

Abstract
Recent reports have shown that several heterotrimeric protein-coupled receptors that signal through Gαq can induce Rho-dependent responses, but the pathways that mediate the interaction between Gαq and Rho have not yet been identified. In this report we present evidence that Gαq expressed in COS-7 cells coprecipitates with the Rho guanine nucleotide exchange factor (GEF) Lbc. Furthermore, Gαq expression enhances Rho-dependent responses. Coexpressed Gαq and Lbc have a synergistic effect on the Rho-dependent rounding of 1321N1 astrocytoma cells. In addition, serum response factor-dependent gene expression, as assessed by the SRE.L reporter gene, is synergistically activated by Gαq and Rho GEFs. The synergistic effect of Gαq on this response is inhibited by C3 exoenzyme and requires phospholipase C activation. Surprisingly, expression of Gαq, in contrast to that of Gα12 and Gα13, does not increase the amount of activated Rho. We also observe that Gαq enhances SRE.L stimulation by activated Rho, indicating that the effect of Gαq occurs downstream of Rho activation. Thus, Gαq interacts physically and/or functionally with Rho GEFs; however this does not appear to lead to or result from increased activation of Rho. We suggest that Gαq-generated signals enhance responses downstream of Rho activation.
Heterotrimeric G proteins are activated by exchange of GDP for GTP in response to agonist occupation of G protein-coupled receptors (GPCRs).1 In contrast, low molecular weight or small G proteins are activated by guanine nucleotide exchange factors (GEFs). Until fairly recently there was no direct evidence or molecular mechanism for GPCR agonists activating small G proteins. Pathways for activation of the small G protein Ras in response to receptor tyrosine kinases and GPCRs have now been clearly delineated (reviewed in Ref. 1). In addition, the observation that GPCR agonists such as thrombin, bombesin, and lysophosphatidic acid (LPA) activate cytoskeletal responses through pathways involving the small G protein Rho (2) has now been explored mechanistically (3–9). Several lines of evidence suggest that GPCR-stimulated, Rho-dependent responses are mediated through activation of the heterotrimeric G proteins G12 and G13 (reviewed in Refs. 10 and 11), which in turn associate with and may activate Rho GEFs (12, 13).
The Rho family of small G proteins mediate numerous cytoskeletal responses, including stress fiber formation, focal adhesion assembly, and neurite retraction. In addition, many Rho family small G proteins have been demonstrated to activate downstream kinases and to stimulate gene transcription (14–17). Studies using the c-fos promoter demonstrate that Rho can activate transcription through the serum response element (SRE), independent of effects on the ternary complex factor (16). The ternary complex factor-independent SRE (SRE.L) has thus been frequently used as a readout for Rho-mediated transcriptional responses. The Rho effector which links Rho to the SRE has not yet been identified; however several lines of evidence indicate that Rho kinase is not required (18–20) and that protein kinase N (21) or protein kinase C (PKC)-related serine/ threonine kinase 2 (22) may contribute significantly to this response.
Most of the GPCRs that have been shown to activate Rho-dependent cytoskeletal rearrangements, gene expression, or cellular contractility can couple to Gαq and thereby activate phospholipase C (PLC) and PKC (9, 23, 24). Several reports indicate that activation of Gαq and its downstream effectors is insufficient or unnecessary to induce Rho-mediated signals (3, 9, 25–29). For example, LPA and thrombin induce Rho-mediated responses in Gαq/11-deficient cells (9, 23). On the other hand, the α1-adrenergic, M1 muscarinic, and metabotropic glutamate receptors were reported to require Gαq/11 to induce Rho-mediated changes in SRE.L-reporter gene expression (23, 24). In addition, some studies have shown that expression of activated mutants of Gαq leads to Rho-mediated responses such as cytoskeletal rearrangements (30, 31), transcriptional activation of the atrial natriuretic factor gene (24), and SRE.L response element activation (23, 32). These studies suggest that stimulation of Gαq, like Gα12/13, might result in Rho activation. There are, however, no published studies demonstrating that Gαq signaling activates Rho or examining how Gαq pathways regulate Rho-dependent responses.
It has been proposed that Gα12/13-coupled receptors activate Rho via interaction of the Gα subunit with Rho-specific GEFs such as p115RhoGEF (13, 30, 32; also see Refs. 10 or 11 for review). Two Rho GEFs, p115RhoGEF and PDZ-RhoGEF, were shown to coprecipitate with Gα12/13 (12, 13, 33). Furthermore, the exchange activity of purified p115RhoGEF was activated by Gα13 (12), and downstream signaling to cytoskeletal and transcriptional responses was enhanced by coexpression of Gα13 with p115RhoGEF (32). In addition, catalytically inactive mutants of p115RhoGEF (32) and PDZ-RhoGEF (13) have been shown to attenuate SRE.L responses due to Gα12 and Gα13, thrombin, or LPA. Likewise, the catalytically inactive mutants of the Rho GEFs lymphoid blast crisis (Lbc) and p115RhoGEF blocked the cytoskeletal response induced by Gα12 or thrombin (30). These findings support a model in which the α subunits of G proteins interact with Rho GEFs to activate Rho-mediated signaling pathways.
The studies reported here were initiated to elucidate the mechanism by which Gαq signaling stimulates Rho-dependent responses. Based on the findings of direct interaction of Gα12/13 with Rho GEFs, we hypothesized that Gαq might also interact with Rho GEFs. In this report we demonstrate both a physical and a functional interaction between Gαq and the Rho GEF Lbc. Surprisingly, we report that these interactions do not lead to or result from increased activation of Rho. Rather, the interaction of Gαq with Rho-dependent pathways requires generation of PLC-mediated second messengers and occurs downstream of Rho activation.
EXPERIMENTAL PROCEDURES
Plasmids
Plasmids encoding the G αq, Gα12, and Gα13 subunits were the generous gifts of M. Simon and J. Exton. The GαsRC plasmid was provided by M. Farquar. The SRE.L-luciferase reporter plasmid was provided by K. Kaibuchi. The plasmid encoding Green Lantern green fluorescent protein (GFP), which was used to normalize transfections, was acquired from Life Technologies, Inc. The nuclear GFP plasmid used in microinjection studies was a gift from G. Wahl. The cDNA plasmids for onco- and proto-Lbc are as described previously (34). Proto-Lbc was c-Myc-tagged by subcloning the proto-Lbc cDNA into the pJ3M vector which contains a 5′ c-Myc tag. The resultant cDNA construct was subcloned into pMT3 for efficient mammalian expression. The c-Myc-tagged p115RhoGEF was provided by M. Hart. L. Heasley kindly provided the PLCβCT (35), which we subcloned into the pBJ mammalian expression vector using standard techniques. pGEX-2T vector encoding a glutathione S-transferase (GST) fusion protein containing amino acids 7–89 of Rhotekin, which is the Rho-binding domain (RBD), was kindly provided by M. Schwartz (36).
