

Abstract
Thrombin activates protease-activated receptor-1 (PAR-1) and engages signaling pathways that influence the growth and survival of cardiomyocytes as well as extracellular matrix remodeling by cardiac fibroblasts. This study examines the role of Shc proteins in PAR-1-dependent signaling pathways that influence ventricular remodeling. We show that thrombin increases p46Shc/p52Shc phosphorylation at Tyr239/Tyr240 and Tyr317 (and p66Shc-Ser36 phosphorylation) via a pertussis toxin-insensitive epidermal growth factor receptor (EGFR) trans-activation pathway in cardiac fibroblasts; p66Shc-Ser36 phosphorylation is via a MEK-dependent mechanism. In contrast, cardiac fibroblasts express β2-adrenergic receptors that activate ERK through a pertussis toxin-sensitive EGFR transactivation pathway that does not involve Shc isoforms or lead to p66Shc-Ser36 phosphorylation. In cardiomyocytes, thrombin triggers MEK-dependent p66Shc-Ser36 phosphorylation, but this is not via EGFR transactivation (or associated with Shc-Tyr239/Tyr240 and/or Tyr317 phosphorylation). Importantly, p66Shc protein expression is detected in neonatal, but not adult, cardiomyocytes; p66Shc expression is induced (via a mechanism that requires protein kinase C and MEK activity) by Pasteurella multocida toxin, a Gαq agonist that promotes cardiomyocyte hypertrophy. These results identify novel regulation of individual Shc isoforms in receptor-dependent pathways leading to cardiac hypertrophy and the transition to heart failure. The observations that p66Shc expression is induced by a Gαq agonist and that PAR-1 activation leads to p66Shc-Ser36 phosphorylation identifies p66Shc as a novel candidate hypertrophy-induced mediator of cardiomyocyte apoptosis and heart failure.
Cardiac hypertrophy and the progression to heart failure is the result of a complex array of signaling events that lead to structural remodeling of the ventricle. The literature has focused primarily on signaling mechanisms that regulate cardiomyocyte growth and survival. However, cardiac fibroblasts (activated by fibrogenic growth factors and pro-inflammatory cytokines) also play an important role in influencing the architecture and mechanical behavior of the heart by regulating collagen matrix remodeling and by elaborating growth factors that promote cardiomyocyte hypertrophy. Extensive cardiac fibro-blast-induced structural remodeling disrupts the normal transmission of electrical impulses, impairs contractile performance, and critically influences clinical heart failure syndromes.
Protease-activated receptor-1 (PAR-1)2 is a G protein-coupled receptor that is proteolytically activated by thrombin and other pro-inflammatory proteases. We previously demonstrated that chronic/persistent PAR-1 activation leads to cardiomyocyte hypertrophy and cardiac fibroblast proliferation (1). PAR-1-dependent growth responses are mediated by distinct signaling mechanisms in cardiomyocytes and cardiac fibroblasts. PAR-1 evokes a robust increase in ERK, p38-MAPK, and AKT and increases DNA synthesis via an epidermal growth factor receptor (EGFR) transactivation pathway in cardiac fibroblasts, whereas PAR-1 activates ERK (and induces only a relatively minor increase in AKT) via a mechanism that does not require EGFR kinase activity in cardiomyocytes. The contextual nature of PAR-1 signaling emphasizes the need to directly examine PAR-1 actions in physiologically relevant cardiac preparations.
Shc proteins are molecular adapters that link surface receptors to mitogenic responses. Shc is expressed as three isoforms that are encoded by a single gene locus (2, 3). p46Shc and p52Shc are ubiquitous isoforms that arise through the use of alternative translation initiation sites within the same transcript. In contrast, p66Shc expression is restricted to certain cell types and stimulatory conditions through epigenetic modifications of its distinct promoter (4). All three Shc isoforms share a common domain organization consisting of a C-terminal SH2 domain, an N-terminal PTB domain (slightly truncated in p46Shc), and a central CH1 with phosphorylation sites at Tyr239/Tyr240 and Tyr317 (nomenclature based on human p52Shc). Phosphorylation at Tyr239/Tyr240 and Tyr317 creates docking sites for the Grb2-SH2 domain, which constitutively associates with the Ras exchange factor SOS, activates the Ras-ERK pathway, and induces mitogenic responses. This function is particularly important for p52Shc, which is hyperphosphorylated in many tumors and oncogenic when overexpressed in fibroblasts (5).
p66Shc also contains the three sites for growth factor-dependent tyrosine phosphorylation (and it is tyrosine-phosphorylated and binds Grb2 in cells treated with EGF or subjected to oxidative stress). However, the subcellular targeting and signaling functions of p66Shc and p46/p52Shc are quite different. p66Shc does not couple to Ras/ERK activation and it does not transform rodent fibroblasts. Rather, p66Shc inhibits Ras/ERK signaling and blunts mitogenic responses, at least in part by competing with p52Shc for a limited pool of Grb2 (6). p66Shc also has emerged as a pivotal regulator of oxidative stress responses and life span in mammals. p66Shc sensitizes cells to apoptosis through a mechanism that involves its unique N-terminal CH2 domain (not present in p46/p52Shc) and specifically a phosphorylation event at Ser36 (stimulated by PMA, UV irradiation, or H2O2 (7)). p66Shc links stress activation of p53 to increased intracellular reactive oxygen species accumulation and apoptosis (8). p66Shc also inhibits FOXO transcription factors; because FOXOs induce antioxidant defense enzymes, p66Shc-dependent FOXO inhibition impairs reactive oxygen species detoxification and enhances apoptosis (9). Finally, p66Shc localizes to mitochondria where it participates in membrane depolarization during oxidative stress (10, 11). Although these studies identify p66Shc as a critical transducer of oxidative stress signals (and a recent study implicates p66Shc in the deleterious cardiac remodeling induced by subpressor doses of angiotensin II), the mechanisms that control Shc protein expression or activation in the heart have largely been neglected in the literature (12, 13).
