

Abstract
Pasteurella multocida toxin (PMT) stimulates and subsequently uncouples phospholipase C (PLC) signal transduction through its selective action on the Gαq subunit. This review summarizes what is currently known about the molecular action of PMT on Gq and the resulting cellular effects. Examples are presented illustrating the use of PMT as a powerful tool for dissecting the molecular mechanisms involving pertussis toxin (PT)-insensitive heterotrimeric G proteins.
Introduction
Protein toxins have long been known to constitute important virulence determinants for pathogenic bacteria. The anthrax bioterrorism events of 2001 and the emergence of antibiotic-resistant, toxin-producing bacteria have provided strong impetus to increase our understanding of toxin-mediated disease processes (Wilson and Salyers 2002). The growing relevance of toxin-mediated effects on host cells in developing alternative antitoxin strategies heightens the need to better understand the structure-function relationships of protein toxins produced by pathogenic bacteria. Over the past couple of decades, our understanding of toxin action has expanded tremendously. Information learned from these efforts has enabled scientists to exploit their noxious properties for beneficial applications in studying problems in cell biology, physiology, and pharmacology.
Many protein toxins share the common feature of being highly specialized enzymes, capable of entering eukaryotic cells and catalyzing reactions that interfere with normal signal transduction and physiological processes, often resulting in morphological changes, cellular damage or cell death. Because of their highly specific action in cells, bacterial protein toxins can be used as selective and efficient tools for studying molecular mechanisms controlling signal transduction and other physiological processes (Schiavo and Van Der Goot 2001). For example, a number of toxins, including the clostridial toxins C2, C3, toxA and toxB, and the E. coli toxins CNF1 and CNF2, among others, have been used as selective modulators of cytoskeletal function through their action on small GTPases (Aktories et al. 2000). The clostridial neurotoxins have been enormously beneficial in uncovering molecular mechanisms of neurotransmitter release and essential aspects of neuronal physiology (Lalli et al. 2003; Schiavo et al. 2000). These neurotoxins are increasingly being put to beneficial use in medicine for the treatment of human diseases characterized by hyperfunction of nerves (Rossetto et al. 2001).
Heterotrimeric G proteins constitute a large family of pivotal regulatory GTPases that are responsible for transducing external (e.g., hormonal) signals from ligand-bound receptors to intracellular responses. They are made of α, β, and γ subunits and are distinguished into four main classes: Gs, Gi, G12, and Gq. The first toxins used to define the molecular mechanisms of heterotrimeric G proteins and adenylate cyclase-mediated signaling pathways were cholera toxin (CT) and pertussis toxin (PT): CT activates Gs proteins, while PT inhibits Gi/o proteins (Casey and Gilman 1988). PT was used to further distinguish a group of PT-insensitive G proteins (see Table 1) (Fields and Casey 1997). Of these G proteins that are refractory to PT treatment, a subgroup, later identified as the Gq family, were found to play critical roles as regulators of phospholipase C (PLC) signaling (Fields and Casey 1997; Rhee and Choi 1992; Sternweis and Smrcka 1992).
Table 1.
Signaling of heterotrimeric G-proteins and their modulating toxins
| Subfamily | Gα | Signalinga | Modulating toxin | Toxin effect on Gα | Referencesb |
|---|---|---|---|---|---|
| Gi subfamily | Gi1 | ↓AC | PT | Inhibition | (a) (b) |
| Gi2 | ↓AC | PT | Inhibition | (a) (b) | |
| Gi3 | ↓AC | PT | Inhibition | (a) (b) | |
| Go | ↓Ca2+ channels | PT | Inhibition | (a) (b) | |
| Gt | ↑cGMP-PDE | PT | Inhibition | (a) (b) (c) | |
| CT | Activation | (a) (b) (d) | |||
| Ggust | ↑PDE | PT | ? | (e) | |
| Gz | ↓AC | ? | ? | (f) (g) | |
| Gs subfamily | Gs | ↑AC | CT | Activation | (a) (b) |
| Golf | ↑AC | CT | Activation | (h) | |
| Gq subfamily | Gq | ↑PLCβ, ↑Rho | PMT | Activation/Inhibitionc | (i) (j) (k) |
| G11 | ↑PLCβ, ↑Rho | ? | ? | (k) | |
| G14 | ↑PLCβ | ? | ? | ||
| G15 | ↑PLCβ | ? | ? | ||
| G16 | ↑PLCβ | ? | ? | ||
| G12 subfamily | G12 | ↑Rho | ? | ? | |
| G13 | ↑Rho | ? | ? |
AC: adenylate cyclase; PDE: phosphodiesterase; PLC: phopholipase C
(a) (Fields and Casey 1997), (b) (Casey and Gilman 1988), (c) (Van Dop et al. 1984a), (d) (Van Dop et al. 1984b), (e) (Gilbertson et al. 2000), (f) (Casey et al. 1990), (g) (Ho and Wong 2001), (h) (Jones et al. 1990), (i) (Wilson et al. 1997), (j) (Zywietz et al. 2001), (k) (Vogt et al. 2003)
Initial activation, followed by uncoupling of Gq signaling. Prolonged treatment with PMT results in inhibition
Receptors for many hormones, neurotransmitters and growth factors are coupled to Gq proteins. Stimulation of Gq-coupled receptors results in transient elevation of intracellular Ca2+ and increased levels of inositol trisphosphate (IP3) and diacylglycerol (DAG). These second messengers control vital cellular processes, including fertilization, cell growth, transformation, secretion, muscle contraction, metabolism and sensory perception (Berridge 1993). IP3 and DAG are generated from phosphatidylinositol 4,5-bisphosphate (PIP2) through the action of PLC, of which γ-isoforms are activated through tyrosine kinase-linked receptors and β-isoforms are activated through G-protein-coupled receptors (Rhee and Choi 1992; Sternweis and Smrcka 1992). Ligands and G-protein-coupled receptors that activate PLCβ (Deckmyn et al. 1993; Quick et al. 1994; Rebecchi and Pentyala 2000; Rhee and Choi 1992; Sternweis and Smrcka 1992) can be distinguished by their sensitivity to PT (Berstein et al. 1992a, b; Camps et al. 1992; Wu et al. 1993). The βγ subunits (but not α subunits) of PT-sensitive Gi/o proteins preferentially stimulate PLCβ2>PLCβ3>PLCβ1, whereas the α subunits of the PT-insensitive Gq proteins stimulate PLCβ1≧PLCβ3≫PLCβ2 (Hepler et al. 1993; Park et al. 1993; Rhee and Choi 1992; Smrcka and Sternweis 1993).
Until recently, there had been no specific modulating reagent available for studying the role of Gq proteins in hormonal communication and signal transduction. The dermonecrotic toxin produced by Pasteurella multocida (PMT) can now be added to the list of bacterial protein toxins that modulate G proteins (Table 1). PMT stimulates Ca2+ and IP3 signaling by activating Gq-dependent PLCβ1 (Wilson et al. 1997). PMT facilitation of Gαq-protein coupling to PLCβ1 causes the same cellular responses elicited by Gq-protein-linked receptors, such as the muscarinic (M1, M3, M5), bombesin, vasopressin, endothelin, thyrotropin-releasing hormone (TRH), and adrenergic receptors (α1AR). Understanding the bio-chemical mechanism of PMT action may thus afford unique insight into Gq-mediated molecular signaling events and enables the use of PMT as a tool for studying these processes.
Cellular effects of PMT
PMT is the major virulence factor produced by P. multocida that is responsible for atrophic rhinitis, pneumonia-like respiratory disease, and dermonecrosis (Foged 1992). PMT is secreted as a monomeric, 1285-amino acid protein (Mr 146-kDa) (Lax and Chanter 1990; Petersen 1990; Petersen and Foged 1989). PMT binds to ganglioside-type receptors and enters mammalian cells via receptor-mediated endocytosis (Dudet et al. 1996; Pettit et al. 1993; Rozengurt et al. 1990) and acts intracellularly to initiate a number of signaling pathways leading to DNA synthesis and cellular proliferation (Higgins et al. 1992; Rozengurt et al. 1990; Seo et al. 2000; Wilson et al. 1997, 2000). Purified PMT alone is sufficient to experimentally induce progressive atrophic rhinitis in swine and symptoms of pneumonia in rabbits (Chrisp and Foged 1991; Foged 1992; Lax and Chanter 1990). Recombinant PMT is indistinguishable from native toxin, and both are equally potent at pi-comolar concentrations (Lax and Chanter 1990; Rozengurt et al. 1990; Wilson et al. 1997). Vaccination against PMT protects against challenge with P. multocida and development of atrophic rhinitis (Foged 1992).
