

Abstract
Mitochondrial thioredoxin (mtTrx) can be oxidized in response to inducers of oxidative stress; yet the functional consequences of the oxidation have not been determined. This study evaluated the redox status of mtTrx and its association to oxidant-induced apoptosis. Results showed that mtTrx was oxidized after exposure to peroxides and diamide. Overexpression of mtTrx protected against diamide-induced oxidation and cytotoxicity. Oxidation of mtTrx was also achieved by knocking down its reductase; and lead to increased susceptibility to cell death. The data indicate that the redox status of mtTrx is a regulatory mechanism underlying the vulnerability of mitochondria to oxidative injury.
Keywords: thioredoxin, mitochondria, oxidative stress, redox, thioredoxin reductase
Abbreviations used: AMS, 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid; mtΔψ; mitochondrial membrane potential; mtTrx, mitochondrial thioredoxin; PS, phosphatidylserine; tBH, tert-butylhydroperoxide; TMRM, tetramethylrhodamine methyl ester; TR, thioredoxin reductase; Trx1, thioredoxin 1
1. INTRODUCTION
The redox status of protein sulfhydryl groups is essential for mammalian cell functions [1–3]. Thiol-dependent antioxidant proteins are key component of the redox regulation system. They function by eliminating reactive oxygen species (ROS) and thereby controlling the concentrations of reactive signaling intermediates. Some of these antioxidant proteins, such as thioredoxin (Trx) and glutaredoxin, also directly reduce oxidized proteins and reverse the oxidative stress-induced changes in their activities [4,5].
Trx family proteins are conserved redox proteins containing the invariant active center -Cys-Gly-Pro-Cys- [6]. Functioning as electron donors for various redox reactions in vivo, Trxs play fundamental roles in diverse cellular events by regulating DNA binding activities of a number of transcription factors as well as interacting with other protein substrates. Trxs participate in ROS clearance by providing electrons to peroxiredoxins (Prxs) which detoxify hydrogen peroxide into water. In mammals there are at least two complete Trx systems, the cytosolic Trx system (Trx 1, thioredoxin reductase 1 (TR1) and NADPH) and the mitochondrial Trx system (mtTrx, thioredoxin reductase 2 (TR2) and NADPH) [7–10]. The protein components of these two systems are functional homologues but are encoded by different genes. Both systems are dependent on NADPH that serves as the ultimate electron donor. Both Trx 1 and mtTrx are capable of reducing insulin in vitro and are good substrates for either TR1 or TR2 [7–9].
The majority of the functions of Trxs rely on their thiol reductase activities [6]. In the present study, we showed that, in response to various conditions of oxidative stress, mtTrx was more extensively oxidized and slower in recovery than the cytosolic Trx 1. Using overexpression and siRNA approaches, we showed that the redox status of mtTrx controlled the sensitivity to oxidative stress-induced apoptosis. Our data suggest that, in addition to the abundant ROS production in the mitochondria, the sensitivity of mitochondrial proteins per se may provide another mechanism that accounts for the vulnerability of mitochondria to oxidative injury. Oxidation of the mitochondrial protein thiols may lead to increased susceptibility to apoptotic cell death.
2. MATERIALS AND METHODS
2.1. Cell culture
Human 143 B osteosarcoma cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). Cells stably transfected with full-length human mtTrx were described previously [10]. All cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS) in a humidified CO2 incubator at 37°C. For all experiments, 80–90% confluent cells were used for treatments.
2.2. Protein expression, purification and antibody production
The truncated form of human mtTrx that lacks the N-terminal mitochondrial localization signal [10] was subcloned into pET15b expression vector (EMD Biosciences, Inc, San Diego, CA) and transformed into E. coli strain BL21 (EMD Biosciences). The induction and purification were performed as described elsewhere with modifications [7]. Briefly, the expression of recombinant protein was induced with 1 mM of isopropyl-beta-D-thiogalactopyranoside at 37°C for 3 h. Cells were harvested by centrifugation at 6,000 g for 10 min and the pellet was resuspended with BugBuster™ Protein Extraction Reagent (EMD Biosciences). The lysates were loaded onto a HiTrap affinity column (Amersham pharmacia Biotech Inc, Piscataway, NJ) and the wash and elution steps were performed following the manufacturer’s instructions. The homogeneity of purified protein was confirmed with silver stained-SDS/PAGE. The protein was injected into New Zealand White rabbits for the antiserum production (Covance, Princeton, NJ). The obtained antibody showed specific binding to human mtTrx and no cross-reactivity to human Trx 1 on Western blot analyses (data not shown).
