other hydrophobic compounds by proteins could affect nitrosation because of the increased efficiency of micellar catalysis.
In vivo, protein-mediated catalysis of ⋅NO oxidation is likely to compete with a similar process within lipid membranes (25, 27). It remains to be determined which reaction (and under what conditions) contributes most to the formation of primary nitrosative species at the cellular and subcellular levels.
As we show here, BSA and RNAP provide the environment for effective nitrosation of not only their own nucleophiles but also external molecules such as ANSA. In the separate study (R.R., O. Rafikova, and E.N., unpublished work), we demonstrate that albumin significantly stimulates formation of vasoactive low-molecular-weight nitrosothiols via the mechanism of micellar catalysis. QNO for BSA/H2O was determined to be ≈20. Taken together, these data suggest that the hydrophobic phase formed by plasma protein serves as a major reservoir of ⋅NO and its reactive species and plays an important role in maintaining the pool of RS-NO in vivo.
Here we show that the ability of a protein to accelerate nitrosation can be directed toward its own inactivation. ANSA molecules with the nucleophilic NH2 group can be used as a suicide-nitrosative substrate for different proteins and thus can serve as a paradigm for the design of a new class of antibiotics. The high selectivity and specificity of such compounds can be determined by the peptide attached to the NH2 group (Fig. 4B). Peptidyl-ANSA molecules, which are resistant to activation by nitrosation until they have been transported into the cell and the peptide has been cleaved by a particular protease or peptidase, can be synthesized. Because high concentrations of ⋅NO are associated with host defense systems against microbial infections and also some tumor cells, such molecules would preferentially target those cells. Peptide transport and protease activities are species- or tissue-specific, such that the precise design of peptidyl-ANSA molecules allows the exciting possibility for new kinds of selective therapy.
Acknowledgments
We thank Bruce Freeman, Joseph Beckman, and Warren Jelinek for valuable comments. This work was funded by the Searle Scholar Award and a grant (to E.N.) from the National Institutes of Health.
Abbreviations
- RS
thiols
- GSH
glutathione
- ANSA
5-aminonaphthaline-sulfonamides
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.250398197.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.250398197
References
- 1.Moncada S, Palmer R M, Higgs E A. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 2.Kerwin J F, Jr, Lancaster J R, Jr, Feldman P L. J Med Chem. 1995;38:4343–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 3.Gow A J, Stamler J S. Nature (London) 1998;391:169–173. doi: 10.1038/34402. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y Y, Xu A M, Nomen M, Walsh M, Keaney J F, Jr, Loscalzo J. J Biol Chem. 1996;271:14271–14279. [PubMed] [Google Scholar]
- 5.Stamler J S, Simon D I, Osborne J A, Mullins M E, Jaraki O, Michel T, Singel D J, Loscalzo J. Proc Natl Acad Sci USA. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nunoshiba T, deRojas-Walker T, Wishnok J S, Tannenbaum S R, Demple B. Proc Natl Acad Sci USA. 1993;90:9993–9997. doi: 10.1073/pnas.90.21.9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stamler J S, Toone E J, Lipton S A, Sucher N J. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 8.Hausladen A, Privalle C T, Keng T, DeAngelo J, Stamler J S. Cell. 1996;86:719–729. doi: 10.1016/s0092-8674(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 9.Berendji D, Kolb-Bachofen V, Zipfel P F, Skerka C, Carlberg C, Kroncke K D. Mol Med. 1999;5:721–730. [PMC free article] [PubMed] [Google Scholar]
- 10.Lander H M, Ogiste J S, Pearce S F, Levi R, Novogrodsky A. J Biol Chem. 1995;270:7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- 11.Xu L, Eu J P, Meissner G, Stamler J S. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 12.Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher A M, Mulsch A, Dimmeler S. J Biol Chem. 1999;274:6823–6826. doi: 10.1074/jbc.274.11.6823. [DOI] [PubMed] [Google Scholar]
- 13.Bauer P M, Fukuto J M, Buga G M, Pegg A E, Ignarro L J. Biochem Biophys Res Commun. 1999;262:355–358. doi: 10.1006/bbrc.1999.1210. [DOI] [PubMed] [Google Scholar]
- 14.Kroncke K D, Fehsel K, Kolb-Bachofen V. Nitric Oxide. 1997;1:107–120. doi: 10.1006/niox.1997.0118. [DOI] [PubMed] [Google Scholar]
- 15.Grisham M B, Jourd'Heuil D, Wink D A. Am J Physiol. 1999;276:315–321. doi: 10.1152/ajpgi.1999.276.2.G315. [DOI] [PubMed] [Google Scholar]
- 16.Beckman J S, Koppenol W H. Am J Physiol. 1996;271:1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 17.Espey M G, Miranda K M, Feelisch M, Fukuto J, Grisham M B, Vitek M P, Wink D A. Ann N Y Acad Sci. 2000;899:209–221. doi: 10.1111/j.1749-6632.2000.tb06188.x. [DOI] [PubMed] [Google Scholar]
- 18.Williams D L. Nitric Oxide. 1997;1:522–527. doi: 10.1006/niox.1997.0159. [DOI] [PubMed] [Google Scholar]
- 19.Stamler J S, Hausladen A. Nat Struct Biol. 1998;5:247–249. doi: 10.1038/nsb0498-247. [DOI] [PubMed] [Google Scholar]
- 20.Liu Z, Rudd M A, Freedman J E, Loscalzo J. J Pharmacol Exp Ther. 1998;284:526–534. [PubMed] [Google Scholar]
- 21.Jourd'Heuil D, Hallen K, Feelisch M, Grisham M B. Free Radical Biol Med. 2000;28:409–417. doi: 10.1016/s0891-5849(99)00257-9. [DOI] [PubMed] [Google Scholar]
- 22.Mulsch A, Mordvintcev P, Vanin A F, Busse R. FEBS Lett. 1991;294:252–256. doi: 10.1016/0014-5793(91)81441-a. [DOI] [PubMed] [Google Scholar]
- 23.Stamler J S. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 24.Gow A J, Buerk D G, Ischiropoulos H. J Biol Chem. 1997;272:2841–2845. doi: 10.1074/jbc.272.5.2841. [DOI] [PubMed] [Google Scholar]
- 25.Gordin V A, Nedospasov A A. FEBS Lett. 1998;424:239–242. doi: 10.1016/s0014-5793(98)00164-1. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Miller M J S, Joshi M S, Thomas D D, Lancaster J R., Jr Proc Natl Acad Sci USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goss S P, Singh R J, Hogg N, Kalyanaraman B. Free Radical Res. 1999;31:597–606. doi: 10.1080/10715769900301171. [DOI] [PubMed] [Google Scholar]
- 28.Kazantsev A G, Kuznetsov N V, Iakhimovich A D, Sharina I G, Nezavibat'ko V N, Nedospasov A A. Biochemistry. 1994;59:1139–1144. [PubMed] [Google Scholar]
- 29.Nudler E, Avetissova E, Markovtsov V, Goldfarb A. Science. 1996;273:211–217. doi: 10.1126/science.273.5272.211. [DOI] [PubMed] [Google Scholar]
- 30.Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K. Protein Eng. 1999;12:439–446. doi: 10.1093/protein/12.6.439. [DOI] [PubMed] [Google Scholar]
- 31.Zhang G, Campbell E A, Minakhin L, Richter C, Severinov K, Darst S A. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]