PAR-1 increases Shc tyrosine phosphorylation (and Shc is implicated in PAR-1-dependent mitogenic responses) in non-cardiac model systems (14, 15). However, studies to date have generally been confined to experiments that use anti-phospho-Tyr antibodies (and do not resolve phosphorylation at the Tyr239/Tyr240 and Tyr317 sites), focusing generally on phosphorylation of the abundant p52Shc isoform (or heterologously overexpressed Shc proteins). Mechanisms that regulate Shc phosphorylation in cardiac fibroblasts or cardiomyocytes have never been examined. This report takes advantage of phosphorylation site-specific antibodies (PSSAs) to track thrombin-dependent phosphorylation of native Shc proteins (at physiologic expression levels) in the heart. We demonstrate that thrombin promotes p46/p52Shc-Tyr239/Tyr240 and -Tyr317 (and p66Shc-Ser36) phosphorylation via an EGFR transactivation pathway in cardiac fibroblasts and that thrombin increases p66Shc-Ser36 phosphorylation (without increasing p46/p52Shc-Tyr239/Tyr240 and -Tyr317 phosphorylation) through a PKC and MEK (but not EGFR transactivation) pathway in cardiomyocytes. We also show that Gαq activation increases p66Shc expression and Ser36 phosphorylation in cardiomyocytes, identifying p66Shc as a candidate hypertrophy-induced modifier of cardiac apoptosis.
EXPERIMENTAL PROCEDURES
Cardiac Fibroblast and Cardiomyocyte Cultures
Cardiomyocytes were isolated from the hearts of 2-day-old Wistar rats by a trypsin dispersion procedure using a differential attachment procedure to enrich for cardiomyocytes followed by irradiation as described in detail in previous publications (1). The yield of cardiomyocytes typically is 2.5−3 × 106 cells/neonatal ventricle. Cells were plated on protamine sulfate-coated culture dishes at a density of 5 × 106 cells/100-mm dish. Experiments were performed on cultures grown for 5 days in minimum Eagle’s medium (Invitrogen) supplemented with 10% fetal calf serum and then serum-deprived for the subsequent 24 h. Primary cardiac fibroblast cultures were obtained from the cells adherent to the culture dishes during the preplating step as described previously (1).
Measurements of cAMP accumulation and immunoblotting (on lysates or immunoprecipitated proteins) were done according to methods described previously or the manufacturer’s instructions (1). All antibodies in immunoblotting studies were from Cell Signaling Technology, with each panel in each figure representing results from a single gel (exposed for a uniform duration); detection was with enhanced chemiluminescence. Recombinant Pasteurella multocida toxin (PMT) was expressed and purified as described previously (16). All results were replicated in at least four experiments on separate culture preparations.
RESULTS
Shc and ERK Are Downstream Targets of the Thrombin-activated PAR-1 Signaling Cascade in Cardiac Fibroblasts
Fig. 1A shows that thrombin promotes a rapid (maximal at 2–5 min) and transient increase in ERK activity, measured as an increase in phospho-ERK immunoreactivity and decreased ERK protein mobility in SDS-PAGE. Thrombin and PMA induce comparable increases in ERK activity, whereas the magnitude of the ERK response to EGF is ~18% higher (n = 7, p < 0.05).
FIGURE 1. Thrombin activates ERK, promotes p46/p52Shc-Tyr239/Tyr240/Tyr317 phosphorylation, and induces a p66Shc band shift in cardiac fibroblasts.
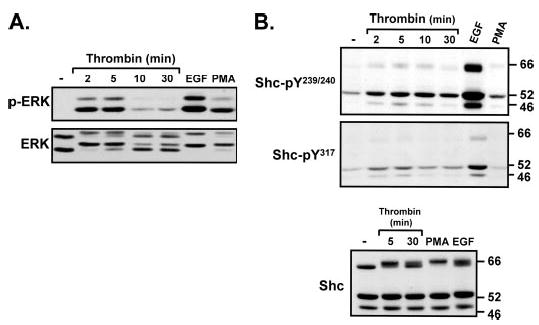
Shown are immunoblots of cell extracts from cardiac fibroblast cultures treated with vehicle, 1 nM thrombin (for the indicated intervals), EGF (100 nM, 5 min), or PMA (300 nM, 20 min).
Thrombin also promotes Shc tyrosine phosphorylation (Fig. 1B). Cardiac fibroblasts express all three Shc isoforms. p52Shc is slightly more abundant than p46Shc and p66Shc; it is the predominant Tyr239/Tyr240- and Tyr317-phosphorylated species in resting cultures. Thrombin promotes p46Shc and p52Shc phosphorylation at Tyr239/Tyr240 and Tyr317; this response is detected at 2–5 min and wanes thereafter (at 30 min, similar to the kinetics of thrombin-dependent ERK activation). In contrast, thrombin induces a very minor increase in p66Shc-Tyr239/Tyr240 phosphorylation, which is variably detected only in some experiments; thrombin-dependent p66Shc-Tyr317 phosphorylation was not detected. Control experiments show that all three Shc isoforms are phosphorylated at Tyr239/Tyr240 and Tyr317 in response to EGF.
Although thrombin does not consistently increase p66Shc tyrosine phosphorylation, thrombin slows the mobility of p66Shc in SDS-PAGE (without altering the mobility of the smaller p46/p52Shc isoforms). This thrombin-dependent mobility shift (characteristic of a post-translational modification) has been attributed to agonist-induced phosphorylation at Thr29, Ser36, and/or Ser138 (in the unique N-terminal extension of p66Shc (17)). Although p66Shc-Ser138 corresponds to p52Shc-Ser29, p52Shc-Ser29 phosphorylation does not lead to an obvious p52Shc mobility shift; p46Shc lacks N-terminal phosphorylation sites and is not regulated through this mechanism.
The thrombin-dependent p66Shc mobility shift is pronounced at 5 min and sustained for at least 30 min. EGF and PMA also slow the mobility of p66Shc. PMA (which promotes p66Shc phosphorylation at all three potential Ser/Thr phosphorylation sites) induces the most pronounced p66Shc mobility shift (17). The smaller mobility shifts induced by thrombin and EGF could be due to phosphorylation at fewer sites on p66Shc (or phosphorylation events at all three sites, but on a limited pool of p66Shc protein).
We previously demonstrated that thrombin stimulates ERK in cardiac fibroblasts primarily via a pertussis toxin (PTX)-insensitive pathway involving EGFR transactivation by Src family kinases; PTX-sensitive Gi proteins also link PAR-1 to ERK activation, but this is a minor pathway in cardiac fibroblasts (1). Although both pathways in theory could increase Shc tyrosine phosphorylation (18), Fig. 2A shows that thrombin-dependent increases in p46/p52Shc-Tyr239/Tyr240 and -Tyr317 phosphorylation are completely blocked by the EGFR kinase inhibitor AG1478 and the Src family kinase inhibitor PP1. EGF-dependent Shc tyrosine phosphorylation and ERK activation also are blocked by AG1478 (but not PP1). Collectively, these results place Src family kinases upstream from EGFRs in the PAR-1-dependent pathway leading to Shc phosphorylation.