Cell Culture and Transfection
COS-7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) in a 37 °C, humidified incubator with 10% CO2. One day prior to transfection cells were set at 4 × 104 cells/ml. Cells were transfected overnight by calcium phosphate precipitation, then washed and incubated in serum-free DMEM supplemented with 1 mg/ml fatty acid-free bovine serum albumin, penicillin, and streptomycin. 1321N1 cells were maintained in DMEM supplemented with 5% fetal calf serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) in a 37 °C, humidified incubator with 10% CO2. For microinjection the cells were set onto 12-mm round glass coverslips at a density of ~1.0 × 104 cells/slip. Where indicated, cells were incubated with recombinant Pasteurella multocida toxin (PMT) (37) for 24 h at doses of 100–200 ng/ml serum-free medium. The Rho kinase inhibitor Y-27632 was provided by Welfide Corp. (Hirakata-shi, Osaka, Japan) and was used at 10 μM for 24 h before the luciferase harvest. Phorbol 12-myristate 13-acetate (PMA) and bisin-doylmaleimide (GF109203X) were purchased from Calbiochem and used at 100 m M and 3 μM, respectively, for 24 h prior to harvesting luciferase.
Coprecipitations
24–48 h after transfection of COS-7 cells with epitope-tagged Lbc, or p115RhoGEF constructs (or vector) and Gα cDNA plasmids, the cells were rinsed with phosphate-buffered saline and then lysed in immunoprecipitation buffer containing: 20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, 0.7% Triton X-100, 1 mM dithiothreitol, 1 mM p-nitrophenyl phosphate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 7.5 μg/ml aprotinin. Anti-FLAG M2 monoclonal (Sigma) or anti-c-Myc (Santa Cruz Biotechnology) and protein G-Sepharose beads (Amersham Pharmacia Biotechnology) were used to precipitate the epitope-tagged Lbc or p115RhoGEF and associated proteins. The precipitates were washed four times and then boiled in Laemmli sample buffer to elute, and the resultant samples were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). Proteins were electrophoretically transferred to Immobilon-P membranes (Millipore), then immunoblotted with Gα-specific antibodies (anti-G αq, Gα12 and Gα13 from Santa Cruz Biotechnology; anti-G αq from Upstate Biotechnology).
Rho Activation Assay
GST-Rhotekin RBD was produced in Escherichia coli (DH5α strain) transformed with pGEX-2T-Rhotekin RBD. Bacterial cultures were grown to A600 = 0.6 and then induced with 0.1 mM isopropyl-β-D-thiogalactopyranoside overnight at room temperature. The bacteria were harvested in lysis buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1% Nonidet P-40, 10% glycerol) containing protease inhibitors and were lysed by sonication (60 × 1 s on ice). After centrifugation (15 min @ 12,000 × g), the GST-Rhotekin-RBD was collected from the clarified supernatant by rocking at 4 °C for 45 min with glutathione-Sepharose 4B (Amersham Pharmacia Biotech). The Sepharose was washed three times with assay buffer, resuspended in fresh buffer, and aliquots snap-frozen in liquid N2 for future use.
4–24 h after transfection cells were rinsed with Tris-buffered saline and lysed in buffer containing: 50 mM Tris HCl, pH 7.4, 10% glycerol, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 100 mM NaCl, 5 mM MgCl2, 10 μ g/ml aprotinin, and 10 μg/ml leupeptin. The lysates were clarified by brief centrifugation and then incubated with the Sepharose-bound GST-Rhotekin-RBD for 45 min at 4 °C. The beads and precipitated proteins were washed four times in cold lysis buffer and then the proteins were eluted by boiling in Laemmli buffer and separated by SDS-PAGE. Proteins were transferred to Immobilon-P membranes (Millipore) and immunoblotted with Rho-specific antibodies (Santa Cruz Biotechnology) followed by detection with horseradish peroxidase-conjugated secondary antibodies. Changes in Rho were quantified using an LKB laser densitometer. The precipitated Rho was normalized to the Rho present in whole cell lysate.
SRE.L-mediated Gene Expression
COS-7 cells were set onto six-well plates and transfected with the plasmids of interest or the appropriate vectors along with the SRE.L-luciferase reporter plasmid. 24–48 h after transfection luciferase expression was assessed as described previously (38). In selected experiments cells were cotransfected with a plasmid encoding the GFP under a constitutive promoter, to allow normalization of transfection efficiency and cell viability. A portion of cell lysate was reserved, and fluorescent emission from these samples was read in a fluorescence plate reader (CTI Data model 7600).
Microinjection and Morphology
1321N1 cells were set on glass coverslips and microinjected as described in Majumdar et al. (30). Injected cells were detected by expression of nuclear GFP and actin morphology assessed by staining with rhodamine-conjugated phalloidin (Molecular Probes).
Inositol Phosphate Accumulation
After calcium phosphate transfection, COS-7 cells were maintained in serum-free medium. 10 h after the transfection reagents were washed out, 1–2 μCi of myo[3H]inositol/ml was added, and the cells were allowed to label overnight. The cells were washed extensively to remove unincorporated myo[3H]inositol and then incubated with 10 mM LiCl for 30 min. Reactions were terminated by the addition of 10% trichloroacetic acid. The acidic lysates were neutralized by ether extraction. Total [3H]inositol (poly)phosphates were eluted by formate Dowex anion exchange chromatography with 1 M NH4COOH, 0.1 M HCOOH.
RESULTS
Gαq Coimmunoprecipitates with Lbc but Not p115RhoGEF
To determine whether the Gαq protein associates with Rho GEFs, we cotransfected COS-7 cells with Myc-tagged proto-Lbc or p115RhoGEF and constitutively active forms of either Gαq, Gα12, or Gα13. Analysis of whole cell lysates by Western blotting with antibodies to specific Gα subunits confirmed that Gαq, Gα12, and Gα13 were overexpressed in the transfected cells (Fig. 1A). The Rho GEFs were immunoprecipitated with anti-Myc antibodies and the precipitated proteins analyzed by Western blotting. Lbc or p115RhoGEF was present in the immunoprecipitates from all samples transfected with the corresponding cDNA (Fig. 1B). Immunoblotting with antibodies to specific Gα proteins demonstrated that Gαq was present in Lbc precipitates (Fig. 1C). In contrast, the levels of Gαq detected in p115 precipitates were not significantly above that of cells transfected with control vector in most experiments. Activated Gα12 and Gα13, were detected at similar levels in either Lbc or p115RhoGEF immunoprecipitates. To further confirm the specificity of these coprecipitations, we examined the ability of Gαs to coprecipitate with Lbc. Although the overexpressed Gαs was detected in transfected COS-7 cells, it did not coprecipitate with Lbc (Fig. 2).