PMT stimulates osteoclastic bone resorption in vitro (Felix et al. 1992; Kimman et al. 1987) and increases osteoclast cell number in vivo (Martineau-Doize et al. 1993). PMT appears to stimulate the differentiation of preosteoclasts into osteoclasts (Jutras and Martineau-Doize 1996) and promotes osteoclast proliferation leading to bone resorption, while apparently inhibiting bone regeneration by osteoblasts (Mullan and Lax 1998; Sterner-Kock et al. 1995). PMT acting on these multiple cell types, including activating mature osteoclasts and inducing preosteoclast proliferation, as well as proliferation/differentiation of periosteal (fibroblastic, osteogenic, and adipogenic) cells, may contribute to the symptoms of atrophic rhinitis (Mullan and Lax 1998; Rozengurt et al. 1990). Recent evidence suggests that immunomodulation of the host may be an additional function of the toxin important in pathogenesis (Jordan et al. 2003).
In cultured epithelial cells, such as calf or monkey (Vero) kidney cells, calf testis, or bovine embryonic lung (EBL) cells, PMT causes primarily morphological changes and cytotoxic effects (Pennings and Storm 1984; Pettit et al. 1993; Rutter and Luther 1984). In cultured mesenchymal cells, such as murine, rat, or human fibroblasts, and in osteoblasts, PMT action is mitogenic and initiates DNA synthesis and cell division (Mullan and Lax 1998; Rozengurt et al. 1990). PMT has also been shown to induce anchorage-independent cell growth of fibroblasts (Higgins et al. 1992), as evidenced by colony formation in soft agar, suggesting that it has the ability to promote a transformed phenotype and leading to the speculation that it could promote tumor formation and cancer (Lax and Thomas 2002). Flow cytometry analysis of cells treated with PMT showed that PMT stimulates cells to move from the G1 phase into and through the S phase, but it does not trigger apoptosis (Wilson et al. 2000). PMT-treated confluent quiescent Swiss 3T3 cells formed dense monolayers over the course of 4–6 days, with a concomitant increase in cell number up to threefold (Wilson et al. 2000). However, cell cycle analysis revealed that after the initial mitogenic response to PMT, cells subsequently arrested primarily in G1 and became unresponsive to further PMT treatment (see Fig. 1), indicating that the mitogenic response was not sustained (Wilson et al. 2000).
Fig. 1.
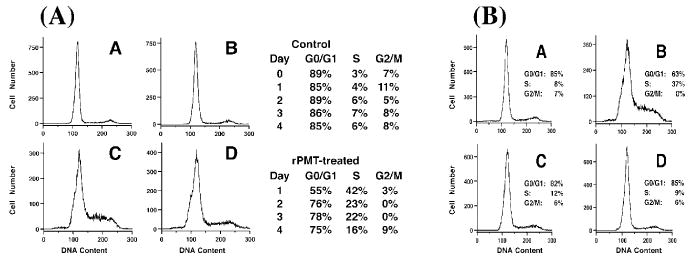
Effect of PMT treatment on cell cycle progression in Swiss 3T3 cells. Shown are results from flow cytometry analyses of confluent Swiss 3T3 cells with or without single or multiple PMT treatments. A Representative DNA histograms from a time course of untreated (a and b) or PMT-treated (c and d) cells from day 1 (a and c) and day 4 (b and d). To the right is the summary of the percentages of untreated (top) or PMT-treated (bottom) cells found in G0/G1, S, and G2/M phases of the cell cycle during the time course. B Representative DNA histograms from multiple PMT treatments: (a) untreated cells analyzed on day 5; (b) cells treated with PMT on day 5 and analyzed on day 6; (c) cells treated with PMT on day 0 and analyzed on day 5; (d) cells treated with PMT on day 0 and again on day 5 and analyzed on day 6. The percentages of cells found in G0/G1, S, and G2/M phases of the cell cycle are also shown.(Reprinted with permission from Wilson et al. 2000.)
Western blot analysis of the effect of PMT on the expression of a number of cell cycle markers, including the proto-oncogene c-Myc; cyclins D1, D2, D3, and E; p21; PCNA; and the Rb proteins, p107 and p130, showed that PMT initially upregulated these markers and stimulated cell cycle progression in Swiss 3T3 cells, yet continued expression of these markers, and hence continued proliferation, was not sustained (Wilson et al. 2000). However, PMT exhibited a differential effect on epithelial-like cells. Confluent Vero cells underwent rapid, dramatic morphological changes upon toxin exposure, but a mitogenic effect was not evident, based on the lack of a PMT-induced increase in cell numbers or in the rate of DNA synthesis, which was further substantiated by flow cytometry analysis. Furthermore, PMT failed to upregulate PCNA or cyclins D3 and E, which is critical for driving cells from G1 into S phase, and hence, little or no cell cycle progression occurred in Vero cells (Wilson et al. 2000).
PMT effects on signal transduction
PMT and Gq-PLC signaling
PMT activates inositol phosphate pathways, Ca2+ mobilization and PKC-dependent phosphorylation in cultured fibroblasts and osteoblasts (Mullan and Lax 1998; Staddon et al. 1990, 1991). PMT also potentiates G protein-coupled receptor responses to bombesin, vasopressin, and endothelin (Murphy and Rozengurt 1992). These effects suggested the involvement of a cellular phosphatidylinositol-specific phospholipase C (PLC) in PMT action (Murphy and Rozengurt 1992). Wilson et al. (Wilson et al. 1997) subsequently demonstrated direct PMT-mediated stimulation of PLCβ1 activity and IP3-induced intracellular Ca2+ mobilization by using voltage-clamped Xenopus oocytes as a model system to monitor the transient Ca2+-dependent Cl− current evoked upon microinjection with PMT. To identify the intracellular targets involved in the PMT-induced IP3 signaling pathway, they examined the effects of specific antibodies against various G-protein and PLC signaling molecules on the PMT-induced Cl− currents in the oocyte system. As showed in Fig. 2, only antibodies directed against PLCβ1, Gαpan, and the C-terminus of Gαq/11 completely blocked the PMT-mediated response in oocytes. In addition, GDPβS, a known inhibitor of Gα subunit-mediated signaling pathways, blocked the PMT-induced response. Further-more, overexpression of mouse Gαq in Xenopus oocytes increased the PMT-induced response, whereas treatment with antisense Gαq mRNA reduced the response. These results established the direct involvement of Gαq protein in PMT-activation of PLCβ1 (Fig. 2).
Fig. 2.
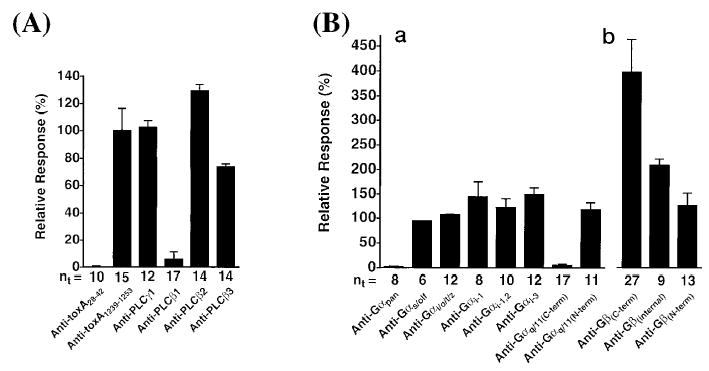
Identification of the intracellular target of PMT as Gq/11 by using specific antibodies against key signaling proteins to block the PMT response in Xenopus oocytes. The peak inward Cl− current was used to measure the effect of specific antibodies on the PMT-induced Ca2+-dependent Cl− currents in oocytes voltage-clamped at a holding potential of –80 mV. Antibodies against PMT (N-terminus or C-terminus) and various PLC isoforms (A) or against various Gα or Gβ subunits (B) were microinjected into the oocytes 3 h prior to microinjection with PMT. (Reprinted with permission from Wilson et al. 1997.)
Antibodies against Gβ subunit did not block, but rather enhanced the PMT-induced response (Fig. 2), distinguishing that PMT action did not involve Gβγ subunit activation of PLCβ2 by a Gi/o-dependent pathway. Rather, by binding to Gβ, the antibodies caused the dissociation of Gαq subunit from the heterotrimeric complex, which could then be acted upon by PMT to give an enhanced response. The PMT-induced response was likewise enhanced by the release of Gαq subunit through sequestration of Gβγ subunits by using PT (Wilson et al. 1997). From these studies, the researchers concluded that the monomeric Gαq subunit is the preferred target of PMT action, which subsequently activates PLCβ1. Mouse knockout cell lines were used to confirm that PMT-induced formation of inositol phosphates was exclusively dependent on Gαq, and not closely-related Gq family members, such as G11, G12, or G13 (other Gq-family members, G14 or G15/16, were not examined) (Zywietz et al. 2001).
