

Abstract
In our ongoing research on the synthesis of constrained analogues of CCK/opioid chimeric peptides, a bicyclic dipeptide mimetic for Nle-Asp was designed and synthesized. Starting from β-allyl substituted aspartic acids, the terminal double bond was oxidized resulting in spontaneous cyclization to form racemic hemiaminals. Allylation of the hemiaminals afforded 5-allyl substituted proline analogues, which on oxidation, Horner–Emmons olefination, asymmetric hydrogenation, and bicyclization afforded bicyclic dipeptide mimetics for Nle-Asp. Constrained CCK/opioid peptide analogues containing bicyclic dipeptide mimetics for Nle-Gly, Nle-Asp, and homoPhe-Gly were then synthesized and analyzed at both the CCK and opioid receptors.
Our group has recently been involved in the design and synthesis of chimeric peptides that interact with both the CCK and opioid receptors.1 Constrained analogues of these peptides have been synthesized via disulfide and lactam cyclizations.2 To further explore the topographical requirements for interaction of these peptides with receptors, bicyclic dipeptide mimetics for Nle-Gly (1a, 1b, 1c), and homoPhe-Gly 2 have been designed and synthesized (Fig. 1).3 In this letter, the design and synthesis of indolizidinone bicyclic dipeptide mimetics for Nle-Asp 3 and the synthesis of peptides containing bicyclic dipeptide mimetics are discussed. The peptides were tested at both the CCK and opioid receptors.
Figure 1.
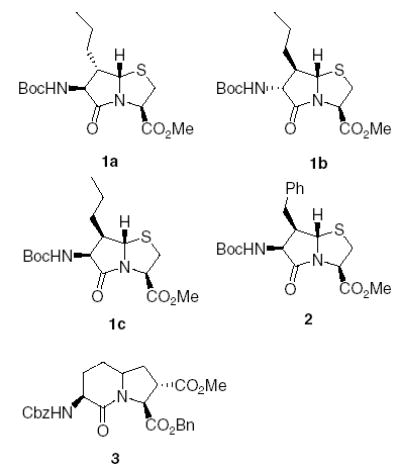
Bicyclic dipeptide mimetics.
The synthesis of the indolizidinone type of bicyclic dipeptide mimetics has been reported by a number of authors.4 In our lab, we have developed the synthesis of these mimetics from analogues of pyroglutamic acid.5 The target compound can be obtained from lactam cyclization of dehydroamino acids derived from Horner–Emmons olefination of allyl substituted proline analogues.
Alkylation of aspartic acid with different electrophiles has been reported by our group3,6 and other authors.7 Alkylation with allyl bromide in the presence of lithium bis(trimethylsilyl)amide (LHMDS) and HMPA resulted in the formation of two β-allyl substituted aspartic acids in a total yield of 57% and a ratio of 4:1 in favor of the (2 S, 3R)-5a isomer (Scheme 1).
Scheme 1.
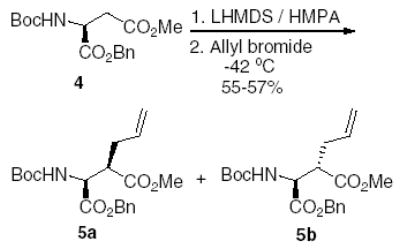
Alkylation of aspartic acid with allyl bromide.
When 5a was subjected to ozonolysis, the resultant aldehyde spontaneously cyclized to the racemic hemiaminal 6 (Scheme 2). The hydroxyl group was then methylated and the resultant compound 7 reacted with BF3·OEt2 and allyl trimethyl silane at −78 °C→rt to give compounds 8a and 8b in 48% yield and a ratio of 1:1. When the minor isomer 5b was subjected to ozonolysis and allylation, only compound 8c was obtained in 51% yield.
Scheme 2.
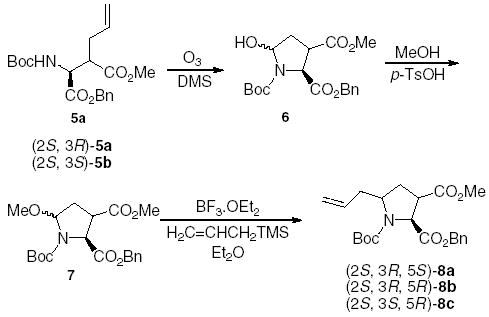
Synthesis of δ-allyl-substituted proline analogues.