2.3. Redox Western analysis
To assess the in vivo redox status of mammalian Trxs, we used a method that was previously described with modifications [11,12]. The assay principle is illustrated in Fig. 1. The reduced cysteine residues were alkylated with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) (Molecular Probes, Eugene, OR) followed by non-reducing Western blot to distinguish between the reduced (alkylated) and oxidized (nonalkylated) proteins. Cells were harvested with ice-cold 10% trichloroacetic acid and collected after centrifugation. Pellets were resuspended with 100% acetone and kept on ice for 15 min. At the end of incubation, cells were centrifuged for 10 min. The pellet was dissolved in lysis buffer (62.5 mM Tris-Cl, pH 6.8, 1 % SDS) with 25 mM AMS. The lysates were incubated for 30 min at 4°C followed by 10 min incubation at 37°C. Samples were resolved with 15% non-reducing SDS-PAGE. Western analysis was performed with antibodies against Trx 1 (American Diagnostica, Inc., Greenwich, CT) and mtTrx. The signals were detected by an Odyssey infrared imaging system (LI-COR, Lincoln, NE). The redox status of protein was presented as the percentage of oxidized proteins.
Fig. 1.
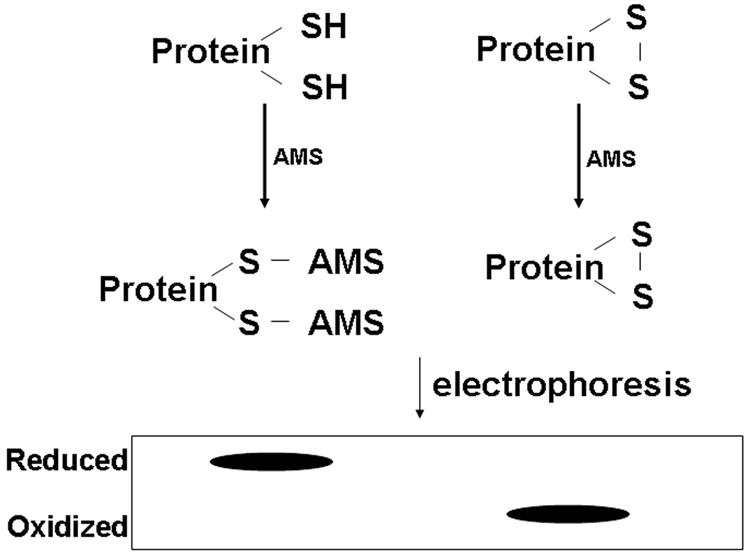
Schematic representation of Redox Western analysis. The alkylating agent AMS reacts with –SH groups and adds ~500 Da in the molecular weight. The reaction conditions were optimized so that AMS will not alkylate amines.
2.4. Knock down TR2 expression by siRNA approach
The siRNA against TR2 (5′-AAG GAG GAC ACG GGC ACC U-3′) was synthesized by Integrated DNA technologies (Coralville, IA). Control scrambled siRNA was purchased from Ambion (Austin, TX). Cells were transfected with the double stranded-siRNA using Lipofectamine 2000 Reagent (Invitrogen). At 36 h after transfection, cells were further treated with peroxide in fresh media for an additional 12 h. All treatments were performed in culture media containing 10% FBS. Cell viability was measured using the LIVE/DEAD viability/cytotoxicity kit (Molecular Probes) followed by flow cytometry (BD Immunocytometry Systems, San Jose, CA) as described [10].