A recent study using a series of chimeras between Gαq and Gα11 in Gαq/11-knockout cells identified a region of the helical domain of Gαq that is important for PMT-induced activation of PLCβ (Orth et al. 2004). Exchange of Glu-105 or Asn-109 of Gα11, each of which is located in the helical domain of the Gα subunit, with the corresponding His residues of Gαq resulted in a mutant Gα11 that was now capable of mediating PMT-induced activation of PLCβ. However, the converse was not true, in that the reciprocal exchange of either His in Gαq with the corresponding Gα11 amino acid did not prevent PMT activation of PLCβ. Whether this differential interaction is due to a difference in PMT recognition of Gαq versus Gα11, due to a difference in Gαq versus Gα11 recognition of PLCβ, or due to a difference in recognition of Gαq versus Gα11 by another unidentified PMT mediator has yet to be determined.
PMT was shown to stimulate tyrosine phosphorylation of Gαq, but a mutant of PMT that does not activate Gq was also found to cause tyrosine phosphorylation of Gq, suggesting that this phosphorylation is not a prerequisite for Gαq activation by PMT (Baldwin et al. 2003). Consequently, although tyrosine phosphorylation of Gq/11 has been reported to regulate Gq/11 activation (Umemori et al. 1997, 1999), the role of tyrosine phosphorylation in PMT action on Gq is not clear.
Repeated microinjection of IP3 into oocytes reproduced transient Ca2+-dependent Cl− currents, indicating that the IP3 pathway is not readily desensitized. After the initial transient response to PMT, additional injection of IP3 still gave a response, but additional injection of PMT had no further response (Wilson et al. 1997). These results confirmed that the target of PMT action is upstream of IP3 release and that PMT uncouples the signaling between Gq and PLCβ1. A model for the intracellular action of PMT based on all of these results is shown in Figure 3.
Fig. 3.
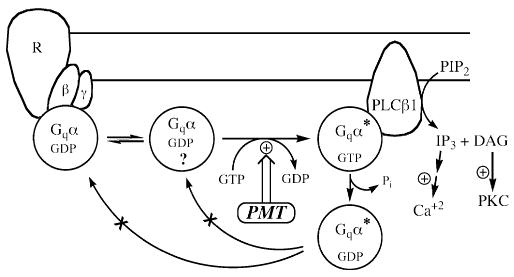
A proposed model for PMT action on Gq-coupled PLC signal transduction. In this model, PMT acts on free, monomeric Gαq, most likely in the GDP-bound form, and converts it into an active form, presumably GTP-bound, which stimulates PLCβ1. The PLCβ1 hydrolyzes PIP2 into IP3 and DAG, leading to Ca2+ mobilization that results in the Ca2+-dependent Cl− current. The PMT-induced response is transient due to GTPase activity of Gαq, which is still intact and is stimulated by interaction with PLCβ1. The presumably modified GDP-bound Gq can be neither acted upon again by PMT nor reassociated with the Gβγ-receptor complex.
PMT and downstream signaling
Some of the intracellular events that occur upon exposure to PMT are: enhanced hydrolysis of inositol phospholipids to increase the total intracellular content of inositol phosphates (Staddon et al. 1991); increased production of DAG (Staddon et al. 1990); mobilization of intracellular Ca2+ pools (Staddon et al. 1991); interconversion of GRP78/BiP (Staddon et al. 1992); and activation of protein kinase phosphorylation (Lacerda et al. 1996; Staddon et al. 1990). It has been suggested that activation of the small Rho GTPase mediates PMT-induced tyrosine phosphorylation of focal adhesion kinase (p125FAK) and paxillin, which results in actin stress fiber formation and focal adhesion assembly (Lacerda et al. 1996; Ohnishi et al. 1998; Thomas et al. 2001). Yet, this tyrosine phosphorylation appears to be independent of PKC activation and Ca2+ mobilization (Lacerda et al. 1996; Ohnishi et al. 1998).
How PMT-mediated activation of the PLC-IP3 signaling pathway promotes cytoskeletal rearrangement is, as of yet, not clear. One hint toward this may be the recent finding that PMT can associate with vimentin (Shime et al. 2002), a component of intermediate filaments in cells. Another possibility is for PMT to act on the actin cytoskeleton through its indirect action on Rho via Gq (Chikumi et al. 2002; Dutt et al. 2002; Katoh et al. 1998; Vogt et al. 2003). Although the stimulation of inositol phosphate signaling by PMT did not occur in Gαq-deficient or Gαq/Gα11-deficient cells, PMT could still stimulate other cellular effects in those knockout cells, including Rho activation, Rho-dependent actin rearrangements and focal adhesions, as well as JNK and Erk mitogenic signaling (Zywietz et al. 2001). These results indicate that certain effects of PMT action may also occur through other signaling pathways, independent of Gq or G11. One possibility is that PMT acts on G12 or G13, both of which are known to activate Rho protein (Fukuhara et al. 1999; Gratacap et al. 2001; Hart et al. 1998; Kozasa et al. 1998; Kurose 2003). Gα12/13 can induce Rho-dependent responses by interaction with Rho-specific guanine nucleotide exchange factors (Sah et al. 2000). Although it is not known if PMT acts on Gα12/13, this may provide a mechanism by which PMT could activate Rho in the absence of Gq and G11.
PMT as a tool for studying signal transduction
PMT as a tool for studying Gq-PLC signaling
A number of investigators have used PMT as a pharmacological tool to study Gq-coupled PLC signaling along the lines of that shown in Figure 3. For example, PMT has been used as a selective activator of Gq-coupled PLC effectors. Gq-coupled adrenergic receptor signaling in cardiomyocytes differs significantly between even closely related animal species, such as mice and rats. In the rat cardiomyocytes, α1-AR and endothelin receptors selectively activated PLC through Gq protein, but these receptors were not functional in mouse cardiomyocytes. PMT was used to show that Gq-PLC signaling pathway was still functional in the mouse cardiomyocytes (Sabri et al. 2000).
In discriminating PT-insensitive G-protein coupling of noradrenaline-induced α1AAR activation in neonatal rat cardiomyocytes, α1AAR was found to couple specifically to Gq/11 and not G12/13 proteins by showing that overexpression of Gq/11-specific RGS4, but not G12/13-specific Lsc-RGS blocked α1AAR activation of both PLC and phospholipase D (PLD) (Gosau et al. 2002). In addition, this study showed that PLD activation occurred subsequent to Gq-activation of PLCβ and novel, Ca2+-independent PKC isoforms δ and ε. The importance of Gq and not G11 in α1AAR activation of both PLC and PLD was further demonstrated by using PMT, which mimicked the α1AAR response.
Histamine induces catecholamine secretion from bovine adrenal chromaffin cells. PMT treatment caused a substantial additive increase in basal and histamine-stimulated inositol phosphate levels, but did not increase or prevent basal or histamine-stimulated secretion of the catecholamines, adrenaline and noradrenaline (Donald et al. 2002). This study showed that the secretion occurs through a PLC-independent membrane depolarization. The results obtained with PMT were consistent with other data, which showed that the PLC inhibitor ET-18-OCH3 blocked inositol phosphate formation without inhibiting catecholamine secretion. This histamine-induced catecholamine secretion also does not involve Ca2+ mobilization, since IP3-receptor inhibitors, such as 2-aminoethoxydiphenylborate (2-APB) or ryanodine plus caffeine, or thapsigargin-depletion of intracellular Ca2+ stores, had no effect.
PMT was used to discriminate between Gq-dependent and Gq-independent signaling induced by saccharin in isolated rod taste cells from frogs (Okada et al. 2001). Data had suggested a role for G-protein-mediated release of IP3 rather than cAMP in the saccharin- induced cationic conductance. However, treatment with PMT did not induce a response in the frog taste cells, suggesting that it may be a Gβγ-coupled PLCβ2 isoform rather than a Gαq-coupled PLCβ1 or PLCβ3 isoform that is involved in saccharin taste transduction. Although this conclusion has since been substantiated by others (Imendra et al. 2002), a cautionary note might be warranted here, regarding interpreting negative results with the use of PMT as a reagent. The investigators had found that PMT did not elicit any current response in the cells (Okada et al. 2001); however, unlike the previous studies mentioned above, they had not verified as a control that PMT in their system was still able to evoke an IP3 response.