The structure of compound 8a was confirmed by X-ray crystallography (Fig. 2).
Figure 2.
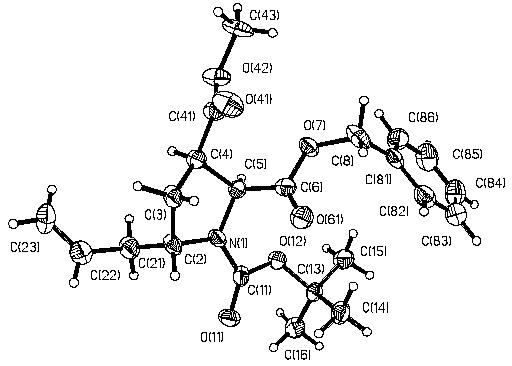
X-ray crystal structure of compound 8a.
Compound 8a was then subjected to ozonolysis and the resultant aldehyde subjected to a Horner–Emmons olefination8 to give the dehydroamino acid 9a in 58% yield. When osmylation was used for the oxidation of 8a, the dehydroamino acid was obtained in 71% yield (Scheme 3). Oxidation of compound 8b by ozonolysis followed by Horner–Emmons olefination gave the desired compound and an unidentified compound in a total yield of 38% and a ratio of 1:1. The reaction was optimized by changing the protocol to oxidation by osmylation followed by olefination to give the desired dehydroamino acid 9b in 35% yield.
Scheme 3.
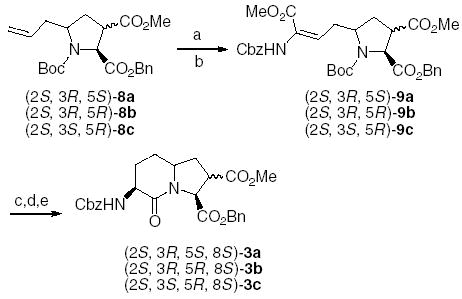
Synthesis of Nle-Gly bicyclic dipeptide mimetic. Reagents and conditions: (a) OsO4, NaIO4; (b) (MeO)2POCH(NHCbz)CO2Me, DBU; (c) (S,S)-COD-EtDUPHOSRh(I)OTf, H2; (d) 20% TFA/DCM, (e) pyridine, 50 °C.
Osmylation and Horner–Emmons olefination of 8c gave the dehydroamino acid 9c in 40% yield. Asymmetric hydrogenation of the dehydroamino acids 9a, 9b, and 9c gave the saturated analogues, which after deprotection of the Boc group were cyclized by heating in pyridine at 50 °C for 4 days to give the bicyclic dipeptide mimetics 3a, 3b, and 3c, respectively.
The three bicyclic dipeptide mimetics were then characterized by NOE measurements (Fig. 3). A strong NOE value was observed for H3 and H6 in compounds 3b and 3c (4.2% and 4.0%, respectively) signifying a cis relationship between the two hydrogens. For compound 3a, the NOE value between H3 and H6 was relatively low (1.9%) due to the trans relationship between the hydrogens. The cis relationship between H8 and H9 in 3a and 3b resulted in a strong NOE value of 4.8% compared with the weak value of 1.9% observed for compound 3c.
Figure 3.
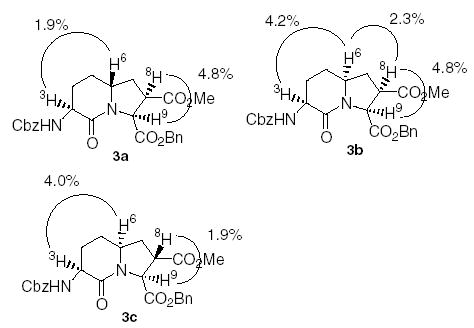
NOE data for Nle-Asp bicyclic dipeptide mimetics.
To introduce the bicyclic dipeptide mimetics 1a, 1b, 1c, and 2 into the peptides, the methyl ester was hydrolyzed and the Nα-Boc group deprotected using TFA. The amino group was then Fmoc-protected. For compound 3a, both the carboxyl and amino groups were deprotected by hydrogenation and the amino group Fmoc-protected. Fmoc/t-Bu solid phase peptide synthesis method was used for the synthesis of the peptides. The peptides JMN1-5 (Fig. 4) were consequently synthesized and analyzed at both the CCK and opioid receptors.
Figure 4.