FIGURE 2. Thrombinpromotesp46/p52Shc-Tyr239/Tyr240/Tyr317phosphorylation and decreases the electro-phoretic mobility of p66Shc via an EGFR transactivation pathway in cardiac fibroblasts.
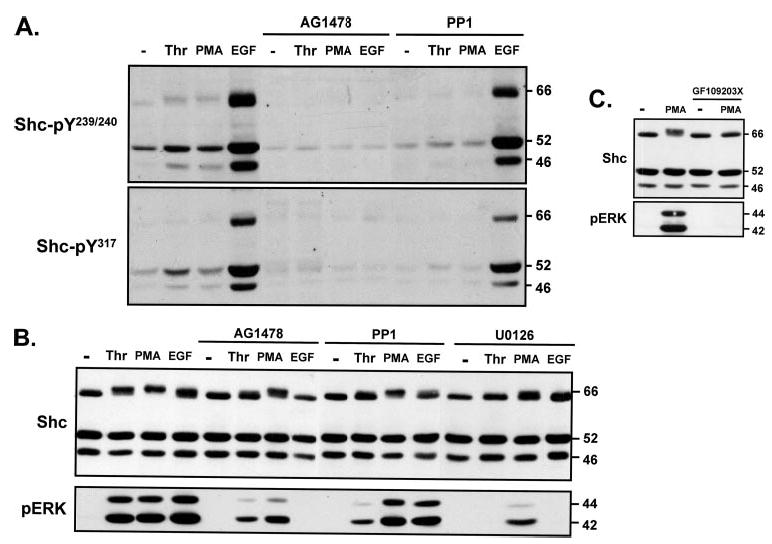
Shown are immunoblots of cell extracts from cardiac fibroblast cultures pretreated for 45 min with vehicle, AG1478 (2 μM), PP1 (10 μM) (A and B), U0126 (5 μM) (B), or GF109203X (5 μM) (C), followed by stimulations with thrombin (1 nM), PMA (300 nM), or EGF (100 nM) as indicated.
EGFR transactivation (and the MEK-ERK cascade) also underlies the thrombin-dependent p66Shc mobility shift, because the thrombin-dependent p66Shc band shift is blocked by AG1478, PP1, and the MEK inhibitor U0126 (Fig. 2B) (under conditions where AG1478 and PP1 markedly attenuate and U0126 completely inhibits ERK activation by thrombin). EGF also slows the mobility of p66Shc via a mechanism that is blocked by AG1478 and U0126, further implicating p66Shc as a downstream target of the EGFR-MEK-ERK pathway.
The effects of PMA to alter p66Shc mobility and activate ERK are markedly attenuated by U0126, but not by AG1478 or PP1 (Fig. 2B). Although these results implicate MEK as an upstream component of the p66Shc-Ser/Thr phosphorylation pathway, Fig. 2B provides surprising evidence that a minor component of the PMA response (both the p66Shc band shift and ERK activation) is resistant to U0126 (under conditions where U0126 completely abrogates ERK activation by thrombin and EGF). In contrast, PMA responses are completely blocked by GF109203X (which completely inhibits phorbol ester-sensitive PKC isoforms, Fig. 2C). These results suggest that PKC-dependent p66Shc-Ser/Thr phosphorylation occurs via a MEK-independent pathway. Of note, MEK-independent ERK activation mechanisms have been noted in other cell types (and recently attributed to a PKCε-Lck pathway in primary human Schwann cells (19–22)).
PAR-1 Promotes p66Shc-Ser36 Phosphorylation via EGFR Transactivation in Cardiac Fibroblasts
The next series of studies used a PSSA to determine whether thrombin promotes p66Shc phosphorylation at Ser36 (a site uniquely implicated in oxidative stress responses). Preliminary studies show that this PSSA identifies p66Shc-Ser36 phosphorylation in lysates from thrombin-, PMA-, and H2O2-treated cultures, but not in post-immunoprecipitation lysates that have been cleared of Shc proteins (Fig. 3A). Although these results validate the selectivity of this PSSA, the results show relatively high background nonspecific immunoreactivity in Western blots on cell lysates with this PSSA. Therefore, p66Shc-Ser36 phosphorylation was examined using an immunoprecipitation approach; Shc proteins were immunoprecipitated from resting and activated cultures and then probed with the anti-p66Shc-pSer36 PSSA and an anti-Shc protein antibody (to validate equal protein recovery and loading). Fig. 3B shows that p66Shc-Ser36 phosphorylation is not detected in resting cardiac fibroblasts; it is induced by thrombin, PMA, and (to a lesser extent) EGF.
FIGURE 3. Thrombin promotes p66Shc-Ser36 phosphorylation via EGFR transactivation of the MEK-ERK pathway in cardiac fibroblasts.
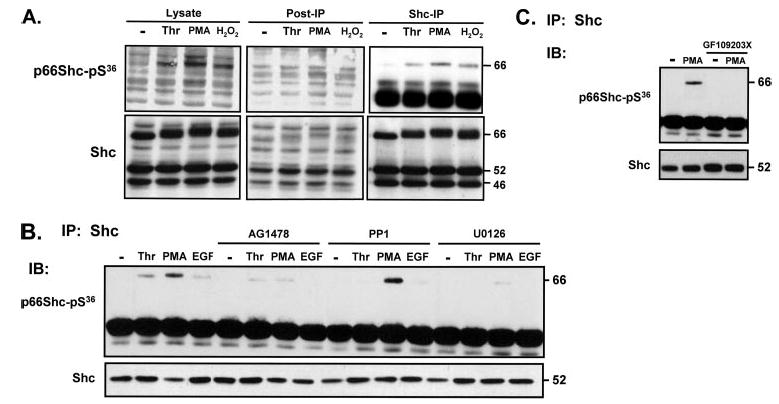
Cardiac fibroblasts were treated as described in the legend to Fig. 2. Immunoblots (IB) are of cell lysates, post-immunoprecipitation lysates (cleared of Shc proteins) (A), and immunoprecipitated (IP) p66Shc protein (A–C). Equal amounts of immunoprecipitated protein (derived from 600 μg of starting cell extract) were probed for p66Shc-pSer36 immunoreactivity and p66Shc protein (A). In panels B and C (and all subsequent figures in this article), immunoblotting for Shc (to verify equal protein recovery and loading) was performed on protein immunoprecipitated from 100 μg of starting cell extract due to limiting amounts of sample material from primary cultures. At this lower level of protein loading, only p52Shc is consistently detected.