Fig. 1. Selective precipitation of Gαq with Lbc.
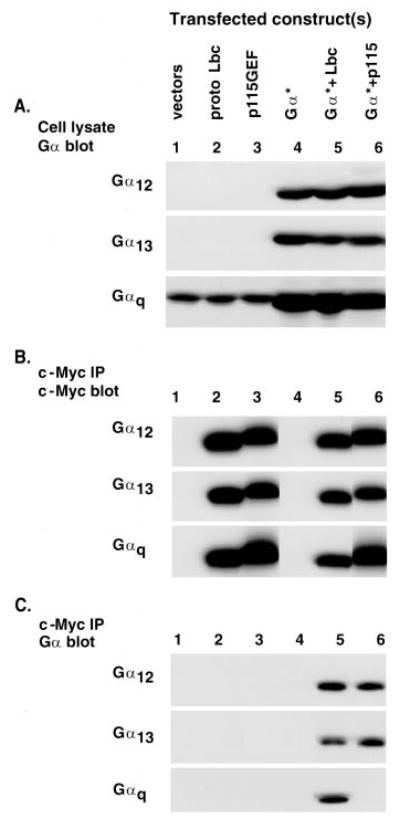
COS-7 cells were transfected with empty vector, plasmid encoding Myc-tagged proto-Lbc, or plasmid for Myc-tagged p115RhoGEF and with empty vector or plasmid encoding the GTPase-deficient Gα subunit of either Gαq, Gα12, or G α13. A, whole cell lysates from the transfected COS-7 cells were prepared and Western blotted for the appropriate Gα subunit, to ensure protein expression. B, cell extracts were immunoprecipitated (IP) with anti-Myc (9E10 mouse monoclonal) antibody and then Western blotted with c-Myc antibody to visualize the epitope-tagged proto-Lbc and p115RhoGEF. C, immunoprecipitates, as in B, were blotted with antibodies against the appropriate Gα subunit. Blots are representative of two to three experiments, each performed in duplicate.
Fig. 2. Gαs does not coprecipitate with Lbc.

COS-7 cells were transfected with empty vector or plasmid encoding Myc-tagged proto-Lbc along with empty vector or plasmid encoding the GTPase-deficient Gα subunit of either G αq or Gαs. A, whole cell lysates from the transfected COS-7 cells were prepared and Western blotted for the appropriate Gα subunit, to ensure protein expression. B, cell extracts were immunoprecipitated (IP) with anti-Myc (9E10 mouse monoclonal) antibody, which recognizes the epitope-tagged proto-Lbc, and then Western blotted with antibodies against the appropriate Gα subunit. Blots are representative of two experiments, each performed in duplicate.
Gαq Does Not Activate Rho
To examine effects of Gαq on Rho activation we expressed the Gαq subunit in COS-7 cells and used the RBD of a Rho effector (Rhotekin) to affinity precipitate active Rho (36, 39). In initial studies we compared the ability of the RBD of Rhotekin to precipitate constitutively active RhoA versus wild-type RhoA expressed in COS-7 cells. As shown in Fig. 3A, constitutively active RhoA was efficiently affinity-precipitated, while wild-type RhoA was not.
Fig. 3. Gαq does not induce activation of RhoA.
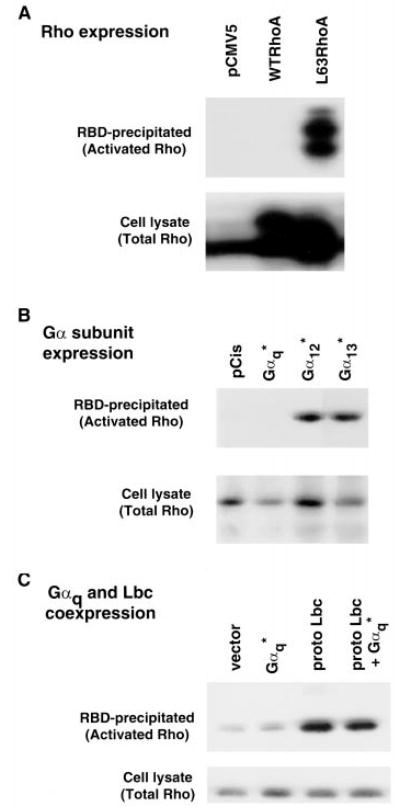
A, COS-7 cells were transfected with hemagglutinin-tagged wild-type RhoA, hemag-glutinin-tagged L63RhoA or empty vector (pCMV5). Cell lysates were prepared, and a portion of this whole cell lysate was subjected to SDS-PAGE and Western blotting with anti-RhoA antibody to measure total Rho (lower panel). The remaining portion of the lysates was affinity-precipitated with the RBD of Rhotekin and Western blotted with anti-RhoA. Only activated Rho binds to the Rhotekin RBD (top panel). The doublet pattern is present because the hemagglutinin-tagged RhoA migrates slower than the endogenous RhoA. B, COS-7 cells were transfected with pCis vector or various Gα subunits. The cell extracts were analyzed as in A. C, after transfection of COS-7 with Gαq, proto-Lbc or both active or total Rho was detected in cell extracts as in A.
Expression of constitutively active Gα12 or Gα13 in COS-7 cells led to increases in the amount of RBD-precipitable, active Rho (Fig. 3B). In contrast expression of constitutively active Gαq did not increase activation of Rho. The inability of Gαq to activate Rho could be due to a limiting concentration of its cognate exchange factor, suggested by our previous experiments to be Lbc. We therefore cotransfected Lbc with Gαq and assessed the amount of active Rho. Lbc led to Rho activation (Fig. 3C) as did other Rho GEFs (p115RhoGEF and PDZ-Rho-GEF; data not shown). However Gαq did not increase the ability of Lbc to activate Rho (Fig. 3C). A range of concentrations of the Gαq and Lbc plasmids were tested, but at no concentrations did Gαq enhance Lbc-mediated Rho activation (data not shown). In prior studies we have established that [35S]GTPγS binding can be used to assess activation of Rho (40). Expression of Gαq alone or in combination with Lbc failed to promote increased [35S]GTPγS binding to Rho in lysates from COS-7 cells (data not shown).