PMT has been used to explore the pathways that mediate interaction between endogenous Gαq and Rho signaling. In COS-7 cells, Gαq coimmunoprecipitated with the Rho guanine nucleotide exchange factor (Lbc), and Gαq and Lbc synergistically activated serum response element (SRE.L)-dependent gene expression in a PLC- and Rho-dependent manner (Sagi et al. 2001). In this study, the ability of PMT to synergize with Lbc in stimulating SRE.L-mediated gene expression confirmed that Gαq at endogenous levels also interacts with Rho-GEF-regulated pathways. However, it is still not clear how Gαq activation leads to Rho activation since, unlike for G12/13, expression of Gαq alone did not activate Rho, but did enhance Rho-dependent responses.
PMT has also been used to help define the signaling processes that control maturation of dendritic cells (DCs) (Bagley et al. 2004). In these studies, the investigators were interested in determining the role of PLC and Ca2+ signaling in activation of monocyte-derived DCs. To show the involvement of Ca2+ signaling in DC maturation, they used a number of different agonists, including lipopolysaccharide, CT, dibutyryl-cAMP, prostaglandin E2, and the Ca2+ ionophore A23187, all of which induced maturation. PMT was employed as a control to validate the involvement of PLC signaling in DC maturation. They further showed that this activation by PMT was inhibited by xestospongin, an inhibitor that blocks Ca2+ release from IP3-gated intracellular stores.
PMT as a tool for studying circadian rhythms
PMT has recently been used as a tool to study cholinergic regulation of the circadian clock in the hypothalamic suprachiasmatic nucleus (SCN) of the rat brain. In a brain slice model, the SCN clock is subject to muscarinic regulation through the Gq-coupled M1 mAChR, with sensitivity exhibited only during the night phase of the clock’s 24-h cycle (Gillette et al. 2001). It was found that the effect of 1-h treatment with PMT on circadian clock resetting mimicked the advance of the clock phase induced by carbachol-stimulated M1 mAChR signaling and IP3-mediated Ca2+ release (L. Artinian, W. Yu, B.A. Wilson, E. Gratton, M.U. Gillette, unpublished data). Inhibitors of IP3-mediated Ca2+ release, such as xestospongin, blocked this PMT-induced phase shift. Thus, PMT-mediated activation of the Gq-PLC-IP3-induced Ca2+ release resets the clock in the same direction as activation of muscarinic receptors and cGMP signaling in early night.
PMT as a tool for studying endothelial permeability
PMT has been used to show that Rho activation and resulting cell retraction plays an important role in increased endothelial permeability (Essler et al. 1998). Disruption of endothelial integrity by PMT involves Rho-dependent activation of Rho kinase (ROKα), which in turn inactivates myosin light chain (MLC) phosphatase PP1 and thereby increases MLC phosphorylation and actin reorganization, followed by cell retraction and concomitant rise in endothelial permeability (Fig. 4). PMT-induced actin rearrangement could be blocked by microinjection of the Rho GTPase inhibitor C3 transferase from C. botulinum or microinjection of the Rho-binding domain (RBD) or pleckstrin homology (PH) domain of ROKα, which interfere with ROKα interaction with its regulators. These results have led to the speculation that PMT-mediated Rho activation is responsible for the observed vascular effects of PMT in bite wounds (Aepfelbacher and Essler 2001).
Fig. 4.
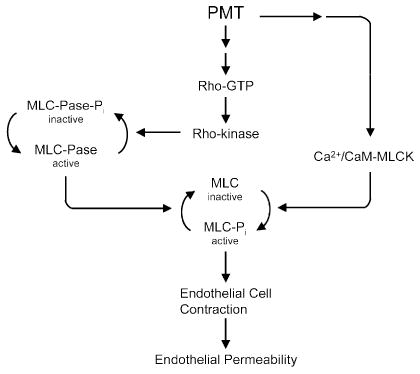
A proposed model for PMT action on Rho-dependent vascular permeability. In this model, PMT stimulates the conversion of Rho protein into its active GTP-bound state. Activated Rho then stimulates Rho kinase to phosphorylate and thereby inactivate myosin light chain phosphatase (MLC-Pase), which in turn prevents the dephosphorylation of MLC, keeping it in its active phosphorylated state. PMT concomitantly causes the release of Ca2+ and activation of Ca2+/calmodulin-dependent MLC kinase (Ca2+/CaM-MLCK), which phosphorylates and activates MLC, resulting in endothelial cell contraction and consequent endothelial permeability. (Adapted from Essler et al. 1998.)
PMT as a tool for studying GIRK signaling
After the initial stimulatory response, PMT effectively uncouples Gq-PLC signaling and prevents any further activation through Gq (Wilson et al. 1997, 2000). Several investigators have demonstrated the effectiveness of using prolonged treatment with PMT to down-regulate Gq-mediated signaling. In heart tissue and in various neuronal and endocrine cells, cellular excitability is regulated by G-protein-coupled inward rectifying K+ (GIRK) channels through selective hormonal stimulation (Sadja et al. 2003). As illustrated in Figure 5, GIRKs are activated in a PT-sensitive manner by receptors coupled to G-proteins of the Go/i family, such as the Gi-coupled A1 adenosine receptor. This activation results from binding of the Gi-protein βγ subunits to the channel. On the other hand, GIRK currents can be inhibited in a PT-insensitive manner either by Gi/o-coupled receptors, such as M2/M4 mAChR, or by Gq-coupled receptors, such as α1AAR or M1/M3 mAChR. However, it was not clear whether the inhibition of GIRKs by these two different receptor types occurred through the same mechanism (Fig. 5).
Fig. 5.
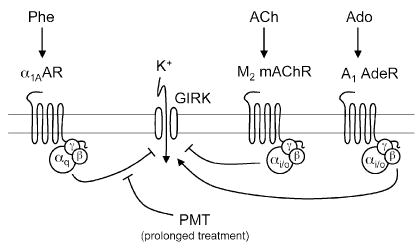
A proposed model for PMT action on GIRK channel inhibition by Gi/o- and Gq-coupled receptors. In this model, opposing sets of G protein-coupled receptors modulate GIRK channels. Stimulation of Gi/o–coupled receptors, such as A1 adenosine receptors, M2 mAChR muscarinic acetylcholine receptors, or serotonin (5-HT1A) receptors, causes the release of Gβγ subunits, which directly interact with and activate GIRK channels to increase K+ currents. Rapid desensitization of the GIRK channel results from subsequent inhibition via the Gαi/o subunits. Inhibition of GIRK currents by the Gq-coupled receptors, such as α1A-AR, ETAR, M1/M3 mAChAR, or thyrotropin-releasing hormone (TRH) receptors, involves activation of PLCβ1 by the released Gαq to cause depletion of PIP2 from the membrane, which results in inactivation of the GIRK channel. Prolonged treatment with PMT uncouples the Gq-signaling pathway and prevents inhibition of GIRK by Gq-coupled receptors, but not desensitization through Gαi/o. ACh, acetylcholine; Phe, phenylephrine; Ado, adenosine.
To address this question, PMT was used to demonstrate the existence of two different regulatory pathways for PT-insensitive inhibition of GIRK channels, one involving PMT- insensitive Gi/o and the other involving PMT-sensitive Gq (Fig. 5) (Buenemann et al. 2000). The researchers examined the effect of prolonged PMT pretreatment on GIRK inhibition in human embryonic kidney HEK293 cells transfected with GIRK1/4, A1 adenosine receptors, and Gq-dependent or Gi/o-dependent inhibitory receptors. They found that pretreatment with PMT did not prevent the Gβγ-mediated GIRK activation by stimulatory A1 adenosine receptors. Likewise, PMT treatment did not affect the Gi/o-coupled M2 mAChR-mediated inhibition of GIRK. In contrast, PMT completely blocked the inhibition of GIRK by the Gαq-coupled α1AAR receptor.