The pharmacologic profiles for the agonist-dependent p66Shc mobility shift and p66Shc-Ser36 phosphorylation are identical (compare Figs. 2B and 3C). The thrombin-dependent increase in p66Shc-Ser36 phosphorylation is blocked by AG1478, PP1, and U0126 (i.e. it is via an EGFR transactivation mechanism involving Src family kinases and MEK). The EGF-dependent increase in p66Shc-Ser36 phosphorylation also is blocked by AG1478 and U0126, whereas the PMA-dependent increase in p66Shc-Ser36 phosphorylation is attenuated by U0126 (and completely blocked only when incubations are performed in the presence of GF109203X).
Shc Proteins Are Not Components of the β2-Adrenergic Receptor (AR)-ERK Pathway in Cardiac Fibroblasts
Previous studies identified a β2-AR-dependent pathway leading to EGFR transactivation, ERK stimulation, and increased DNA synthesis in serially passaged adult rat cardiac fibroblast cultures (23), but a potential role for Shc proteins as components of β2-AR signaling responses was not considered. Fig. 4A shows that isoproterenol induces a transient increase in ERK activity, but there are key differences in the signaling responses to isoproterenol and thrombin: 1) isoproterenol weakly activates ERK compared with the very robust increases in ERK activity triggered by thrombin, angiotensin II, or EGF; 2) isoproterenol increases cAMP accumulation, whereas thrombin does not alter cAMP levels in cardiac fibroblasts (Fig. 4B); 3) thrombin stimulates AKT, whereas β-ARs activate ERK without increasing AKT (Fig. 4C) (4). PAR-1 and β2-ARs both activate ERK via an EGFR transactivation pathway (that is completely abrogated by AG1478) but via different G proteins (Fig. 4C). β2-ARs activate ERK via a PTX-sensitive Gi protein mechanism, whereas the PAR-1-dependent ERK activation pathway is largely PTX-insensitive (5). Isoproterenol-dependent β2-AR activation does not detectably increase Shc-Tyr239/Tyr240 phosphorylation or p66Shc-Ser36 phosphorylation; β2-ARs do not decrease the electrophoretic mobility of p66Shc (Fig. 4, D and E).
FIGURE 4. β2-ARs activate ERK via an EGFR transactivation mechanism that does not involve Shc in cardiac fibroblasts.
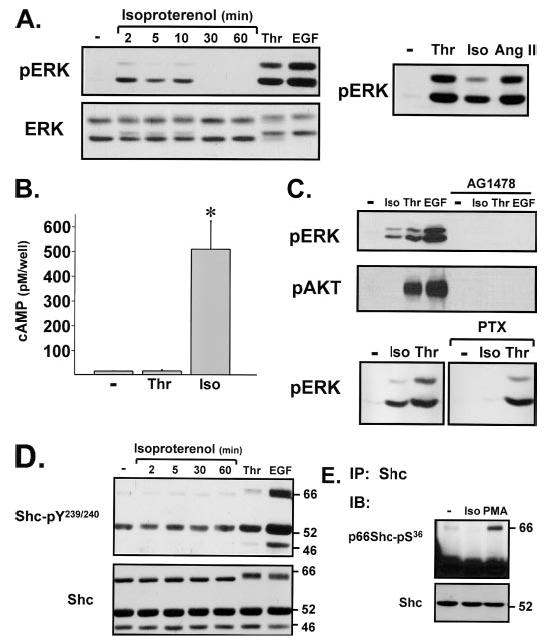
Cultures were pretreated with AG1478 (2 μM, 45 min) or PTX (100 ng/ml, 24 h) as indicated prior to stimulation with isoproterenol (Iso) (5 μM), thrombin (1 nM), angiotensin II (Ang II) (0.1 μM), EGF (100 nM), or PMA (300 nM), all for 5 min unless otherwise indicated. Immunoblotting (IB) (for pERK, ERK protein, pAKT, and Shc-pTyr239/Tyr240) (A, C, and D) measurements of cAMP accumulation (B) and immunoprecipitations (IP) and immunoblotting for p66Shc-Ser36 phosphorylation (E) were as described under “Experimental Procedures.”
Shc Isoform Regulation in Cardiomyocytes
We previously demonstrated that PAR-1 activates ERK via an EGFR transactivation pathway in cardiac fibroblasts, but PAR-1 activates ERK via a distinct mechanism that does not require EGFR kinase activity in cardiomyocytes. Therefore, the next series of experiments examined whether Shc phosphorylation constitutes an additional cell-specific component of the PAR-1-ERK signaling pathway in the heart.
Fig. 5 shows that neonatal cardiomyocyte cultures express all three Shc isoforms, that Tyr239/Tyr240-phosphorylation is detected only on p52Shc at rest, and that thrombin increases ERK phosphorylation without detectably increasing Shc-Tyr239/Tyr240 phosphorylation in cardiomyocytes. Control experiments show that EGF and H2O2 increase Shc-Tyr239/Tyr240 phosphorylation in this preparation.
FIGURE 5. Thrombin increases p66Shc-Ser36, but not Shc-Tyr239/Tyr240, phosphorylation via a PKC- and MEK-dependent mechanism in cardiomyocytes.
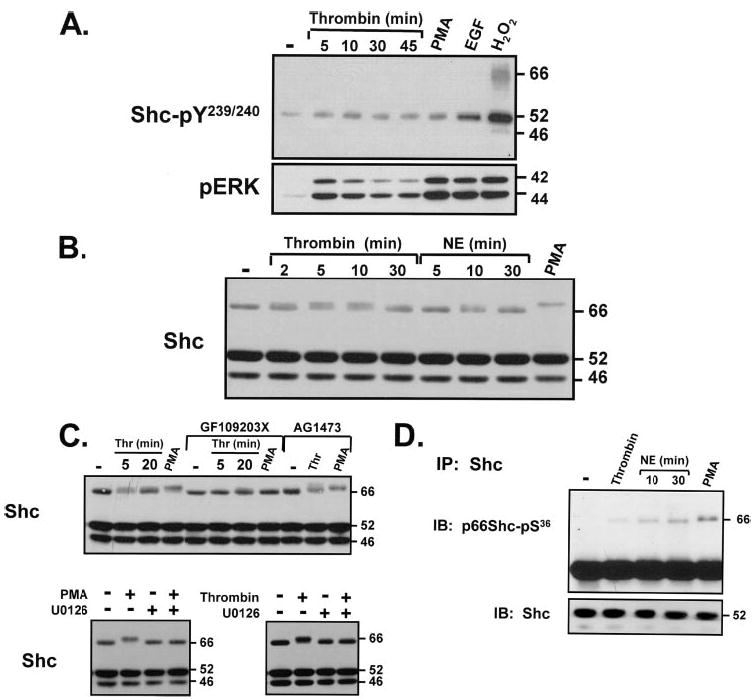
Stimulations were with thrombin (1 nM), PMA (300 nM), EGF (100 nM), H2O2 (5 mM), or norepinephrine (1 μM), all for 5 min unless otherwise indicated. Inhibitor protocols were as described in the legend to Fig. 2. Immunoblotting for Shc-pTyr239/Tyr240 and Shc protein (A–C ), pERK (A), and Shc immunoprecipitations followed by p66Shc-pSer36 immunoblotting (D) were according to methods.