Gαq and RhoGEFs Synergistically Activate Rho-dependent SRE.L-mediated Gene Transcription
The literature provides compelling evidence that Gαq signals through Rho-dependent pathways. A commonly used measure of Rho-dependent signaling is stimulation of SRE.L-mediated gene expression as originally described by Treisman’s laboratory (16). Both activated Gαq and Lbc induced ~5-fold increases in transcription of luciferase from an SRE.L-luciferase reporter gene in COS-7 cells. Strikingly, when GαqRC and proto-Lbc were coexpressed the SRE.L-luciferase was synergistically activated (~25-fold). The data in Fig. 4 also demonstrate that the cooperative effect of Gαq was selective, since neither Gα12 nor Gα13 synergized with Lbc to induce SRE.L-luciferase expression. This differential effect of Gαq was maintained over a wide range of doses of the Gα subunit plasmids (from 0.5–2.5 μg/well; data not shown) and thus was not dependent on high levels of Gα subunit expression. Notably, Gαq also synergized with p115RhoGEF to activate the SRE.L response (Fig. 4) despite its minimal ability to coimmunoprecipitate with this Rho GEF. Conversely, Gα12 and Gα13 association with Lbc (Fig. 1C) did not lead to synergistic SRE.L activation (Fig. 4).
Fig. 4. Gαq synergizes with Rho GEFs to induce Rho-dependent SRE.L-mediated gene expression.
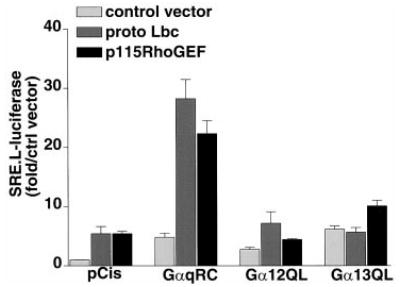
COS-7 cells were transfected with an SRE.L-luciferase reporter plasmid along with the empty pCis vector or with pCis containing cDNA for the different Gα subunits as on the x axis. Cells were cotransfected with empty vector (light gray), proto Lbc (medium gray), or p115RhoGEF (black). Luciferase was quantified 24–48 h after transfection. Data are averages ± S.E. of the fold over vector control for three experiments, each performed in triplicate.
To determine whether activation of endogenous Gαq could induce transcriptional activation of the SRE.L reporter, we used a toxin that specifically activates Gαq. PMT has been demonstrated to cause transient activation of Gαq and downstream effectors in Xenopus oocytes (41). Furthermore, this toxin has been used to investigate Gαq-mediated signaling pathways in mammalian cells (42, 43). Treatment of COS-7 cells with 100–200 ng/ml PMT for 24 h activated PLC as evidenced by a 5-fold increase in total inositol phosphate formation. The ability of PMT to stimulate SRE.L-mediated gene expression and to synergize with Lbc (Fig. 5) confirmed that endogenous Gαq can also cooperate with Rho GEF-activated pathways.
Fig. 5. Activation of endogenous Gαq synergizes with proto-Lbc on SRE.L-mediated gene expression.
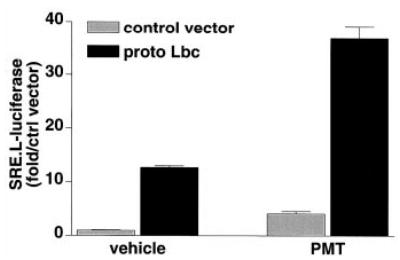
COS-7 cells were trans-fected with a control vector or proto-Lbc along with an SRE.L-luciferase reporter plasmid. Following transfection cells were maintained in serum-free medium (vehicle) or in serum-free medium containing 100 ng/ml recombinant PMT for 24 h, then harvested and assayed for luciferase activity. Data are representative of three experiments, each performed in triplicate.
The Rho dependence of Gαq effects on SRE.L-mediated gene expression was examined by using C3 exoenzyme to inhibit Rho function. Expression of C3 exoenzyme (EFC3) completely inhibited the induction of SRE.L-luciferase by Gαq or Lbc. The synergy between Gαq and Lbc was also abolished, implicating Rho function in mediating this synergistic response (Fig. 6). To confirm that the inhibitory effect of C3 did not reflect its cytotoxicity, luciferase expression was normalized to that of co-transfected GFP. These experiments demonstrated that expression of C3 exoenzyme did not alter the cellular accumulation of GFP (data not shown). In addition, the effect of another Rho family small G protein, Cdc42, on SRE.L-luciferase expression was not attenuated by C3. These data argue against nonspecific effects of C3 exoenzyme on cell viability or on luciferase expression.
Fig. 6. The synergistic SRE.L responses to Gαq and Lbc in COS-7 cells are Rho-dependent.
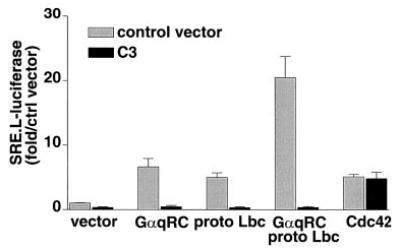
COS-7 cells were transfected with an SRE.L-luciferase reporter plasmid and the expression plasmids for the proteins indicated on the x axis. Black bars indicate cotransfection with an expression plasmid for C3 exoenzyme; gray bars are vector-transfected controls. Luciferase was quantified 24–48 h after transfection. Data are averages ± S.E. of the fold increase over vector control for three experiments, each performed in triplicate.
Activated Gαq and Lbc Cooperate to Induce Cytoskeletal Changes
We previously reported that 1321N1 human astrocytoma cells show a marked cytoskeletal response to thrombin in which the cell processes retract and the cell bodies become rounded. We also showed that activated Gα subunits or Rho GEFs could induce cell rounding (30). Although we reported that the effect of thrombin was Rho- and Gα12/13-dependent (30), we observed a significant response to Gαq. We used this preparation to demonstrate interactions between Gαq and Lbc in another cell system and with a different functional readout. Cells were microinjected with plasmid vectors containing cDNA for GαqRC, proto-Lbc, or both and coinjected with cDNA for nuclear GFP to identify injected cells. We established concentrations of GαqRC or proto-Lbc that alone induced rounding in ~20% of the injected cells (versus 8% of cells injected with pCis vector; Fig. 7). When these concentrations of GαqRC and proto-Lbc were coinjected their effect was more than additive, with nearly 50% of the injected cells manifesting the rounded morphology (Fig. 7). In contrast to what was observed with Gαq, microinjected Gα12QL, at a concentration that elicited 20% rounding alone, did not synergize with proto-Lbc (Fig. 7).
Fig. 7. Lbc selectively synergizes with Gαq to induce astrocytoma cell rounding.