PMT has also been used to mediate uncoupling of Gq signaling in cardiomyocytes to specifically block GIRK inhibition induced by phenylephrine and endothelin-1 (Meyer et al. 2001). In these studies, the investigators provided strong evidence for use of PMT as a superior tool to the commonly used aminosteroid PLC inhibitor U73122 for demonstrating the involvement of Gq-coupled PLCβ activity in mediating GIRK inhibition through PLCβ-mediated depletion of PIP2. These conclusions were further substantiated by results in which PMT was used to examine GIRK channel regulation in HEK293 cells coexpressing GIRK1/4 with the Gi/o-coupled 5-HT1A serotonin or Gq-coupled thyrotropin-releasing hormone (TRH) receptors (Lei et al. 2001). Both TRH and constitutively active Gαq inhibited GIRK. On the other hand, the inhibition of GIRK by TRH was shutdown by prolonged pretreatment with PMT, as well as by treatment with other known inhibitors of Gq signaling, RGS2 and PLCβ1-ct, which bind to Gαq and interfere with Gαq-effector interaction, or by treatment with agents known to lower plasma membrane PIP2 levels via cleavage of PIP2 with 5′-phosphatidylinositol-phosphatase or via sequestration of PIP2 with PLCδ-PH.
PMT as a tool for studying mitogenic signaling
Because PMT acts on Gq, PMT can now be used to study the role of Gq-mediated signaling in hormonal-stimulated mitogenesis. PMT stimulation of Erk signaling was shown to occur via Gq-dependent transactivation of the epidermal growth factor (EGF) receptor in a Ras-dependent manner in some cells, but via a PKC-dependent, Ras-independent pathway in other cells (Fig. 6). In one study (Seo et al. 2000), the mechanism of PMT-mediated Erk activation was compared to that of endogenous Gq/11-protein-coupled α-thrombin receptors in HEK-293 cells. Both PMT and the endogenous Gq-coupled receptors were found to induce Ras-dependent Erk activation via a PKC-independent transactivation of the EGF receptor. For both PMT and the α-thrombin receptor, expression of two inhibitors of Gq signaling, a dominant-negative mutant of the G-protein-coupled receptor kinase (GRK2) and a C-terminal peptide of Gαq (Gαq305–359), blocked Erk activation. Erk activation by PMT was insensitive to a PKC inhibitor (GF109203X), but was blocked by an EGF receptor-specific inhibitor tyrphostin (AG1478), as well as by dominant-negative inhibitors of mSos1 and Ha-Ras. The results suggested that PMT-activated Gαq transactivates the EGF receptor. In the other study involving cardiac fibroblasts, PMT was also found to stimulate Erk activation via EGF receptor transactivation (Sabri et al. 2002). However, in cardiomyocytes novel PKC isoforms mediated PMT activation of Erk1/2, as well as p38-MAPK, and JNK, and the EGF receptor appeared to have no role in this activation (Fig. 6).
Fig. 6.
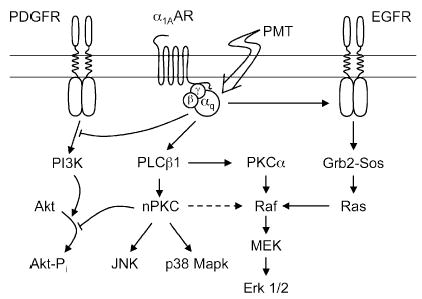
A proposed model for PMT action on Gq-mediated mitogenic signaling. In this model, activation of Gq protein by either PMT or Gq-coupled receptors, such as α1A-AR or α-thrombin receptor, results in activation of PLCβ1, as well as transactivation of the EGF receptor via Gαq subunit and inhibition of the PDGF receptor. In HEK-293 cells and cardiac fibroblasts, subsequent activation of the Erk1/2 cascade is mediated predominantly via Gαq transactivation of the Ras-dependent EGF receptor tyrosin kinase pathway, with no significant contribution derived from the PLCβ1-dependent activation of the PKC pathway. In cardiomyocytes, PMT stimulates via the Gαq-PLCβ1 pathway both Ca2+-dependent PKCα and novel nPKCs (PKCδ and PKCε), which in turn lead to activation of the Erk1/2, as well as p38 MAPK and JNK cascades. PMT both inhibits PDGF receptor-dependent PI 3-kinase (PI3K) activation and prevents subsequent activation of the Akt-dependent survival pathway.
Another example of PMT-mediated transactivation of tyrosine kinase signaling via Gq activation was reported for the generation of inositol phosphoglycans, second messengers of insulin signaling (Sleight et al. 2002). In rat liver membranes, PMT stimulated the production of inositol phosphoglycans, as measured by release of myoinositol and chiroinositol after acid hydrolysis, in a manner similar to what occurs upon insulin stimulation. Interaction between the Gq signaling pathway and the insulin receptor tyrosine kinase pathway was further supported by immunogold-labeling experiments showing colocalization of the insulin receptor β subunit (IRβ) and Gαq/11 in partially purified rat liver membranes, enriched in PLCβ1, clathrin, and caveolin-1 (Sleight et al. 2002). Furthermore, direct interaction of Gαq/11 with IRβ was demonstrated in another study through coimmuno-precipitation (Imamura et al. 1999).
PMT as a tool for studying apoptosis
In neonatal rat cardiomyocytes, PMT induced cardiac hypertrophy (i.e., cardiomyocyte enlargement, sarcomeric organization, and atrial natriuretic factor expression) in a manner similar to that which occurs upon norepinephrine stimulation (Sabri et al. 2002). PMT also activated Erk, and to a lesser extent p38 MAPK and JNK, via activation of PLC and novel PKC isoforms. PMT decreased basal Akt activation by preventing Akt phosphorylation through the activation by EGF or insulin-like growth factor-1 (IGF-1), and consequently enhanced cardiomyocyte susceptibility to apoptotic agents such as H2O2. PMT initially stimulates cardiac hypertrophy in a manner similar to moderate Gq stimulation, yet inhibits the Akt survival pathway and thereby enhances cardiomyocyte susceptibility to apoptosis in a manner similar to what occurs under intense, prolonged stimulation of Gq-coupled receptors (Adams et al. 1998). This suggests that hypertrophy and apoptosis may represent two phases of the same process leading eventually to cardiac decompensation and heart failure and further suggests that PMT might serve as an excellent tool to study this process.
A connection between Gq-coupled α1AAR receptor activation and augmented UV-induced apoptosis through inhibition of phosphatidylinositol 3-kinase (PI3K) and Akt in response to platelet-derived growth factor (PDGF), as well as insulin and insulin-like growth factor 1 (IGF-1), has also come to light (Ballou et al. 2000, 2001). In Rat-1 fibroblasts, PI3K signaling induced by PDGF was also inhibited by treatment with PMT in a manner similar to α1AAR (Fig. 6) (Lin et al. 2003). In this study, PMT pretreatment reduced the amount of phospho-Tyr751 on the PDGF receptor β subunit, thereby eliminating the docking site for the p85 subunit of PI3K. In addition, PMT pretreatment significantly inhibited the PDGF-induced Akt phosphorylation at Ser473.
Conclusions and future prospects
Discriminating among the various G-proteins involved in signal transduction processes has always been a challenge for cell biologists, physiologists, and pharmacologists. The discovery that certain toxins produced by bacteria can selectively act on different G-proteins has provided researchers with a growing repertoire of agents that can be used to manipulate signaling pathways for elucidating cellular functions of various signaling molecules. Until recently, studying the signaling of Gq GTPases was relatively intractable due to the lack of effective molecular tools that were specific for them; in fact, this family was referred to simply as the PT-insensitive G-proteins. While we do not yet know the precise biochemical basis for PMT action on Gq, there is considerable and convincing evidence that PMT can be used as a highly selective agent that targets Gq protein-coupled PLC signal transduction. Because initial exposure to PMT results in activation of Gq signaling, but prolonged treatment subsequently uncouples Gq-dependent PLC signaling, PMT can be used as both an activator and a downregulator of Gq-PLC signaling, depending upon the length of toxin treatment. PMT has already been shown to decipher a number of important signaling pathways involving Gq signaling. With the introduction of PMT as a Gq-selective molecular tool for studying PLC-mediated signaling, we can now begin to discern the different roles that Gq family members play in other signal transduction pathways and physiological processes. An important question that still remains to be answered is whether the other Gq family members, which are not coupled to PLC, might also be targets of PMT. Indeed, there is strong evidence for other PMT effects that are independent of Gq-PLC signaling in Gq- and Gq/11-knockout cells.
Acknowledgments
Some of the work reported here was supported by grants from the National Institutes of Health (NIAID/AI38396) and from the United States Department of Agriculture (NRI/1999–02295) (to B.A.W.).