2.5. Measurement of Mitochondrial Depolarization
Diamide (1,1′-Azobis(N,N-dimethylformamide) was obtained from Sigma ( St. Louis, MO) and was prepared as 1,000 X stock in water. Treatments were performed in culture media containing 10% FBS. Cells were harvested by gentle trypsinization. To measure mitochondrial membrane potential (mtΔψ), cells were stained with 200 nM tetramethylrhodamine methyl ester (TMRM) in culture media for 15 minutes at 37 ºC. After washing with phosphate-buffered saline (PBS), the TMRM fluorescence was analyzed by flow cytometry.
2.6. Apoptosis Assays
Apoptosis was measured by phosphatidylserine (PS) exposure and caspase activation. After diamide exposure, cells were labeled with fluorescein-conjugated Annexin V (Molecular Probes) in binding buffer supplied by the manufacturer. The labeling reactions were performed at room temperature for 20 minutes and the samples were then kept on ice until analyzed by flow cytometry. To measure caspase activation, cells were harvested after treatments and incubated with 10 μM FITC-VAD-FMK (Promega, Madison, WI) for 30 minutes at 37 ºC. Cells were then washed once with PBS and analyzed by flow cytometry.
3. RESULTS
3.1. Oxidation of Trxs was an early event in oxidative stress
To determine the redox status of mtTrx and Trx 1 in vivo, we used AMS to alkylate thiols of the protein cysteine residues followed by the Western blot analysis. Alkylation with AMS adds ~500 Da molecular weight to every sulfhydryl group. When run on non-reducing SDS-PAGE, proteins in their reduced (alkylated) and oxidized (non-alkylated) forms can be resolved by the increase in molecular weight due to the addition of AMS (Fig. 1).
H2O2 and t-butylhydroperoxide (tBH), a relatively stable peroxide with potent mitochondrial effects [13], were used to generate oxidative stress in 143B human osteosarcoma cells. The same cell line was used in our previous studies of mtTrx [10]. Both agents induced oxidation of mtTrx and Trx 1 in a dose-dependent manner (Fig. 2); however, mtTrx and Trx 1 showed different sensitivities. Treatment with tBH, the peroxide inducing mitochondrial oxidative stress [13], induced over 80% oxidation of mtTrx at 50 μM concentration within 10 min. The oxidation is indicated by the appearance of the faster moving band on the SDS-PAGE (Fig. 2A, labeled as mtTrxo). The extent of oxidation of mtTrx was not further increased at higher concentrations (Fig. 2B). The maximum oxidation of Trx 1 was achieved at 300 μM tBH but was only ~40% (Fig. 2B). Similar to mtTrx, oxidized Trx1 moved faster than its reduced form (Fig. 2A). In contrast to tBH, H2O2 induced maximum oxidation of both mtTrx and Trx 1 at concentrations over 300 μM (Fig. 2C). Again, mtTrx was oxidized to a much greater extent.
Fig. 2.
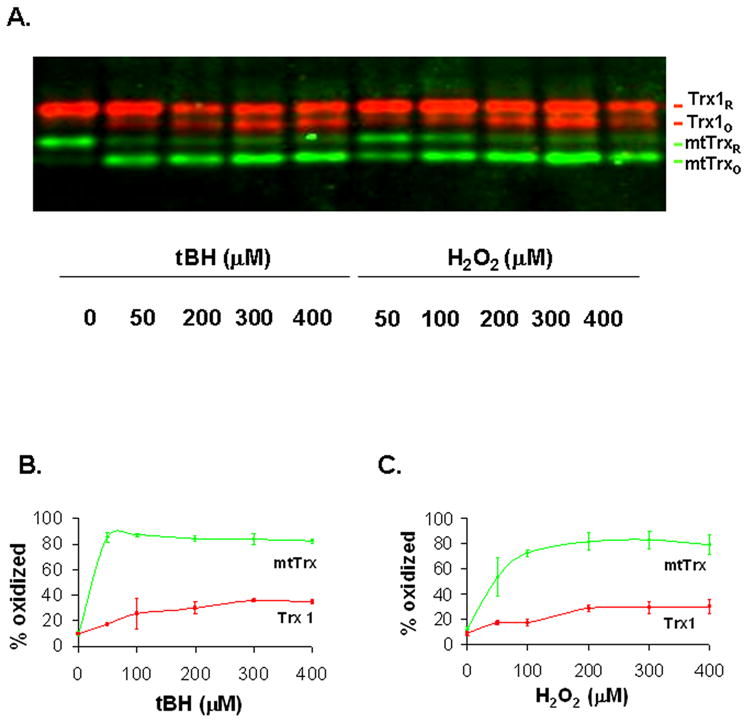
Dose-dependent changes of the redox status of Trx 1 and mtTrx in response to peroxides. (A) The 143 B human osteosarcoma cells were treated with different concentrations of tBH and H2O2 for 10 min. Cells were then harvested and prepared for redox measurement as described in “Materials and Methods”. Data presented are representative of three separate experiments. (B) and (C) Quantification of the Redox Western data after exposure to tBH and H2O2, respectively. The redox status of each protein was presented as the percentage of oxidized proteins. The data presented are the average of three separate experiments (mean ± SEM).