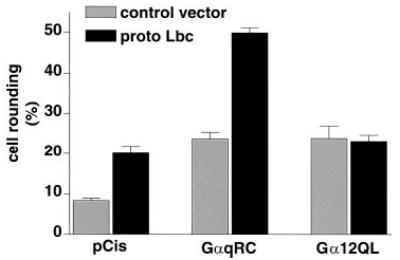
1321N1 human astrocytoma cells were microinjected with empty vector (pCis) or with Gα subunits as indicated on the x axis along with control vector or plasmid encoding proto-Lbc. All cells were coinjected with plasmid for nuclear GFP to allow identification of injected cells. Actin was visualized by staining with rhodamine-conjugated phalloidin. Data shown are the percent of microinjected cells that have a rounded appearance. Values are the average ± S.E. from at least three experiments with a minimum of 50 microinjected cells per condition per experiment.
Gαq Synergizes with Rho
Because Gαq synergized with Rho GEFs but did not elicit Rho activation, we considered the possibility that Gαq cooperates downstream of Rho activation in effecting Rho-dependent responses. Activated Gαq, Gα12, Gα13, or constitutively active RhoA was expressed in COS-7 cells, and SRE.L-mediated gene expression was assessed. Each of these Gα subunits or activated RhoA induced SRE.L-luciferase expression. Neither Gα12 nor Gα13 synergized with cotransfected RhoA. In contrast, Gαq showed substantial synergy with activated RhoA (Fig. 8). These data indicate that the site at which Gαq synergizes with the Rho pathway to enhance SRE.L-luciferase expression is at or downstream of Rho activation.
Fig. 8. Gαq synergizes with Rho to induce SRE.L-mediated gene expression.
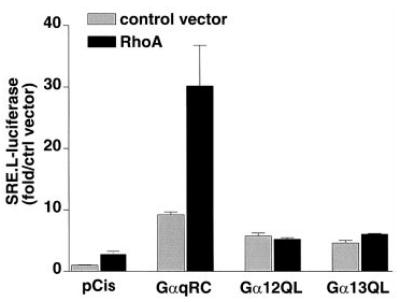
COS-7 cells were transfected with an SRE.L-luciferase reporter plasmid and the expression plasmids for constitutively active Gα subunits along with control vector or plasmid encoding activated RhoA. Luciferase was quantified 24 h after transfection. Values are the average ± S.E. of the fold increase over vector control for three experiments, each performed in triplicate.
Although many Rho effectors have been identified, effectors mediating Rho-dependent SRE.L activation have not been clearly delineated. To determine whether Rho kinase activity is required for the SRE.L response to Gαq, we treated COS-7 cells with the Rho kinase inhibitor Y-27632, which our laboratory has shown to inhibit DNA synthesis, cell migration, and morphological responses to Rho (30, 40). We observed no attenuation of the Gαq-stimulated SRE.L response and no reduction in the response to Lbc or in the synergistic response to coexpressed Gαq and Lbc.
Synergy between Gαq and Rho Pathways Requires Activation of PLC, but Not PKC
To determine whether the activation state of Gαq was critical for synergy with Rho pathways, we compared the effects of wild-type Gαq to those of the constitutively active Gαq. In COS-7 cells wild-type Gαq did not significantly activate PLC (as assessed by [3H]inositol phosphate formation), in contrast to the marked stimulatory effect of GαqQL (Table I). In parallel with this differential effect on PLC activity, the wild-type Gαq did not induce SRE.L-mediated gene expression and did not enhance the activation elicited by Lbc (Fig. 9A). These data suggested that activation of PLC was required for Gαq cooperation with the Rho pathway.
Table I.
[3H]Inositol phosphate (IP) formation due to various mutants of Gαq and PLCβCT
COS-7 cells were transfected overnight then allowed 24 h to recover. During this time 1–2 μCi of myo[3H]inositol/ml was added overnight. The accumulation of inositol phosphates in the presence of 10 mM LiCl for 30 min was assessed. Dash indicates that no inhibitor was introduced. Data are shown as fold increase over empty vector transfectants ± S.E. from two to three experiments, each performed in triplicate.
| Stimulus | Inhibitor | IP accumulation |
|---|---|---|
| GαqWT | — | 1.3 ± 0.1 |
| GαqQL | — | 18.5 ± 2.0 |
| GαqQL/DNE | — | 1.7 ± 0.3 |
| GαqRC | — | 8.4 ± 0.5 |
| GαqRC | PLCβCT | 3.9 ± 0.2 |
Fig. 9. SRE.L activation is dependent on PLC activation and independent of PKC.
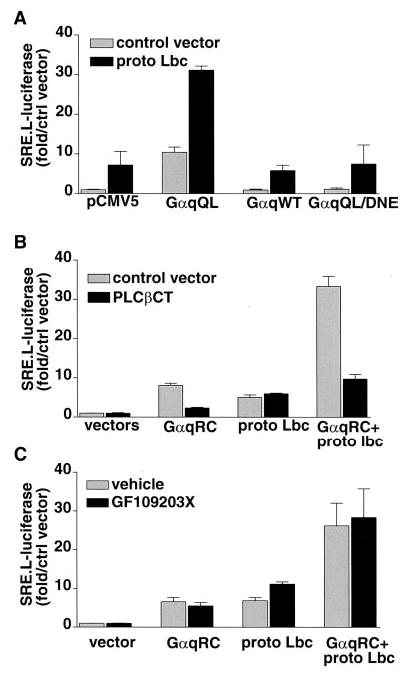
COS-7 cells were transfected with the SRE.L-luciferase reporter plasmid, and luciferase was determined as described. A, cells were transfected with empty pCMV5 vector or one of the following Gαq constructs: GTPase-deficient Gαq (GαqQL), wild-type Gαq (GαqWT), or GTPase-deficient Gαq that cannot activate PLCβ (GαqQL/DNE) along with control vector or proto-Lbc. B, COS-7 cells were transfected with a GTPase-deficient Gαq (GαqRC) and/or Lbc along with control vector plasmid encoding the C terminus of PLCβ1 (PLCβCT). C, COS-7 cells were transfected with a GTPase-deficient Gαq (GαqRC) and/or Lbc. After overnight transfection, cells were treated with vehicle or with 3 μ M GF109203X (to inhibit PKC) for 24 h prior to harvest. Data are average ± S.E. of the fold increase over vector control for two or three experiments performed in triplicate.