References
- Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW., 2nd Enhanced Gαq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aepfelbacher M, Essler M. Disturbance of endothelial barrier function by bacterial toxins and atherogenic mediators: a role for Rho/Rho kinase. Cell Microbiol. 2001;3:649–658. doi: 10.1046/j.1462-5822.2001.00145.x. [DOI] [PubMed] [Google Scholar]
- Aktories K, Schmidt G, Just I. Rho GTPases as targets of bacterial protein toxins. Biol Chem. 2000;381:421–426. doi: 10.1515/BC.2000.054. [DOI] [PubMed] [Google Scholar]
- Bagley KC, Abdelwahab SF, Tuskan RG, Lewis GK. Calcium signaling through phospholipase C activates dendritic cells to mature and is necessary for the activation and maturation of dendritic cells induced by diverse agonists. Clin Diagn Lab Immunol. 2004;11:77–82. doi: 10.1128/CDLI.11.1.77-82.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin MR, Pullinger GD, Lax AJ. Pasteurella multocida toxin facilitates inositol phosphate formation by bombesin through tyrosine phosphorylation of G alpha q. J Biol Chem. 2003;278:32719–32725. doi: 10.1074/jbc.M303524200. [DOI] [PubMed] [Google Scholar]
- Ballou LM, Cross ME, Huang S, McReynolds EM, Zhang BX, Lin RZ. Differential regulation of the phosphatidylinositol 3-kinase/Akt and p70 S6 kinase pathways by α(1A)-adrenergic receptor in rat-1 fibroblasts. J Biol Chem. 2000;275:4803–4809. doi: 10.1074/jbc.275.7.4803. [DOI] [PubMed] [Google Scholar]
- Ballou LM, Tian PY, Lin HY, Jiang YP, Lin RZ. Dual regulation of glycogen synthase kinase-3β by the α1A-adrenergic receptor. J Biol Chem. 2001;276:40910–40916. doi: 10.1074/jbc.M103480200. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol triphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Berstein G, Blank JL, Jhon DY, Exton JH, Rhee SG, Ross EM. Phospholipase C-β1 is a GTPase-activating protein for Gq/11, its physiologic regulator. Cell. 1992a;70:411–418. doi: 10.1016/0092-8674(92)90165-9. [DOI] [PubMed] [Google Scholar]
- Berstein G, Blank JL, Smrka AV, Higashijima T, Sternweis PC, Exton JH, Ross EM. Reconstitution of agonist-stimulated phosphatidylinositol 4,5-bisphosphate hydrolysis using purified m1 muscarinic receptor, Gq/11, and phospholipase Cβ-1. J Biol Chem. 1992b;267:8081–8088. [PubMed] [Google Scholar]
- Buenemann M, Meyer T, Pott L, Hosey M. Novel inhibition of Gβγ-activated potassium currents induced by M2 muscarinic receptors via a pertussis toxin-insensitive pathway. J Biol Chem. 2000;275:12537–12545. doi: 10.1074/jbc.275.17.12537. [DOI] [PubMed] [Google Scholar]
- Camps M, Carozzi A, Schnabel P, Scheer A, Parker PJ, Gierschik P. Isozyme-selective stimulation of phospholipase C-β2 by G protein βγ-subunits. Nature. 1992;360:684–686. doi: 10.1038/360684a0. [DOI] [PubMed] [Google Scholar]
- Casey PJ, Gilman AG. G protein involvement in receptor-effector coupling. J Biol Chem. 1988;263:2577–2580. [PubMed] [Google Scholar]
- Casey PJ, Fong HK, Simon MI, Gilman AG. Gz, a guanine nucleotide-binding protein with unique biochemical properties. J Biol Chem. 1990;265:2383–2390. [PubMed] [Google Scholar]
- Chikumi H, Vazquez-Prado J, Servitja JM, Miyazaki H, Gutkind JS. Potent activation of RhoA by Gα q and Gq-coupled receptors. J Biol Chem. 2002;277:27130–27134. doi: 10.1074/jbc.M204715200. [DOI] [PubMed] [Google Scholar]
- Chrisp CE, Foged NT. Induction of pneumonia in rabbits by use of a purified protein toxin from Pasteurella multocida. Am J Vet Res. 1991;52:56–61. [PubMed] [Google Scholar]
- Deckmyn H, Van Geet C, Vermylen J. Dual regulation of phospholipase C activity by G proteins. News Physiol Sci. 1993;8:61–63. [Google Scholar]
- Donald AN, Wallace DJ, McKenzie S, Marley PD. Phospholipase C-mediated signalling is not required for histamine-induced catecholamine secretion from bovine chromaffin cells. J Neurochem. 2002;81:1116–1129. doi: 10.1046/j.1471-4159.2002.00915.x. [DOI] [PubMed] [Google Scholar]
- Dudet LI, Chailler P, Dubreuil JD, Martineau-Doize B. Pasteurella multocida toxin stimulates mitogenesis and cytoskeleton reorganization in Swiss 3T3 fibroblasts. J Cell Physiol. 1996;168:173–182. doi: 10.1002/(SICI)1097-4652(199607)168:1<173::AID-JCP21>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Dutt P, Kjoller L, Giel M, Hall A, Toksoz D. Activated Gαq family members induce Rho GTPase activation and Rho-dependent actin filament assembly. FEBS Lett. 2002;531:565–569. doi: 10.1016/s0014-5793(02)03625-6. [DOI] [PubMed] [Google Scholar]
- Essler M, Hermann K, Amano M, Kaibuchi K, Heesemann J, Weber PC, Aepfelbacher M. Pasteurella multocida toxin increases endothelial permeability via Rho kinase and myosin light chain phosphatase. J Immunol. 1998;161:5640–5646. [PubMed] [Google Scholar]
- Felix R, Fleisch H, Frandsen PL. Effect of Pasteurella multocida toxin on bone resorption in vitro. Infect Immun. 1992;60:4984–4988. doi: 10.1128/iai.60.12.4984-4988.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields TA, Casey PJ. Signalling functions and biochemical properties of pertussis toxin-resistant G-proteins. Biochem J. 1997;321:561–571. doi: 10.1042/bj3210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foged NT. Pasteurella multocida toxin. The characterisation of the toxin and its significance in the diagnosis and prevention of progressive atrophic rhinitis in pigs. APMIS Suppl. 1992;25:1–56. [PubMed] [Google Scholar]
- Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS. A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J Biol Chem. 1999;274:5868–5879. doi: 10.1074/jbc.274.9.5868. [DOI] [PubMed] [Google Scholar]
- Gilbertson TA, Damak S, Margolskee RF. The molecular physiology of taste transduction. Curr Opin Neurobiol. 2000;10:519–527. doi: 10.1016/s0959-4388(00)00118-5. [DOI] [PubMed] [Google Scholar]
- Gillette MU, Buchanan GF, Artinian L, Hamilton SE, Nathanson NM, Liu C. Role of the M1 receptor in regulating circadian rhythms. Life Sci. 2001;68:2467–2472. doi: 10.1016/s0024-3205(01)01040-2. [DOI] [PubMed] [Google Scholar]
- Gosau N, Fahimi-Vahid M, Michalek C, Schmidt M, Wieland T. Signalling components involved in the coupling of α1-adrenoceptors to phospholipase D in neonatal rat cardiac myocytes. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:468–476. doi: 10.1007/s00210-002-0546-x. [DOI] [PubMed] [Google Scholar]
- Gratacap MP, Payrastre B, Nieswandt B, Offermanns S. Differential regulation of Rho and Rac through heterotrimeric G-proteins and cyclic nucleotides. J Biol Chem. 2001;276:47906–47913. doi: 10.1074/jbc.M104442200. [DOI] [PubMed] [Google Scholar]
- Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- Hepler JR, Kozasa T, Smrcka AV, Simon MI, Rhee SG, Sternweis PC, Gilman AG. Purification from Sf9 cells and characterization of recombinant Gq α and G11 α. Activation of purified phospholipase C isozymes by G α subunits. J Biol Chem. 1993;268:14367–14375. [PubMed] [Google Scholar]
- Higgins TE, Murphy AC, Staddon JM, Lax AJ, Rozengurt E. Pasteurella multocida toxin is a potent inducer of anchorage-independent cell growth. Proc Natl Acad Sci USA. 1992;89:4240–4244. doi: 10.1073/pnas.89.10.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho MK, Wong YH. G(z) signaling: emerging divergence from G(i) signaling. Oncogene. 2001;20:1615–1625. doi: 10.1038/sj.onc.1204190. [DOI] [PubMed] [Google Scholar]