To study the time course of oxidation of Trxs, the 143 B cells were treated with 300 μM of either tBH or H2O2, which induced maximum oxidation of both mtTrx and Trx 1, and monitored for up to 3 h. Both oxidants induced rapid oxidation of mtTrx and Trx 1, with the maximal oxidation occurred within 5 min of treatment (Fig. 3A and 3B). The response of Trx 1 was found to be transient and lasted for 30 and 60 min, in cells exposed to H2O2 and tBH, respectively. In contrast, the majority of mtTrx (~70 %) remained oxidized at the end of 3 h incubation with tBH. In H2O2-treated cells, the redox status of mtTrx returned to base line after 60 min. The differences in the redox status of mtTrx were associated with different extents of mitochondrial damage. Loss of mtΔψ was observed in cells treated tBH which induced persistent oxidation; but was not detected in cells exposed to H2O2 which only induced a transient oxidation of mtTrx (Fig. 3C). During the incubation time, no significant changes in the total protein level of either Trx 1 or mtTrx protein were detected (data not shown).
Fig. 3.
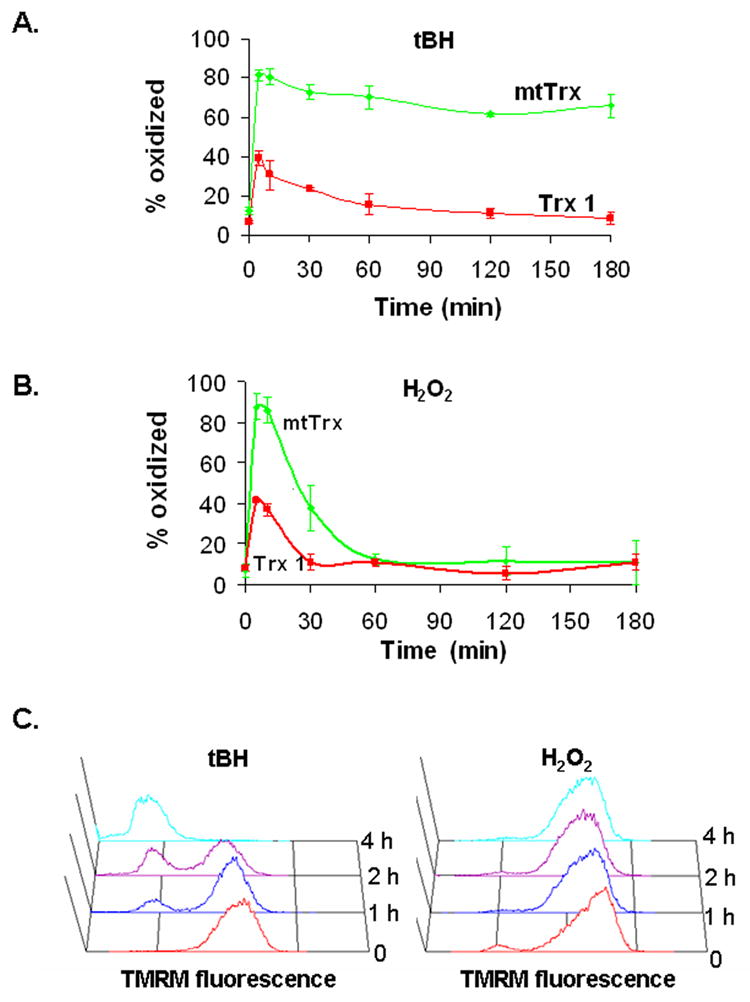
Time courses of peroxide-induced oxidation and mitochondrial depolarization. Cells were treated with either 300 μM of tBH (A) or 300 μM of H2O2 (B) for the indicated times. The redox status is presented as the percentage of the oxidized proteins. Results are the average of four separate experiments (mean ± SEM). (C) The mtΔψ was measured by TMRM staining and flow cytometry. Time-dependent changes in TMRM fluorescence were shown. Data presented are the representative of three separate experiments with similar results.