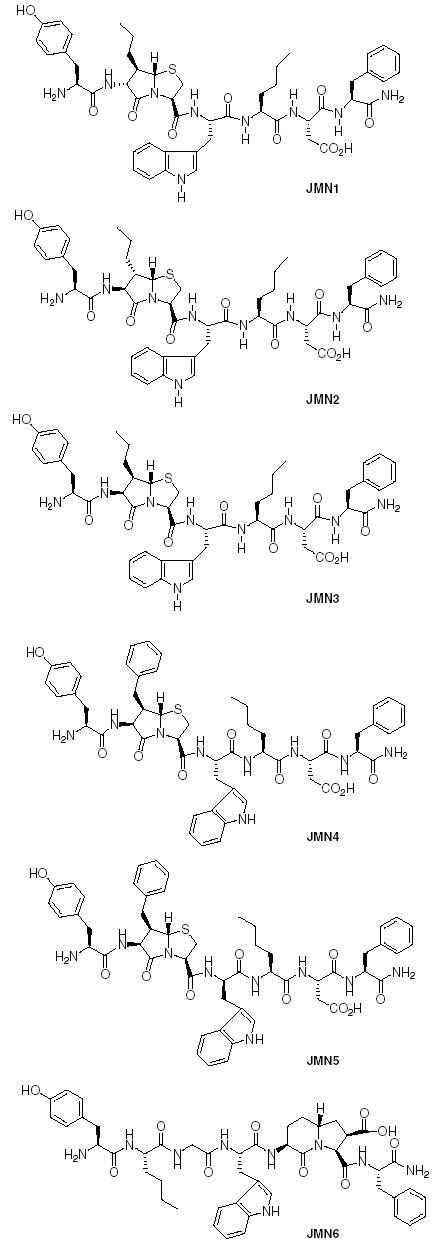
Novel bicyclic dipeptide mimetic containing CCK/opioid chimeric peptides.
When the peptides were evaluated at the opioid receptors they showed weak activity at both the δ- and μ-opioid receptors (Table 1). Peptide JMN2, however, showed low micromolar binding affinity for both the δ- and μ-opioid receptors. The only peptide that showed an agonist effect at the opioid receptors is peptide JMN5 though it had low binding affinity.
Table 1.
Biological evaluation at the opioid receptors
| GTP bindinga |
Competitionb |
|||
|---|---|---|---|---|
| Drug | hDOR EC50 (nM) | rMOR EC50 (nM) | hDOR IC50 (nM) | rMOR IC50 (nM) |
| SNF9007 | n/d | n/d | 250 | 5200 |
| RSA501 | 1000 | 1800 | 74 | 1000 |
| JMN1 | n/d | n/d | 1850 | 9300 |
| JMN2 | NA | NA | 337 | 220 |
| JMN3 | NA | NA | 1700 | 3500 |
| JMN4 | NA | NA | 1400 | 8600 |
| JMN5 | 460 | 770 | 2700 | 8100 |
| JMN6 | NA | 880 | 1500 | 3100 |
n/d = not determined, NA = no activity at 10−5 M.
[35S]GTP-γ-S binding assay.
Competitive binding assays against radiolabeled [3H]DPDPE at hDOR and [3H]DAMGO at rMOR. hDOR and rMOR were expressed from CHO cell lines.
At the CCK receptors, peptides JMN1-4 showed low micromolar binding affinities and biological activities at the CCK-B receptors (Table 2). This can be attributed to the C-terminal tetrapeptide (Trp-Nle-Asp-Phe), which has been shown to be the minimum sequence required for activity at CCK-B receptors. Introduction of a bicyclic dipeptide mimetic for Nle-Asp (see JMN6) led to loss of activity at the CCK receptors possibly due to interference with the tetrapeptide unit. Substitution of D-Trp for Trp in JMN4 to give JMN5 led to loss of activity at the CCK receptors even though there was improved activity at the opioid receptors. However, unlike in most other CCK peptides where substitution of D-Trp leads to antagonistic properties,1 JMN5 retained agonist properties. The peptides had relatively low binding profile at the CCK-A receptors and showed no agonist biological activity.
Table 2.