Although thrombin does not increase Shc-Tyr239/Tyr240 phosphorylation in cardiomyocytes, thrombin decreases the mobility of p66Shc (Fig. 5B). This effect is maximal at 5–10 min and wanes with longer incubation intervals. α1-AR activation with norepinephrine (NE) also leads to a subtle and transient p66Shc mobility shift; the mobility of p66Shc is markedly slowed by PMA. Fig. 5C shows that the thrombin- and PMA-dependent p66Shc band shifts are abrogated by inhibitors of PKC (GF109203X) or MEK (U0126). Of note, AG1478 attenuates the thrombin-dependent p66Shc mobility shift in cardiac fibroblasts, but AG1478 does not block the thrombin-dependent p66Shc mobility shift in cardiomyocytes (compare Figs. 2B and 5C). Collectively, these results identify p66Shc phosphorylation downstream from the PAR-1-dependent ERK activation pathway, both in cardiac fibroblasts (where ERK activation is via EGFR transactivation) and in cardiomyocytes (where ERK is activated via an EGFR-independent mechanism).
The studies next examined whether agonist-dependent p66Shc band shifts in cardiomyocytes can be attributed to p66Shc-Ser36 phosphorylation. Fig. 5D shows that thrombin modestly increases p66Shc-Ser36 phosphorylation and that stronger hypertrophic agonists, such as NE or PMA, induce even greater increases in p66Shc-Ser36 phosphorylation. Of note, at 30 min, NE increases p66Shc-Ser36 phosphorylation without significantly reducing the mobility of the protein (Fig. 5, compare B and D). These results indicate that a p66Shc band shift is not necessarily a reliable surrogate marker of p66Shc-Ser36 phosphorylation in all cell types.
The importance of an agonist-dependent pathway leading to p66Shc-Ser36 phosphorylation is contingent upon the expression of the p66Shc isoform. In this context, Fig. 6 provides important evidence that p66Shc expression is highly regulated in cardiomyocytes. p66Shc immunoreactivity is quite prominent in neonatal cardiomyocyte cultures, in part because of the induction of p66Shc expression during cardiac culture (Fig. 6, compare lanes 5 and 6); p66Shc is detected at lower levels in post-natal day 2 ventricles (lane 3). In contrast, p66Shc is below the limits of detection in adult cardiac preparations (Fig. 6, lanes 2 and 4). These changes in p66Shc expression are paralleled by similar culture- and age-dependent changes in p46Shc expression. In contrast, p52Shc expression is similar in neonatal cardiomyocyte cultures and ventricle and only modestly reduced in the adult preparations. These results establish that p66Shc (like many other signaling proteins that regulate growth and apoptosis) is down-regulated during normal growth and maturation of the ventricle.
FIGURE 6. Developmental and culture-dependent regulation of p66Shc expression in cardiomyocytes.
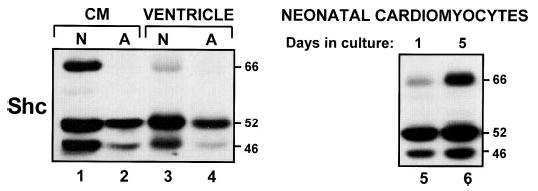
Lysates from day 1 (lane 5) or day 5 (lanes 1 and 6) neonatal (N) cardiomyocyte (CM) cultures, isolated adult (A) cardiomyocytes (lane 2), and post-natal day 2 or adult ventricular myocardial tissue (lanes 3 and 4) were subjected to immunoblot analysis for Shc isoform expression.
Because many signaling proteins that are down-regulated during development are re-expressed during cardiac hypertrophy, we tested the hypothesis that hypertrophic agonists induce p66Shc expression. These studies took advantage of PMT, a direct activator of endogenous free monomeric Gαq subunits; we previously demonstrated that PMT activates PKC isoforms, stimulates ERK, and induces hypertrophy in cardiomyocytes (24). Fig. 7A shows that PMT induces a marked increase in the abundance of p66Shc in association with the activation of ERK. PMT also increases p46Shc and p52Shc, although increases in p52Shc are best appreciated in gels with substantially reduced protein loading (data not shown). Fig. 7B shows that PMT also increases p66Shc-Ser36 phosphorylation in cardiomyocyte cultures to the high level observed in cultures acutely stimulated with PMA. In contrast, PMT triggers a modest increase in ERK activity without increasing Shc isoform expression in cardiac fibroblasts (not significant, n = 3).
FIGURE 7. Gαq protein activation by rPMT increases p66Shc expression and ERK activity via a PKC- and MEK-dependent mechanism in cardiomyocytes.
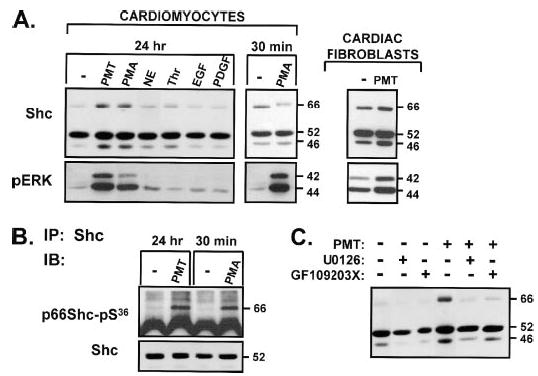
Neonatal rat cardiomyocytes were cultured in 10% fetal calf serum for 24 h and then switched to serum-free medium-containing vehicle, PMT (400 ng/ml), PMA (300 nM), NE (1 μM), thrombin (1 nM), EGF (50 ng/ml), or platelet-derived growth factor (50 ng/ml) alone or in the presence of U0126 or GF109203X (each at 5 μM, starting 30 min prior to stimulations) for an additional 3 days. Extracts were subjected to immunoblotting (IB) for pERK and Shc protein (A and C ). Shc was immunoprecipitated (IP) to probe for p66Shc-Ser36 phosphorylation (B).