- Imamura T, Vollenweider P, Egawa K, Clodi M, Ishibashi K, Nakashima N, Ugi S, Adams JW, Brown JH, Olefsky JM. Gα-q/11 protein plays a key role in insulin-induced glucose transport in 3T3-L1 adipocytes. Mol Cell Biol. 1999;19:6765–6774. doi: 10.1128/mcb.19.10.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imendra KG, Miyamoto T, Okada Y, Toda K. Serotonin differentially modulates the electrical properties of different subsets of taste receptor cells in bullfrog. Eur J Neurosci. 2002;16:629–640. doi: 10.1046/j.1460-9568.2002.02107.x. [DOI] [PubMed] [Google Scholar]
- Jones DT, Masters SB, Bourne HR, Reed RR. Biochemical characterization of three stimulatory GTP-binding proteins. The large and small forms of Gs and the olfactory-specific G-protein, Golf. J Biol Chem. 1990;265:2671–2676. [PubMed] [Google Scholar]
- Jordan RW, Hamilton TD, Hayes CM, Patel D, Jones PH, Roe JM, Williams NA. Modulation of the humoral immune response of swine and mice mediated by toxigenic Pasteurella multocida. FEMS Immunol Med Microbiol. 2003;39:51–59. doi: 10.1016/S0928-8244(03)00201-3. [DOI] [PubMed] [Google Scholar]
- Jutras I, Martineau-Doize B. Stimulation of osteoclast-like cell formation by Pasteurella multocida toxin from hemopoietic progenitor cells in mouse bone marrow cultures. Can J Vet Res. 1996;60:34–39. [PMC free article] [PubMed] [Google Scholar]
- Katoh H, Aoki J, Yamaguchi Y, Kitano Y, Ichikawa A, Negishi M. Constitutively active Gα12, Gα13, and Gαq induce Rho-dependent neurite retraction through different signaling pathways. J Biol Chem. 1998;273:28700–28707. doi: 10.1074/jbc.273.44.28700. [DOI] [PubMed] [Google Scholar]
- Kimman TG, Lowik CW, van de Wee-Pals LJ, Thesingh CW, Defize P, Kamp EM, Bijvoet OL. Stimulation of bone resorption by inflamed nasal mucosa, dermonecrotic toxin-containing conditioned medium from Pasteurella multocida, and purified dermonecrotic toxin from P. multocida. Infect Immun. 1987;55:2110–2116. doi: 10.1128/iai.55.9.2110-2116.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- Kurose H. Gα12 and Gα13 as key regulatory mediator in signal transduction. Life Sci. 2003;74:155–161. doi: 10.1016/j.lfs.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Lacerda HM, Lax AJ, Rozengurt E. Pasteurella multocida toxin, a potent intracellularly acting mitogen, induces p125FAK and paxillin tyrosine phosphorylation, actin stress fiber formation, and focal contact assembly in Swiss 3T3 cells. J Biol Chem. 1996;271:439–445. doi: 10.1074/jbc.271.1.439. [DOI] [PubMed] [Google Scholar]
- Lalli G, Bohnert S, Deinhardt K, Verastegui C, Schiavo G. The journey of tetanus and botulinum neurotoxins in neurons. Trends Microbiol. 2003;11:431–437. doi: 10.1016/s0966-842x(03)00210-5. [DOI] [PubMed] [Google Scholar]
- Lax AJ, Chanter N. Cloning of the toxin gene from Pasteurella multocida and its role in atrophic rhinitis. J Gen Microbiol. 1990;136:81–87. doi: 10.1099/00221287-136-1-81. [DOI] [PubMed] [Google Scholar]
- Lax AJ, Thomas W. How bacteria could cause cancer: one step at a time. Trends Microbiol. 2002;10:293–299. doi: 10.1016/s0966-842x(02)02360-0. [DOI] [PubMed] [Google Scholar]
- Lei Q, Talley EM, Bayliss DA. Receptor-mediated inhibition of G protein-coupled inwardly rectifying potassium channels involves Gαq family subunits, phospholipase C, and a readily diffusible messenger. J Biol Chem. 2001;276:16720–16730. doi: 10.1074/jbc.M100207200. [DOI] [PubMed] [Google Scholar]
- Lin HY, Ballou LM, Lin RZ. Stimulation of the α1A adrenergic receptor inhibits PDGF-induced PDGF β receptor Tyr751 phosphorylation and PI 3-kinase activation. FEBS Lett. 2003;540:106–110. doi: 10.1016/s0014-5793(03)00233-3. [DOI] [PubMed] [Google Scholar]
- Martineau-Doize B, Caya I, Gagne S, Jutras I, Dumas G. Effects of Pasteurella multocida toxin on the osteoclast population of the rat. J Comp Pathol. 1993;108:81–91. doi: 10.1016/s0021-9975(08)80230-7. [DOI] [PubMed] [Google Scholar]
- Meyer T, Wellner-Kienitz MC, Biewald A, Bender K, Eickel A, Pott L. Depletion of phosphatidylinositol 4,5-bisphosphate by activation of phospholipase C-coupled receptors causes slow inhibition but not desensitization of G protein-gated inward rectifier K+ current in atrial myocytes. J Biol Chem. 2001;276:5650–5658. doi: 10.1074/jbc.M009179200. [DOI] [PubMed] [Google Scholar]
- Mullan PB, Lax AJ. Pasteurella multocida toxin stimulates bone resorption by osteoclasts via interaction with osteoblasts. Calcif Tissue Int. 1998;63:340–345. doi: 10.1007/s002239900537. [DOI] [PubMed] [Google Scholar]
- Murphy AC, Rozengurt E. Pasteurella multocida toxin selectively facilitates phosphatidylinositol 4,5-bisphosphate hydrolysis by bombesin, vasopressin, and endothelin. Requirement for a functional G protein. J Biol Chem. 1992;267:25296–25303. [PubMed] [Google Scholar]
- Ohnishi T, Horiguchi Y, Masuda M, Sugimoto N, Matsuda M. Pasteurella multocida toxin and Bordetella bronchiseptica dermonecrotizing toxin elicit similar effects on cultured cells by different mechanisms. J Vet Med Sci. 1998;60:301–305. doi: 10.1292/jvms.60.301. [DOI] [PubMed] [Google Scholar]
- Okada Y, Fujiyama R, Miyamoto T, Sato T. Saccharin activates cation conductance via inositol 1,4,5-trisphosphate production in a subset of isolated rod taste cells in the frog. Eur J Neurosci. 2001;13:308–314. [PubMed] [Google Scholar]
- Orth JH, Lang S, Aktories K. Action of Pasteurella multocida toxin depends on the helical domain of Gαq. J Biol Chem. 2004;279:34150–34155. doi: 10.1074/jbc.M405353200. [DOI] [PubMed] [Google Scholar]
- Park D, Jhon DY, Lee CW, Lee KH, Rhee SG. Activation of phospholipase C isozymes by G protein βγ subunits. J Biol Chem. 1993;268:4573–4576. [PubMed] [Google Scholar]
- Pennings AM, Storm PK. A test in vero cell monolayers for toxin production by strains of Pasteurella multocida isolated from pigs suspected of having atrophic rhinitis. Vet Microbiol. 1984;9:503–508. doi: 10.1016/0378-1135(84)90071-3. [DOI] [PubMed] [Google Scholar]
- Petersen SK. The complete nucleotide sequence of the Pasteurella multocida toxin gene and evidence for a transcriptional repressor, TxaR. Mol Microbiol. 1990;4:821–830. doi: 10.1111/j.1365-2958.1990.tb00652.x. [DOI] [PubMed] [Google Scholar]
- Petersen SK, Foged NT. Cloning and expression of the Pasteurella multocida toxin gene, toxA, in Escherichia coli. Infect Immun. 1989;57:3907–3913. doi: 10.1128/iai.57.12.3907-3913.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit RK, Ackermann MR, Rimler RB. Receptor-mediated binding of Pasteurella multocida dermonecrotic toxin to canine osteosarcoma and monkey kidney (vero) cells. Lab Invest. 1993;69:94–100. [PubMed] [Google Scholar]
- Quick MW, Simon MI, Davidson N, Lester HA, Aragay AM. Differential coupling of G protein α subunits to seven-helix receptors expressed in Xenopus oocytes. J Biol Chem. 1994;269:30164–30172. [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Choi KD. Regulation of inositol phospholipid-specific phospholipase C isozymes. J Biol Chem. 1992;267:12393–12396. [PubMed] [Google Scholar]