To further test this possibility we examined the response to GαqDNE, an activated (Q209L) mutant of Gαq that also has three amino acids substituted to prevent coupling to PLC (44). As previously demonstrated by Venkatakrishnan and Exton (44), this mutant did not significantly stimulate inositol phosphate formation (Table I). GαqDNE also failed to stimulate SRE.L-mediated gene expression or synergize with Lbc (Fig. 9A), consistent with a requirement for PLC signaling pathways. We also tested a cDNA that encodes the C terminus of PLCβ1 (PLCβCT), which has been suggested to interact with Gαq and thereby act as a competitive inhibitor blocking activation of endogenous PLC activation (35). When the PLCβCT was expressed along with GαqRC in COS-7 cells, inositol phosphate accumulation was inhibited by approximately 50% (Table I). Expression of PLCβCT led to a dose-dependent inhibition of Gαq-induced SRE.L activation (not shown), with the highest dose almost completely abolishing SRE.L activation by GαqRC (Fig. 9B). The synergistic SRE.L response due to Gαq and Lbc was also markedly inhibited by expression of PLCβCT. Expression of the PLCβCT had no effect on SRE.L activation by Lbc alone. Our findings using GαqQL/DNE and the PLCβCT further support the hypothesis that activation of PLC is required for the synergistic interaction between Gαq and Rho signaling pathways.
One downstream consequence of PLC activation is increased activity of PKC. PKC has been shown to increase transcription from the activator protein-1 (AP-1) response element. To determine whether stimulation of the SRE.L by Gαq could result from PKC activation, we treated COS-7 cells with PMA. Although PMA induced transcription from an AP-1-luciferase reporter, it did not activate the SRE.L or enhance the ability of Lbc to induce SRE.L-mediated transcription (data not shown). In addition, the PKC inhibitor GF109203X failed to affect the SRE.L responses to Gαq and/or Lbc (Fig. 9C), whereas it blocked the PMA-induced activation of AP-1-dependent transcription (data not shown). These observations suggest that activation of PKC is not necessary or sufficient for Gαq to induce transcription from the SRE.L.
DISCUSSION
Many Gq-coupled receptors have been suggested to signal through Rho-dependent pathways, yet no signaling pathway linking Gαq and Rho has been identified. In fact there is conflicting data on the issue of whether and how Gαq might activate Rho (reviewed in Ref. 11). Therefore, our initial goal was to determine whether Gαq activates Rho and if this occurs via interaction with Rho GEFs, as has been suggested for Gα12 and Gα13. In this report we demonstrate that Gαq can associate with Lbc, a Rho GEF, and that Gαq-generated signals cooperate with Rho to enhance two distinct Rho-mediated responses. Although these functional and physical interactions suggest that Gαq signals through a Rho GEF, our data indicate that the interaction between these pathways occurs distal to the activation of Rho.
Two assays demonstrated functional interactions between Gαq and Lbc on Rho-dependent pathways. The first was activation of an SRE.L-luciferase reporter gene, a response that a number of laboratories have utilized as a readout of Rho-dependent signaling (16, 23, 29, 32). In COS-7 cells both Gαq and Lbc activated this reporter. Gαq synergized with either proto-Lbc or p115RhoGEF to activate the SRE.L-luciferase, while Gα12 and Gα13 did not synergize with these Rho GEFs.
Induction of the SRE.L by Lbc was blocked by coexpression of C3 exoenzyme. A less expected observation was that C3 exoen-zyme inhibited the effect of Gαq and abolished the synergistic response elicited by Gαq and Lbc coexpression. Nonspecific effects of the C3 toxin on luciferase expression or cell viability were ruled out, since C3 had no effect on constitutive GFP expression or SRE.L-luciferase expression induced by Cdc42. Thus the effect of C3 indicates that Rho activity is indeed required for Gαq signaling to the SRE.L.
The other functional assay used to look for Gαq and Rho interactions was the morphological response of 1321N1 cells. Since cell rounding is rapid and robust, appearing in less than 3 h after microinjection (4, 30), it represents an early consequence of Rho activation. In our previous studies we demonstrated that proto-Lbc induced C3-sensitive rounding (30). Our current findings reveal that Gαq synergizes with proto-Lbc to stimulate this response. As observed for SRE.L activation, Gα12 did not enhance Lbc-induced rounding. Importantly, the selective enhancement of the cytoskeletal response to Lbc by Gαq indicates that the synergy between Gαq and Lbc is not an artifact of using the SRE.L reporter gene assay as a readout.
In experiments designed to detect interactions of Gαq with Rho GEFs, we asked whether Gαq expressed in COS-7 cells associates with coexpressed Rho GEFs. Two Rho GEFs, Lbc and p115RhoGEF, were tested, and their interactions with Gαq, Gα12, Gα13, and Gαs were compared. The finding that emerged from these studies was that Gαq coprecipitated specifically with Lbc. This was independent of the epitope tag on Lbc and was seen with both the full-length proto form and a truncated, onco, form of Lbc (data not shown). Lbc did not coprecipitate Gαs, confirming that there is Gα subunit specificity, even in the transfected COS-7 cell system. Consistent with previously published findings, Gαq did not associate with p115RhoGEF, although coprecipitation of Gα12 or Gα13 with p115RhoGEF was demonstrable (12, 33). Strikingly, synergy on the SRE.L followed the opposite pattern, that is Gαq synergized with p115RhoGEF, while Gα12 and Gα13 did not. The discrepancy between these findings and those reported by Mao et al. (32) may reflect cell type-specific differences. Nonetheless, our data demonstrate that functional cooperation between Rho GEFs and Gα subunits is not correlated with the physical interaction observed in the coprecipitation studies.
We tested the possibility that the Gαq-Lbc interaction enhances the exchange activity of Lbc, which should result in enhanced activation of Rho in COS-7 cells. Rho activation was observed in cells expressing Lbc or other Rho GEFs, as demonstrated by an increase in the fraction of Rho that bound to the RBD of Rhotekin. Furthermore, the ability of expressed Gα subunits to activate Rho in COS-7 cells was confirmed by the stimulatory effect of both Gα12 and Gα13 on Rho activation. However Rho activation was not observed in cells expressing Gαq nor did we observe a synergistic effect of Gαq on Lbc induced Rho activation. It is of course possible that the rhotekin-RBD assay is not sensitive enough to detect a modest Rho activation by Gαq. However, since Rho activation by Gα12 and Gα13 was detectable, while activation by Gαq was not, at the least significantly less Rho is activated by Gαq. Furthermore, the Rho activation assay was carried out under the same conditions in which both physical and functional interactions of Gαq and Lbc were seen. Thus, it appears that Rho activation neither results from nor explains Gαq synergy with Lbc.
There is evidence in other systems that Rho mediates responses to Gαq. In published studies inhibition of Rho with C3 exoenzyme blocked Gαq-stimulated SRE activation (23) and neurite retraction (31). In addition, depletion of Gαq by anti-sense (45) or Gαq/11-knockout (9) prevented Rho-dependent actin reorganization in smooth muscle cells and fibroblasts. While these observations indicate that Gαq and Rho pathways are interdependent, they do not directly demonstrate that Rho is activated by Gαq expression. Preliminary data from Hall’s laboratory suggests that Gαq expression increases the amount of activated Rho (46). Furthermore agonists that activate Gq-coupled receptors, have been shown to stimulate Rho redistribution (47, 48). While the relationship between Rho activation and its cellular distribution is complex, it is generally believed that inactive Rho is sequestered in the cytosol and translocates to the membrane or cytoskeleton upon stimulation (reviewed in Ref. 11). Interestingly, our preliminary experiments suggest that Gαq increases the amount of Rho in the particulate fraction of COS-7 cell extracts. This altered Rho localization may be distinct from its activation but could contribute to enhancement of Rho-dependent signaling.