The PMT-dependent increase in p66Shc protein abundance is associated with an increase in ERK activity (Fig. 7A). NE, thrombin, EGF, and platelet-derived growth factor do not increase p66Shc or activate ERK under these experimental conditions. These results suggest that Shc expression might be regulated through the MEK-ERK pathway. Fig. 7C provides direct support for this conclusion by showing that the MEK inhibitor U0126 (as well as the PKC inhibitor GF109203X) completely abrogates the PMT-dependent increase in p66Shc expression.
DISCUSSION
These studies implicate Shc isoforms as PAR-1-activated signaling proteins in the heart. We previously demonstrated that PAR-1 couples to an EGFR-dependent MEK-ERK activation pathway in cardiac fibroblasts. These studies identify p52Shc/p46Shc phosphorylation at Tyr239/Tyr240 and Tyr317 as components of this pathway. Although p66Shc shares considerable structural homology with p52Shc and p46Shc (i.e. p66Shc contains the tyrosines that are the targets for growth factor-dependent phosphorylation) and p66Shc is tyrosine-phosphorylated via direct EGFR activation by EGF, we identified little to no thrombin-induced p66Shc-tyrosine phosphorylation. Rather, we demonstrated that PAR-1 increases p66Shc-Ser36 phosphorylation via a mechanism involving EGFR transactivation of the MEK-ERK pathway in cardiac fibroblasts. Of note, thrombin also activates ERK and increases p66Shc-Ser36 phosphorylation in cardiomyocytes, but this is not through an EGFR-dependent pathway and it is not associated with p52/p46Shc tyrosine phosphorylation. However, our results should not be construed as evidence that cardiomyocytes generally lack G protein-coupled receptor-dependent EGFR transactivation pathways leading to Shc tyrosine phosphorylation, because recent studies identify endothelin receptor-dependent activation of ERK via an EGFR transactivation pathway involving Shc tyrosine phosphorylation in cardiomyocyte cultures (25). Studies of the actions of thrombin in this manuscript emphasize the highly contextual nature of PAR-1 signaling in the heart (as schematized in Fig. 8) and the potential pitfalls in extrapolating results across cardiac cell models (or between surrogate model systems and physiologically relevant cell types in the heart).
FIGURE 8. Schematic of Shc isoform regulation in cardiac fibroblasts and cardiomyocytes.
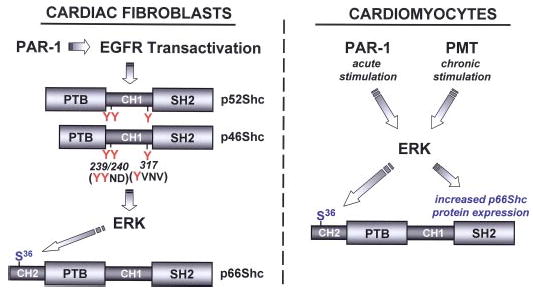
PAR-1 promotes p46/p52Shc phosphorylation at Tyr239/Tyr240 and Tyr317 via an EGFR transactivation pathway that leads to the activation of ERK and p66Shc-Ser36 phosphorylation in cardiac fibroblasts. PAR-1 activates ERK through a pathway that does not involve EGFR transactivation or p46/p52Shc phosphorylation (at Tyr239/Tyr240 or Tyr317) in cardiomyocytes. In cardiomyocytes, acute ERK activation by PAR-1 leads to p66Shc-Ser36 phosphorylation. Chronic Gαq activation (by PMT) leads to increased p66Shc-Ser 36 phosphorylation via a mechanism involving PKC and ERK. See “Discussion” for further details.
Early studies implicated p52/p46Shc phosphorylation events at both Tyr239/Tyr240 and Tyr317 in thrombin-dependent mitogenic responses (15). The focus of most studies was on the similar roles of phospho-Tyr239/Tyr240 and phospho-Tyr317 as docking sites for the Grb2-SH2 domain (26). However, more recent literature suggests that these residues are functionally distinct, with Tyr317 coupling Shc to Fos-dependent activation of the Ras-ERK cascade and proliferation and Tyr239/Tyr240 coupling Shc to Myc-dependent survival pathways. There is evidence that phospho-Tyr239/Tyr240 might even be dispensable for EGFR-dependent Ras/ERK activation. However, it is worth noting that the consequences of Shc-Tyr239/Tyr240 and -Tyr317 phosphorylation generally have been inferred from studies that compare responses to high concentrations of growth factors in cells that heterologously overexpress wild-type or Tyr→Phe-substituted Shc mutants. The relevance of these results to the controls and consequences of Tyr239/Tyr240 and Tyr317 phosphorylation events on native Shc isoforms responding to physiologic stimuli has been questioned (3). Moreover, most studies have been predicated on the assumption that individual phosphorylation events on Shc serve distinct (rather than interdependent) functions. The recent observation that Tyr317 phosphorylation increases the structural rigidity of Shc (and decreases its affinity for binding partners such as the EGFR and protein phosphatase 2A) suggests that phosphorylation events at Tyr239/Tyr240 and Tyr317 impact on many aspects of the adapter function of Shc and play a more complex regulatory role in thrombin-dependent responses (27).
p66Shc inhibits the growth-stimulatory effects of p46/p52Shc and also plays a distinctive role in oxidative stress responses through a mechanism that requires p66Shc-Ser36 phosphorylation. Studies to date have implicated kinase pathways involving MEK (that may or may not involve ERK) or stress-activated protein kinases (such as JNK and p38) in p66Shc-Ser/Thr phosphorylation, depending upon the cell type and inciting stimulus (28, 29). However, many of the conclusions in the literature are based upon studies that track p66Shc mobility shifts. Although this technique provides a reliable readout of overall p66Shc-Ser/Thr phosphorylation, it does not distinguish phosphorylation events at individual Ser/Thr residues. Results reported herein suggest that it is not necessarily a valid surrogate for p66Shc-Ser36 phosphorylation, because the NE-dependent increase in p66Shc-Ser36 phosphorylation in cardiomyocytes is associated with only a paltry p66Shc mobility shift. Conversely, thrombin induces an obvious p66Shc mobility shift in cardiomyocytes; the magnitude of the associated increase in p66Shc-Ser36 phosphorylation is modest in comparison. These results emphasize the importance of studies with PSSAs for unambiguous conclusions. To the best of our knowledge, studies reported herein are the first to directly link activation of a G protein-coupled receptor (PAR-1 in cardiac fibro-blasts and cardiomyocytes, α1-ARs in cardiomyocytes) to p66Shc-Ser36 phosphorylation. The observation that p66Shc-Ser36 phosphorylation is a downstream target of a G protein-coupled receptor-activated pathway involving MEK identifies a novel mechanism to account for cardiac remodeling by MAPKs.