Biological evaluation at CCK receptors
| Functional analysisa |
Binding affinityb (ki,
nM) |
||||
|---|---|---|---|---|---|
| Drug | hCCK-A EC50 (nM) | hCCK-B EC50 (nM) | hCCK-A [125I] CCK8 | hCCK-B [125I] CCK8 | CCK-A agonist activityc |
| SNF9007 | n/d | n/d | 3300 | 2.1 | n/d |
| RSA501 | 790 | 3100 | 140 | 14 | None |
| JMN1 | NA | 4.1 | 740 | 70 | None |
| JMN2 | NA | 6.9 | 1200 | 100 | None |
| JMN3 | NA | 2.3 | 2400 | 16 | None |
| JMN4 | NA | 1.9 | 810 | 11 | None |
| JMN5 | NA | 160 | 1800 | 1400 | None |
| JMN6 | NA | NA | >10,000 | >10,000 | None |
n/d = not determined, NA = no activity at 10−5 M.
Phosphoinositide (PI) hydrolysis assay in hCCK-A and hCCK-B receptors in HEK cell lines.
Competition against [125I] CCK8 (sulfated) in hCCK-A and hCCK-B receptors in HEK cell lines in the presence of naloxone.
Contraction of isolated tissue relative to initial contraction with KCl in the presence of naloxone in GPI/LMMP.
In conclusion, novel bicyclic dipeptide mimetic containing CCK/opioid chimeric peptides was synthesized and evaluated at both the CCK and opioid receptors. Peptides JMN1-5 were active at the CCK-B receptors while JMN5 that contained a D-Trp showed weak opioid activity. To discover peptides that will have agonist properties at the opioid receptors and antagonist properties at the CCK-B receptors, more analogues of these peptides will need to be synthesized. Our first target would be the substitution of D-Trp for Trp on all the peptides, which may lead to improved opioid activity as with JMN5. A combination of D-Trp4 and NMeNle5 may also lead to improved activities at both the opioid and CCK receptors and possibly this may impart CCK antagonistic properties.
Supplementary Material
Acknowledgments
This work is supported by grants from the US Public Health Service DA 12394 and DA 06284.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.tetlet. 2006.01.096.
References
- 1.Hruby VJ, Agnes RS, Davis P, Ma SW, Lee YS, Vanderah TW, Lai J, Porreca F. Life Sci. 2003;73:699–704. doi: 10.1016/s0024-3205(03)00390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unpublished work.
- 3.Ndungu JM, Gu X, Gross DE, Cain JP, Carducci MD, Hruby VJ. Tetrahedron Lett. 2004;45:4139–4142. [Google Scholar]
- 4.Muller R, Revesz L. Tetrahedron Lett. 1994;34:4091–4092. (a) [Google Scholar]; Lombart HG, Lubell WD. J Org Chem. 1996;61:943–946. (b) [Google Scholar]; Gosselin F, Lubell WD. J Org Chem. 1998;63:7463–7471. doi: 10.1021/jo9814602. (c) [DOI] [PubMed] [Google Scholar]; Kim HO, Kahn M. Tetrahedron Lett. 1997;37:6483–6484. (d) [Google Scholar]
- 5.Zhang J, Xiong C, Wang W, Ying J, Hruby V. J Org Lett. 2002;4:4029–4032. doi: 10.1021/ol020160a. (a) [DOI] [PubMed] [Google Scholar]; Zhang J, Xiong C, Ying J, Wang W, Hruby VJ. Org Lett. 2003;5:3115–3118. doi: 10.1021/ol0351347. (b) [DOI] [PubMed] [Google Scholar]; Wang W, Yang J, Ying J, Xiong C, Zhang J, Cai C, Hruby VJ. J Org Chem. 2002;67:6353–6360. doi: 10.1021/jo0203591. (c) [DOI] [PubMed] [Google Scholar]
- 6.Ndungu JM, Gu X, Gross DE, Ying J, Hruby VJ. Tetrahedron Lett. 2004;45:3245–3247. [Google Scholar]
- 7.Baldwin JE, Moloney MG, North M. J Chem Soc, Perkin Trans 1. 1989:833–834. (a) [Google Scholar]; Cotton R, Johnstone ANC, North M. Tetrahedron Lett. 1994;35:8859–8862. (b) [Google Scholar]; Humprey JM, Bridges RJ, Hart JA, Chamberlin AR. J Org Chem. 1994;59:2467–2472. (c) [Google Scholar]; Cotton R, Johnstone ANC, North M. Tetrahedron. 1995;51:8525–8544. (d) [Google Scholar]
- 8.Schmidt U, Griesser H, Leitenberger V, Lieberknecht A, Mangold R, Meyer R, Riedl B. Synthesis. 1992:487–490. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.