The physical interaction between Gα13 and p115RhoGEF has been shown to increase the guanine nucleotide exchange activity of p115RhoGEF in vitro (12). Notably other G protein-Rho GEF interactions have not, to our knowledge, been directly shown to increase Rho activation. Indeed, Gα12 associates with p115RhoGEF but does not stimulate its exchange activity (12). Additionally, while an interaction of Gα12 or Gα13 with PDZ-RhoGEF was shown by coprecipitation and indirectly by assessing SRE.L activation, neither Gα12 nor Gα13 has, to our knowledge, been directly demonstrated to enhance PDZ-Rho-GEF exchange activity or PDZ-RhoGEF-induced Rho activation (13). These data, together with the findings reported here, suggest caution in interpreting physical interactions between Gα subunits and Rho GEFs as evidence that the exchanger is regulated by the Gα protein.
The finding that the activated form of Gαq is required to enhance Rho-mediated signaling, along with experiments using the DNE mutant of activated Gαq and the inhibitory PLCβCT construct, support the conclusion that activation of PLCβ is required for the synergistic interaction of Gαq and Rho. Thus PLC, the primary effector of Gαq signaling, appears to play a central role in a pathway that enhances Rho-mediated responses. Although PKC is downstream of PLC in many pathways, activation of PKC with PMA did not mimic the effects of Gαq on SRE.L-luciferase expression, and inhibition of PKC did not reduce this response. Subsequent experiments will be needed to identify the PLC-dependent signals and effectors that interact with and enhance Rho-mediated responses.
Since most GPCRs thought to induce Rho activation by coupling to Gα12/13 also activate Gαq and PLC, these processes are likely to occur in concert. Interestingly, in 1321N1 cells, thrombin, which stimulates PLC and activates Rho, can induce cell rounding. In contrast, cell rounding is not induced by carbachol, which activates PLC (4, 49) but not Rho, or by LPA, which activates Rho but only weakly stimulates PLC in these cells.2 These findings are consistent with the notion that signals generated by Gαq/PLC may be insufficient to induce Rho activation, but required to enhance responses elicited through Rho signaling pathways.
Footnotes
This work was supported in part by National Institutes of Health Grants GM36927 (to J. H. B.) and AI38396 (to B. A. W.), National Institutes of Health NCI Grant CA62029 (to D. T.), and by an American Heart Association, New England affiliate, grant-in-aid (to D. T.).
The abbreviations used are: GPCR, G protein-coupled receptor; AP-1, activator protein-1; C3, C. botulinum C3 transferase; DMEM, Dulbecco’s modified Eagle’s medium; GEF, guanine nucleotide exchange factor; GFP, green fluorescent protein; GST, glutathione S-transferase; Lbc, lymphoid blast crisis; LPA, lysophosphatidic acid; PAGE, polyacrylamide gel electrophoresis; PKC, protein kinase C; PLC, phospholipase C; PMA, phorbol 12-myristate 13-acetate; PMT, Pasteurella multocida toxin; RBD, Rho-binding domain; SRE, serum response element; GTPγS, guanosine 5′-O-(thio)triphosphate.
S. A. Sagi, T. M. Seasholtz, D. Goldstein, and J. H. Brown, unpublished observations.
References
- 1.van Biesen T, Luttrell LM, Hawes BE, Lefkowitz RJ. Endocr Rev. 1996;17:698–714. doi: 10.1210/edrv-17-6-698. [DOI] [PubMed] [Google Scholar]
- 2.Ridley AJ, Hall A. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 3.Gohla A, Harhammer R, Schultz G. J Biol Chem. 1998;273:4653–4659. doi: 10.1074/jbc.273.8.4653. [DOI] [PubMed] [Google Scholar]
- 4.Majumdar M, Seasholtz TM, Goldstein D, de Lanerolle P, Brown JH. J Biol Chem. 1998;273:10099–10106. doi: 10.1074/jbc.273.17.10099. [DOI] [PubMed] [Google Scholar]
- 5.Bishop AL, Hall A. Biochem J. 2000;348:241–255. [PMC free article] [PubMed] [Google Scholar]
- 6.Narumiya S, Ishizaki T, Watanabe N. FEBS Lett. 1997;410:68–72. doi: 10.1016/s0014-5793(97)00317-7. [DOI] [PubMed] [Google Scholar]
- 7.Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, Kaibuchi K. Science. 1997;275:1308–1311. doi: 10.1126/science.275.5304.1308. [DOI] [PubMed] [Google Scholar]
- 8.Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. J Biol Chem. 1998;273:21867–21874. doi: 10.1074/jbc.273.34.21867. [DOI] [PubMed] [Google Scholar]
- 9.Gohla A, Offermanns S, Wilkie TM, Schultz G. J Biol Chem. 1999;274:17901–17907. doi: 10.1074/jbc.274.25.17901. [DOI] [PubMed] [Google Scholar]
- 10.Seasholtz TM, Majumdar M, Brown JH. Mol Pharmacol. 1999;55:949–956. doi: 10.1124/mol.55.6.949. [DOI] [PubMed] [Google Scholar]
- 11.Sah VP, Seasholtz TM, Sagi SA, Brown JH. Annu Rev Pharmacol Toxicol. 2000;40:459–489. doi: 10.1146/annurev.pharmtox.40.1.459. [DOI] [PubMed] [Google Scholar]
- 12.Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- 13.Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS. J Biol Chem. 1999;274:5868–5879. doi: 10.1074/jbc.274.9.5868. [DOI] [PubMed] [Google Scholar]
- 14.Amano M, Mukai H, Ono Y, Chihara K, Matsui T, Hamajima Y, Okawa K, Iwamatsu A, Kaibuchi K. Science. 1996;271:648–650. doi: 10.1126/science.271.5249.648. [DOI] [PubMed] [Google Scholar]