To further characterize the responses of mtTrx and Trx 1 to oxidative stress, we treated cells with diamide, a dithiol oxidant which oxidizes and crosslinks intracellular thiols such as GSH and protein cysteines [14]. Exposure to diamide resulted in oxidation of mtTrx and Trx 1 at comparable level in a dose-dependent manner (Fig. 4A). Both mtTrx and Trx 1 reached nearly complete oxidation within 10 min after treatment with 300 μM diamide. We next examined the time course of oxidation of mtTrx and Trx 1 in cells incubated with 300 μM diamide for up to 4 h (Fig. 4B). Like peroxides, diamide induced a rapid oxidation of both proteins. Unlike peroxides, the thiol oxidant diamide caused nearly maximal oxidation of both proteins in 5 min; whereas in peroxides treatment, less than 50% of Trx1 was oxidized. Gradual reduction to basal levels of both mtTrx and Trx1 were observed after the maximal oxidation. MtTrx was slower than Trx1 in recovery (Fig. 4B).
Fig. 4.
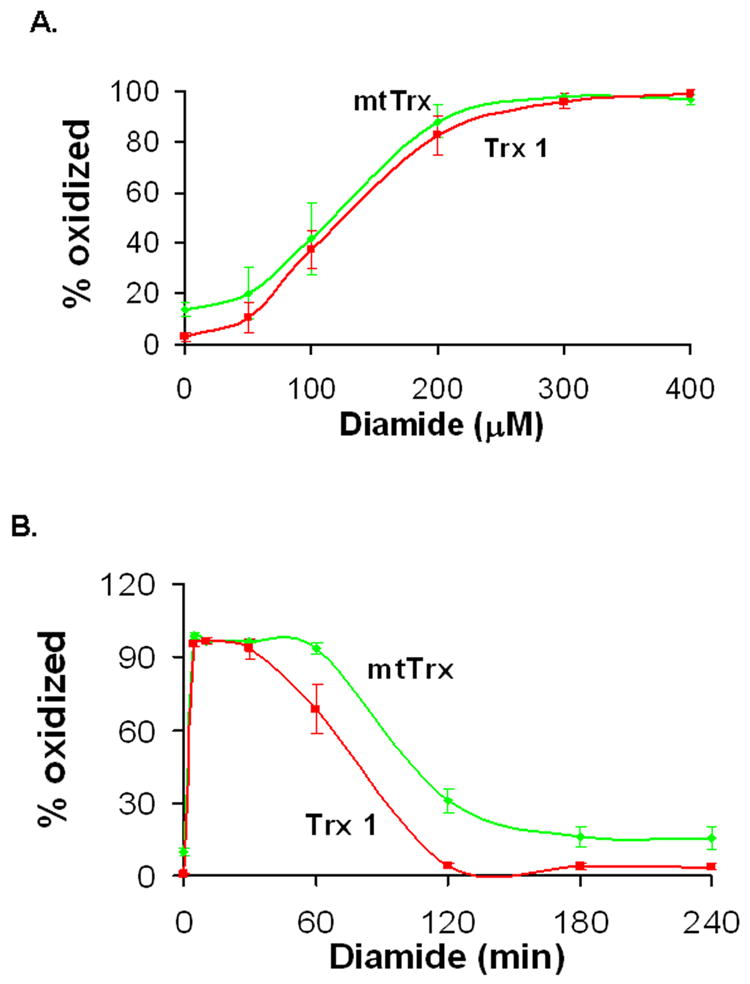
Diamide-induced oxidation of Trx1 and mtTrx in 143B cells. Cells were treated with either different doses of diamide (A) or 300 μM diamide for indicated time (B). The redox status of Trx1 and mtTrx was measured by Western blot analyses. Results are representative of four separate experiments (mean ± SEM).