Chronic catecholamine stimulation leads to molecular and structural remodeling of the ventricle and plays an important role in the pathogenesis of heart failure syndromes. To date, most studies have focused on catecholamine-triggered events that influence contraction, growth, and apoptosis of cardiomyocytes rather than the catecholamine-dependent mechanisms that act on interstitial fibroblasts. Our studies identify a β2-AR pathway involving EGFR transactivation that leads to ERK activation in neonatal cardiac fibroblast cultures. A similar EGFR transactivation pathway is reported to link β2-ARs to increased DNA synthesis in serially passaged adult cardiac fibroblasts (23). At first glance, β2-ARs and PAR-1 might appear to share a similar EGFR transactivation-MEK-ERK pathway in cardiac fibroblasts. However, more detailed studies expose important differences that deserve emphasis. 1) β2-ARs promote a relatively modest increase in ERK activity via a PTX-sensitive pathway. PAR-1 induces a substantially more robust increase in ERK activity that is largely PTX-insensitive (and is presumably mediated by Gq and/or G12/13). Of note, β2-ARs induce a very robust increase in cAMP accumulation, whereas PAR-1 does not couple to cAMP accumulation in cardiac fibroblasts. A potential role for the cAMP/PKA-dependent pathway to limit β2-AR activation of ERK is the focus of ongoing studies. 2) The PAR-1-dependent EGFR transactivation pathway activates AKT; β2-ARs do not activate the anti-apoptotic AKT pathway under these conditions. 3) PAR-1-dependent EGFR transactivation leads to p46/p52Shc tyrosine phosphorylation and p66Shc-Ser36 phosphorylation; β2-ARs activate ERK via a mechanism that does not require the adapter function of Shc and does not lead to p66Shc-Ser36 phosphorylation. These results suggest that PAR-1 and β2-ARs play very distinct roles in the control of cardiac fibroblast-induced ventricular remodeling.
p52Shc is a ubiquitous protein detected in various cardiac cell preparations. In contrast, p66Shc is readily detected in neonatal cardiac preparations, but it is at the limits of detection in the adult ventricle. p66Shc is up-regulated by PMT (as part of a Gαq-dependent hypertrophy program) in neonatal cardiomyoytes, whereas PMT does not lead to a significant change in p66Shc expression in cardiac fibroblasts. These results are consistent with previous publications showing that Shc protein expression is prominent in the developing cardiovascular system during embryogenesis and that p66Shc is essentially undetectable in the adult ventricle. Of note, Cesselli et al. (13) previously identified increased p66Shc expression (in association with an increase in cardiomyocyte apoptosis) in the canine-pacing induced heart failure model. Future studies to determine whether hypertrophic stimuli (and/or pathologic insults) also induce p66Shc expression in adult rodent cardiomyocytes (and human hypertrophy and/or heart failure models) will be critical to assessing the functional relevance of the findings reported in this study performed on neonatal rat cardiomyocytes. In addition, it will be important to identify mechanisms that regulate p66Shc expression in the heart. In this regard, Ventura et al. (4) have reported that p66Shc expression is silenced through histone deacetylation and cytosine methylation. It is important to note that recent studies firmly implicate class II histone deacetylations as negative regulators of cardiac hypertrophy that can be neutralized by phosphorylation events triggered by Gαq-coupled receptors. Although preliminary experiments failed to identify an effect of the histone deacetylation inhibitor trichostatin A to increase p66Shc expression in cardiomyocyte cultures, the relative roles of histone acetylation and promoter methylation as mechanisms to up- or down-regulate p66Shc expression certainly deserve further scrutiny. There also is recent evidence that p38/MAPK-dependent phosphorylation of p66Shc at Ser54 and Thr386 leads to an increase in the stability of the protein and an increase in its abundance (30). Either process could contribute to the Gαq-induced increase in p66Shc expression in cardiomyocytes. Finally, it is worth noting that this study implicates a MEK-ERK pathway in both the induction of p66Shc expression and the stimulus-induced increase in p66Shc-Ser36 phosphorylation. Studies to determine whether these MEK/ERK-dependent effects on p66Shc potentially undermine the otherwise protective effects of the MEK-ERK pathway are ongoing.
There is ample evidence that the natural history of cardiac hypertrophy is to transition to heart failure, at least in part through mechanisms involving cardiomyocyte apoptosis. However, progress toward identifying the molecular signals that execute the apoptosis program in hypertrophied cardiomyocytes has been slow. We previously reported that persistent Gαq activation (by PMT) can result in enhanced cardiomyocyte susceptibility to apoptosis by repressing basal Akt phosphorylation and impairing Akt activation by survival factors (24). These studies expose an alternative mechanism that is predicted to link Gαq stimulation to enhanced cardiomyocyte apoptosis and dilated cardiomyopathy involving an increase in p66Shc expression. This formulation is consistent with recent evidence that p66Shc localizes to mitochondria where it functions as a reactive oxygen species sensor (10, 11). Mitochondrial events play particularly prominent roles in Gαq-dependent apoptosis (31). p66Shc, acting as a candidate hypertrophy-induced effector of the cardiomyocyte Gαq signaling pathway, provides an attractive novel molecular target for heart failure therapy.
Footnotes
This work was supported by United States Public Health Service-NHLBI National Institutes of Health (NIH) Grants HL-64639 and HL77860 (to S. F. S.) and NIAID NIH Grant AI038396 (to B. A. W.).
The abbreviations used are: PAR-1, protease-activated receptor-1; PTX, pertussis toxin; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; SH, Src homology; PMA, phorbol 12-my-ristate 13-acetate; PSSA, phosphorylation site-specific antibodies; AR, adrenergic receptor; NE, norepinephrine; PKC, protein kinase C; PMT, Pasteurella multocida toxin.