- 15.Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. EMBO J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- 16.Hill CS, Wynne J, Treisman R. Cell. 1995;81:1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- 17.Treisman R, Alberts AS, Sahai E. Cold Spring Harbor Symp Quant Biol. 1998;63:643–651. doi: 10.1101/sqb.1998.63.643. [DOI] [PubMed] [Google Scholar]
- 18.Sahai E, Alberts AS, Treisman R. EMBO J. 1998;17:1350–1361. doi: 10.1093/emboj/17.5.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujisawa K, Madaule P, Ishizaki T, Watanabe G, Bito H, Saito Y, Hall A, Narumiya S. J Biol Chem. 1998;273:18943–18949. doi: 10.1074/jbc.273.30.18943. [DOI] [PubMed] [Google Scholar]
- 20.Sahai E, Ishizaki T, Narumiya S, Treisman R. Curr Biol. 1999;9:136–145. doi: 10.1016/s0960-9822(99)80067-0. [DOI] [PubMed] [Google Scholar]
- 21.Morissette MR, Sah VP, Glembotski CC, Brown JH. Am J Physiol. 2000;278:H1769–H1774. doi: 10.1152/ajpheart.2000.278.6.H1769. [DOI] [PubMed] [Google Scholar]
- 22.Quilliam LA, Lambert QT, Mickelson-Young LA, Westwick JK, Sparks AB, Kay BK, Jenkins NA, Gilbert DJ, Copeland NG, Der CJ. J Biol Chem. 1996;271:28772–28776. doi: 10.1074/jbc.271.46.28772. [DOI] [PubMed] [Google Scholar]
- 23.Mao J, Yuan H, Xie W, Simon MI, Wu D. J Biol Chem. 1998;273:27118–27123. doi: 10.1074/jbc.273.42.27118. [DOI] [PubMed] [Google Scholar]
- 24.Sah VP, Hoshijima M, Chien KR, Brown JH. J Biol Chem. 1996;271:31185–31190. doi: 10.1074/jbc.271.49.31185. [DOI] [PubMed] [Google Scholar]
- 25.Tigyi G, Fischer DJ, Sebok A, Yang C, Dyer DL, Miledi R. J Neurochem. 1996;66:537–548. doi: 10.1046/j.1471-4159.1996.66020537.x. [DOI] [PubMed] [Google Scholar]
- 26.Buhl AM, Johnson NL, Dhanasekaran N, Johnson GL. J Biol Chem. 1995;270:24631–24634. doi: 10.1074/jbc.270.42.24631. [DOI] [PubMed] [Google Scholar]
- 27.Vouret-Craviari V, Van Obberghen-Schilling E, Scimeca JC, Van Obberghen E, Pouyssegur J. Biochem J. 1993;289:209–214. doi: 10.1042/bj2890209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jalink K, Moolenaar WH. J Cell Biol. 1992;118:411–419. doi: 10.1083/jcb.118.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coso OA, Montaner S, Fromm C, Lacal JC, Prywes R, Teramoto H, Gutkind JS. J Biol Chem. 1997;272:20691–20697. doi: 10.1074/jbc.272.33.20691. [DOI] [PubMed] [Google Scholar]
- 30.Majumdar M, Seasholtz TM, Buckmaster C, Toksoz D, Brown JH. J Biol Chem. 1999;274:26815–26821. doi: 10.1074/jbc.274.38.26815. [DOI] [PubMed] [Google Scholar]
- 31.Katoh H, Aoki J, Yamaguchi Y, Kitano Y, Ichikawa A, Negishi M. J Biol Chem. 1998;273:28700–28707. doi: 10.1074/jbc.273.44.28700. [DOI] [PubMed] [Google Scholar]
- 32.Mao J, Yuan H, Wu D. Proc Natl Acad Sci U S A. 1998;95:12973–12976. doi: 10.1073/pnas.95.22.12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- 34.Sterpetti P, Hack AA, Bashar MP, Park B, Cheng SD, Knoll JHM, Urano T, Feig LA, Toksoz D. Mol Cell Biol. 1999;19:1334–1345. doi: 10.1128/mcb.19.2.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beekman A, Helfrich B, Bunn PA, Jr, Heasley LE. Cancer Res. 1998;58:910–913. [PubMed] [Google Scholar]
- 36.Ren X-D, Kiosses WB, Schwartz MA. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson BA, Ponferrada VG, Vallance JE, Ho M. Infect Immun. 1999;67:80–87. doi: 10.1128/iai.67.1.80-87.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collins LR, Minden A, Karin M, Brown JH. J Biol Chem. 1996;271:17349–17353. doi: 10.1074/jbc.271.29.17349. [DOI] [PubMed] [Google Scholar]
- 39.Kranenburg O, Poland M, van Horck FPG, Drechsel D, Hall A, Moolenaar WH. Mol Biol Cell. 1999;10:1851–1857. doi: 10.1091/mbc.10.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seasholtz TM, Majumdar M, Kaplan DD, Brown JH. Circ Res. 1999;84:1186–1193. doi: 10.1161/01.res.84.10.1186. [DOI] [PubMed] [Google Scholar]
- 41.Wilson BA, Zhu X, Ho M, Lu L. J Biol Chem. 1997;272:1268–1275. doi: 10.1074/jbc.272.2.1268. [DOI] [PubMed] [Google Scholar]
- 42.Seo B, Choy EW, Maudsley S, Miller WE, Wilson BA, Luttrell LM. J Biol Chem. 2000;275:2239–2245. doi: 10.1074/jbc.275.3.2239. [DOI] [PubMed] [Google Scholar]
- 43.Sabri A, Pak E, Alcott SA, Wilson BA, Steinberg SF. Circ Res. 2000;86:1047–1053. doi: 10.1161/01.res.86.10.1047. [DOI] [PubMed] [Google Scholar]
- 44.Venkatakrishnan G, Exton JH. J Biol Chem. 1996;271:5066–5072. doi: 10.1074/jbc.271.9.5066. [DOI] [PubMed] [Google Scholar]
- 45.Hirshman CA, Emala CW. Am J Physiol. 1999;277:L653–L661. doi: 10.1152/ajplung.1999.277.3.L653. [DOI] [PubMed] [Google Scholar]
- 46.Kjøller L, Hall A. Exp Cell Res. 1999;253:166–179. doi: 10.1006/excr.1999.4674. [DOI] [PubMed] [Google Scholar]
- 47.Fleming IN, Elliot CM, Exton JH. J Biol Chem. 1996;271:33067–33073. doi: 10.1074/jbc.271.51.33067. [DOI] [PubMed] [Google Scholar]
- 48.Keller J, Schmidt M, Hussein B, Rumenapp U, Jakobs KH. FEBS Lett. 1997;403:299–302. doi: 10.1016/s0014-5793(97)00067-7. [DOI] [PubMed] [Google Scholar]
- 49.Post GR, Collins LR, Kennedy ED, Moskowitz SA, Aragay AM, Goldstein D, Brown JH. Mol Biol Cell. 1996;7:1679–1690. doi: 10.1091/mbc.7.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]