- Rossetto O, Seveso M, Caccin P, Schiavo G, Montecucco C. Tetanus and botulinum neurotoxins: turning bad guys into good by research. Toxicon. 2001;39:27–41. doi: 10.1016/s0041-0101(00)00163-x. [DOI] [PubMed] [Google Scholar]
- Rozengurt E, Higgins T, Chanter N, Lax AJ, Staddon JM. Pasteurella multocida toxin: potent mitogen for cultured fibroblasts. Proc Natl Acad Sci USA. 1990;87:123–127. doi: 10.1073/pnas.87.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter JM, Luther PD. Cell culture assay for toxigenic Pasteurella multocida from atrophic rhinitis of pigs. Vet Rec. 1984;114:393–396. doi: 10.1136/vr.114.16.393. [DOI] [PubMed] [Google Scholar]
- Sabri A, Pak E, Alcott SA, Wilson BA, Steinberg SF. Coupling function of endogenous α1- and β-adrenergic receptors in mouse cardiomyocytes. Circ Res. 2000;86:1047–1053. doi: 10.1161/01.res.86.10.1047. [DOI] [PubMed] [Google Scholar]
- Sabri A, Wilson BA, Steinberg SF. Dual actions of the Gαq agonist Pasteurella multocida toxin to promote cardiomyocyte hypertrophy and enhance apoptosis susceptibility. Circ Res. 2002;90:850–857. doi: 10.1161/01.RES.0000016165.23795.1F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadja R, Alagem N, Reuveny E. Gating of GIRK channels: details of an intricate, membrane-delimited signaling complex. Neuron. 2003;39:9–12. doi: 10.1016/s0896-6273(03)00402-1. [DOI] [PubMed] [Google Scholar]
- Sagi SA, Seasholtz TM, Kobiashvili M, Wilson BA, Toksoz D, Brown JH. Physical and functional interactions of Gαq with Rho and its exchange factors. J Biol Chem. 2001;276:15445–15452. doi: 10.1074/jbc.M008961200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah VP, Seasholtz TM, Sagi SA, Brown JH. The role of Rho in G protein-coupled receptor signal transduction. Annu Rev Pharmacol Toxicol. 2000;40:459–489. doi: 10.1146/annurev.pharmtox.40.1.459. [DOI] [PubMed] [Google Scholar]
- Schiavo G, van der Goot FG. The bacterial toxin toolkit. Nature Rev Mol Cell Biol. 2001;2:530–537. doi: 10.1038/35080089. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Matteoli M, Montecucco C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000;80:717–766. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- Seo B, Choy EW, Maudsley S, Miller WE, Wilson BA, Luttrell LM. Pasteurella multocida toxin stimulates mitogen-activated protein kinase via Gq/11-dependent transactivation of the epidermal growth factor receptor. J Biol Chem. 2000;275:2239–2245. doi: 10.1074/jbc.275.3.2239. [DOI] [PubMed] [Google Scholar]
- Shime H, Ohnishi T, Nagao K, Oka K, Takao T, Horiguchi Y. Association of Pasteurella multocida toxin with vimentin. Infect Immun. 2002;70:6460–6463. doi: 10.1128/IAI.70.11.6460-6463.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleight S, Wilson BA, Heimark DB, Larner J. Gq/11 is involved in insulin-stimulated inositol phosphoglycan putative mediator generation in rat liver membranes: co-localization of Gq/11 with the insulin receptor in membrane vesicles. Biochem Biophys Res Commun. 2002;295:561–569. doi: 10.1016/s0006-291x(02)00701-5. [DOI] [PubMed] [Google Scholar]
- Smrcka AV, Sternweis PC. Regulation of purified subtypes of phosphatidylinositol-specific phospholipase C β by G protein α and βγ subunits. J Biol Chem. 1993;268:9667–9674. [PubMed] [Google Scholar]
- Staddon JM, Chanter N, Lax AJ, Higgins TE, Rozengurt E. Pasteurella multocida toxin, a potent mitogen, stimulates protein kinase C-dependent and -independent protein phosphorylation in Swiss 3T3 cells. J Biol Chem. 1990;265:11841–11848. [PubMed] [Google Scholar]
- Staddon JM, Barker CJ, Murphy AC, Chanter N, Lax AJ, Michell RH, Rozengurt E. Pasteurella multocida toxin, a potent mitogen, increases inositol 1,4,5- trisphosphate and mobilizes Ca2+ in Swiss 3T3 cells. J Biol Chem. 1991;266:4840–4847. [PubMed] [Google Scholar]
- Staddon JM, Bouzyk MM, Rozengurt E. Interconversion of GRP78/BiP. A novel event in the action of Pasteurella multocida toxin, bombesin, and platelet-derived growth factor. J Biol Chem. 1992;267:25239–25245. [PubMed] [Google Scholar]
- Sterner-Kock A, Lanske B, Uberschar S, Atkinson MJ. Effects of the Pasteurella multocida toxin on osteoblastic cells in vitro. Vet Pathol. 1995;32:274–279. doi: 10.1177/030098589503200309. [DOI] [PubMed] [Google Scholar]
- Sternweis PC, Smrcka AV. Regulation of phospholipase C by G proteins. Trends Biol Sci. 1992;17:502–506. doi: 10.1016/0968-0004(92)90340-f. [DOI] [PubMed] [Google Scholar]
- Thomas W, Pullinger GD, Lax AJ, Rozengurt E. Escherichia coli cytotoxic necrotizing factor and Pasteurella multocida toxin induce focal adhesion kinase autophosphorylation and Src association. Infect Immun. 2001;69:5931–5935. doi: 10.1128/IAI.69.9.5931-5935.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemori H, Inoue T, Kume S, Sekiyama N, Nagao M, Itoh H, Nakanishi S, Mikoshiba K, Yamamoto T. Activation of the G protein Gq/11 through tyrosine phosphorylation of the alpha subunit. Science. 1997;276:1878–1881. doi: 10.1126/science.276.5320.1878. [DOI] [PubMed] [Google Scholar]
- Umemori H, Hayashi T, Inoue T, Nakanishi S, Mikoshiba K, Yamamoto T. Involvement of protein tyrosine phosphatases in activation of the trimeric G protein Gq/11. Oncogene. 1999;18:7399–7402. doi: 10.1038/sj.onc.1203152. [DOI] [PubMed] [Google Scholar]
- Van Dop C, Yamanaka G, Steinberg F, Sekura RD, Manclark CR, Stryer L, Bourne HR. ADP-ribo-sylation of transducin by pertussis toxin blocks the light-stimulated hydrolysis of GTP and cGMP in retinal photoreceptors. J Biol Chem. 1984a;259:23–26. [PubMed] [Google Scholar]
- Van Dop C, Tsubokawa M, Bourne HR, Ramachandran J. Amino acid sequence of retinal transducin at the site ADP-ribosylated by cholera toxin. J Biol Chem. 1984b;259:696–698. [PubMed] [Google Scholar]
- Vogt S, Grosse R, Schultz G, Offermanns S. Receptor-dependent RhoA activation in G12/G13-deficient cells: genetic evidence for an involvement of Gq/G11. J Biol Chem. 2003;278:28743–28749. doi: 10.1074/jbc.M304570200. [DOI] [PubMed] [Google Scholar]
- Wilson BA, Salyers AA. Ecology and physiology of infectious bacteria—implications for biotechnology. Curr Opin Biotechnol. 2002;13:267–274. doi: 10.1016/s0958-1669(02)00312-9. [DOI] [PubMed] [Google Scholar]
- Wilson BA, Zhu X, Ho M, Lu L. Pasteurella multocida toxin activates the inositol triphosphate signaling pathway in Xenopus oocytes via Gqα-coupled phospholipase C-β1. J Biol Chem. 1997;272:1268–1275. doi: 10.1074/jbc.272.2.1268. [DOI] [PubMed] [Google Scholar]
- Wilson BA, Aminova LR, Ponferrada VG, Ho M. Differential modulation and subsequent blockade of mitogenic signaling and cell cycle progression by Pasteurella multocida toxin. Infect Immun. 2000;68:4531–4538. doi: 10.1128/iai.68.8.4531-4538.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Katz A, Simon MI. Activation of phospholipase C β2 by the α and βγ subunits of trimeric GTP-binding protein. Proc Natl Acad Sci USA. 1993;90:5297–5301. doi: 10.1073/pnas.90.11.5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zywietz A, Gohla A, Schmelz M, Schultz G, Offermanns S. Pleiotropic effects of Pasteurella multocida toxin are mediated by Gq-dependent and -independent mechanisms. Involvement of Gq but not G11. J Biol Chem. 2001;276:3840–3845. doi: 10.1074/jbc.M007819200. [DOI] [PubMed] [Google Scholar]