3.2. Overexpression of mtTrx accelerated its recovery from diamide-induced oxidation and protected cells from the cytotoxicity
We have shown in our previous studies that overexpression of mtTrx protected cells from tBH toxicity [10]. To determine whether similar protection could be achieved against diamide, we used cells overepxressing mtTrx and treated them with 300 μM diamide. The redox status of mtTrx and Trx 1 in vector- and mtTrx-transfected cells were examined (Fig. 5). Although the maximum level of oxidized mtTrx was not influenced by overexpression of mtTrx, increased mtTrx facilitated the recovery of mtTrx from the initial oxidation. After exposure to diamide for 60 min, cells overexpressing mtTrx showed much lower extent of oxidation of mtTrx (Fig. 5A). No statistically significant difference was observed in redox status of Trx 1 (Fig. 5B).
Fig. 5.
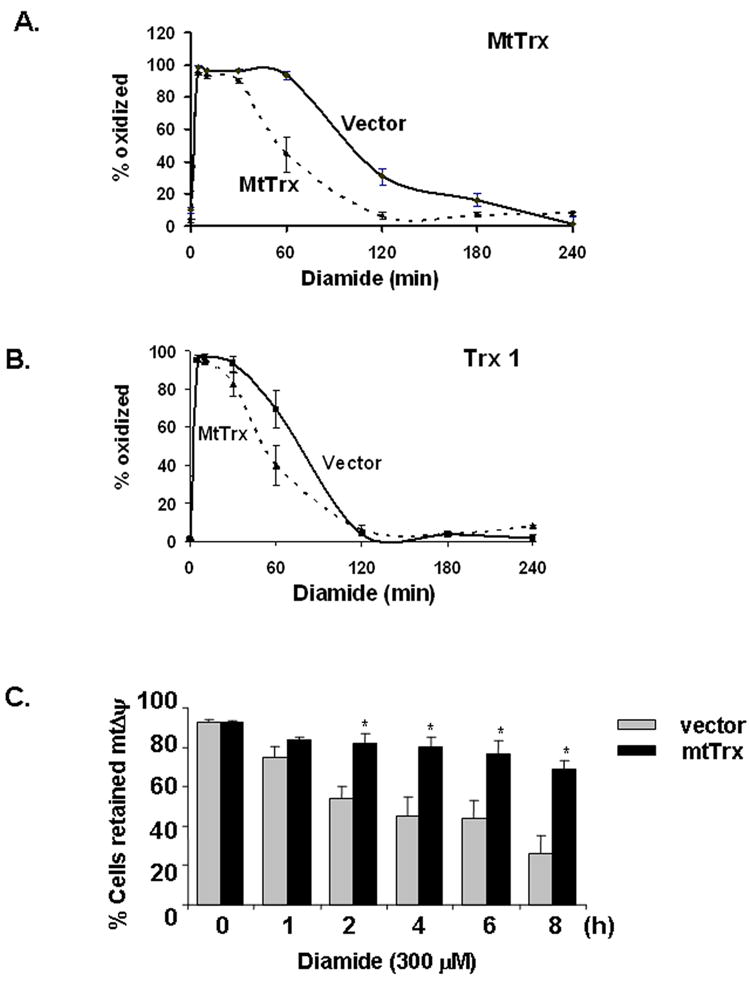
Overexpression of mtTrx in 143 B cells prevented oxidation of mtTrx and mitochondrial depolarization induced by diamide. 143 B cells transfected with empty vector or plasmid expressing mtTrx were treated with 300 μM diamide for indicated times. Cells were analyzed with Redox Western analysis to evaluate the redox status of mtTrx (A) and Trx 1 (B). The data were presented as the averages of five separate experiments (mean ± SEM). (C) Vector control and mtTrx-overexpressing cells were treated with 300 μM diamide for indicated time. MtΔψ was measured by TMRM staining followed by flow cytometry. The data presented are the average of five separate experiments (mean ± SEM). Significant differences from vector-transfected cells, determined by Student’s t-Test, are indicated as: *, P<0.05.