References
- 1.Sabri A, Short J, Guo J, Steinberg SF. Circ Res. 2002;91:532–539. doi: 10.1161/01.res.0000035242.96310.45. [DOI] [PubMed] [Google Scholar]
- 2.Pellegrini M, Pacini S, Baldari CT. Apoptosis. 2005;10:13–18. doi: 10.1007/s10495-005-6057-8. [DOI] [PubMed] [Google Scholar]
- 3.Ravichandran KS. Oncogene. 2001;20:6322–6330. doi: 10.1038/sj.onc.1204776. [DOI] [PubMed] [Google Scholar]
- 4.Ventura A, Luzi L, Pacini S, Baldari CT, Pelicci PG. J Biol Chem. 2002;277:22370–22376. doi: 10.1074/jbc.M200280200. [DOI] [PubMed] [Google Scholar]
- 5.Pelicci G, Lanfrancone L, Salcini AE, Romano A, Mele S, Grazia BM, Segatto O, Di Fiore PP, Pelicci PG. Oncogene. 1995;11:899–907. [PubMed] [Google Scholar]
- 6.Pacini S, Pellegrini M, Migliaccio E, Patrussi L, Ulivieri C, Ventura A, Carraro F, Naldini A, Lanfrancone L, Pelicci P, Baldari CT. Mol Cell Biol. 2004;24:1747–1757. doi: 10.1128/MCB.24.4.1747-1757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 8.Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, Milia E, Padura IM, Raker VA, Maccarana M, Petronilli V, Minucci S, Bernardi P, Lanfrancone L, Pelicci PG. Oncogene. 2002;21:3872–3878. doi: 10.1038/sj.onc.1205513. [DOI] [PubMed] [Google Scholar]
- 9.Nemoto S, Finkel T. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 10.Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, Martin-Padura I, Pelliccia G, Trinei M, Bono M, Puri C, Tacchetti C, Ferrini M, Mannucci R, Nicoletti I, Lanfrancone L, Giorgio M, Pelicci PG. J Biol Chem. 2004;279:25689–25695. doi: 10.1074/jbc.M401844200. [DOI] [PubMed] [Google Scholar]
- 11.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 12.Graiani G, Lagrasta C, Migliaccio E, Spillmann F, Meloni M, Madeddu P, Quaini F, Padura IM, Lanfrancone L, Pelicci P, Emanueli C. Hypertension. 2005;46:433–440. doi: 10.1161/01.HYP.0000174986.73346.ba. [DOI] [PubMed] [Google Scholar]
- 13.Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Circ Res. 2001;89:279–286. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- 14.Collins LR, Ricketts WA, Olefsky JM, Brown JH. Oncogene. 1997;15:595–600. doi: 10.1038/sj.onc.1201220. [DOI] [PubMed] [Google Scholar]
- 15.Ricketts WA, Brown JH, Olefsky JM. Mol Endocrinol. 1999;13:1988–2001. doi: 10.1210/mend.13.12.0385. [DOI] [PubMed] [Google Scholar]
- 16.Wilson BA, Ponferrada VG, Vallance JE, Ho M. Infect Immun. 1999;67:80–87. doi: 10.1128/iai.67.1.80-87.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faisal A, El-Shemerly M, Hess D, Nagamine Y. J Biol Chem. 2002;277:30144–30152. doi: 10.1074/jbc.M203229200. [DOI] [PubMed] [Google Scholar]
- 18.van Biesen T, Hawes BE, Luttrell DK, Krueger KM, Touhara K, Porfiri E, Sakaue M, Luttrell LM, Lefkowitz RJ. Nature. 1995;376:781–784. doi: 10.1038/376781a0. [DOI] [PubMed] [Google Scholar]
- 19.Jorgensen K, Skrede M, Cruciani V, Mikalsen SO, Slipicevic A, Florenes VA. Biochem Biophys Res Commun. 2005;329:266–274. doi: 10.1016/j.bbrc.2005.01.143. [DOI] [PubMed] [Google Scholar]
- 20.Bapat S, Verkleij A, Post JA. FEBS Lett. 2001;499:21–26. doi: 10.1016/s0014-5793(01)02511-x. [DOI] [PubMed] [Google Scholar]
- 21.Grammer TC, Blenis J. Oncogene. 1997;14:1635–1642. doi: 10.1038/sj.onc.1201000. [DOI] [PubMed] [Google Scholar]
- 22.Tapinos N, Rambukkana A. Proc Natl Acad Sci U S A. 2005;102:9188–9193. doi: 10.1073/pnas.0501196102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Eckhart AD, Eguchi S, Koch WJ. J Biol Chem. 2002;277:32116–32123. doi: 10.1074/jbc.M204895200. [DOI] [PubMed] [Google Scholar]
- 24.Sabri A, Wilson BA, Steinberg SF. Circ Res. 2002;90:850–857. doi: 10.1161/01.RES.0000016165.23795.1F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kodama H, Fukuda K, Takahashi T, Sano M, Kato T, Tahara S, Hakuno D, Sato T, Manabe T, Konishi F, Ogawa S. J Mol Cell Cardiol. 2002;34:139–150. doi: 10.1006/jmcc.2001.1496. [DOI] [PubMed] [Google Scholar]
- 26.Gotoh N, Toyoda M, Shibuya M. Mol Cell Biol. 1997;17:1824–1831. doi: 10.1128/mcb.17.4.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suenaga A, Kiyatkin AB, Hatakeyama M, Futatsugi N, Okimoto N, Hirano Y, Narumi T, Kawai A, Susukita R, Koishi T, Furusawa H, Yasuoka K, Takada N, Ohno Y, Taiji M, Ebisuzaki T, Hoek JB, Konagaya A, Kholodenko BN. J Biol Chem. 2004;279:4657–4662. doi: 10.1074/jbc.M310598200. [DOI] [PubMed] [Google Scholar]
- 28.Okada S, Kao AW, Ceresa BP, Blaikie P, Margolis B, Pessin JE. J Biol Chem. 1997;272:28042–28049. doi: 10.1074/jbc.272.44.28042. [DOI] [PubMed] [Google Scholar]
- 29.Le S, Connors TJ, Maroney AC. J Biol Chem. 2001;276:48332–48336. doi: 10.1074/jbc.M106612200. [DOI] [PubMed] [Google Scholar]
- 30.Khanday FA, Yamamori T, Mattagajasingh I, Zhang Z, Bugayenko A, Naqvi A, Santhanam L, Nabi N, Kasuno K, Day BW, Irani K. Mol Biol Cell. 2005 doi: 10.1091/mbc.E05-06-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams JW, Pagel AL, Means CK, Oksenberg D, Armstrong RC, Brown JH. Circ Res. 2000;87:1180–1187. doi: 10.1161/01.res.87.12.1180. [DOI] [PubMed] [Google Scholar]