The time course of mtΔψ changes were measured in both vector- and mtTrx-transfected cells after exposure to 300 μM diamide. Results (Fig. 5C) showed that diamide caused time-dependent mitochondrial depolarization which started as early as 2 hours. Thus, similar to tBH treatment, diamide-induced oxidation of mtTrx was associated with mitochondrial damage. Overexpression of mtTrx protected cells from loss of mtΔψ (Fig. 5C).
Diamide caused dose-dependent cell death in 143B cells. PS exposure and caspase activation were measured after cells were exposed to 300 μM diamde for different time. Results showed that PS exposure (Fig. 6A) and caspase activation (Fig. 6B) occurred early and both were inhibited by overexpression of mtTrx. When measured for viability at 16 h, cells treated with 300 μM diamide had only 40% viability (Fig. 6C). Overexpression of mtTrx rendered cells more resistant to diamide toxicity at both 300 μM and 400 μM. Because the viability assay was performed at a relatively late time point as compared to the caspase activation and mitochondrial depolarization, it is unlikely that increased mtTrx only delayed cell death. Thus, increased mtTrx showed protective effects in both the redox status of mtTrx and oxidant-induced apoptosis.
Fig. 6.
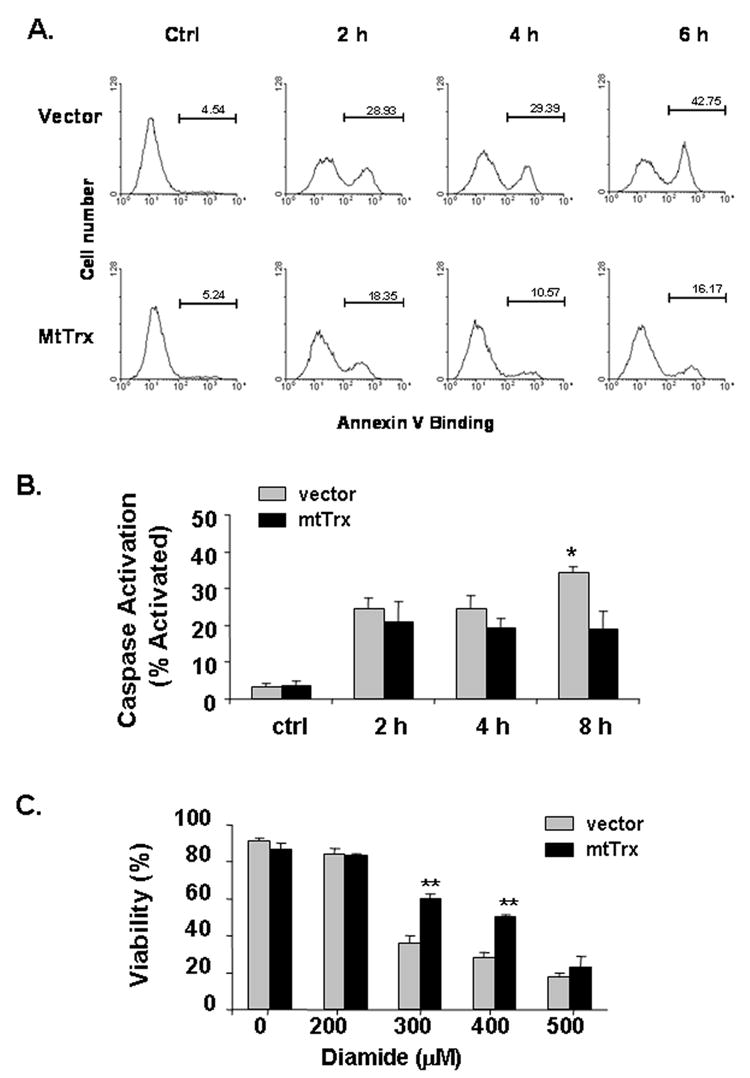
Protection against diamide-induced apoptosis by overexpressing mtTrx. Cells stably transfected with vector or mtTrx were treated with 300 μM diamide for indicated time. (A) PS exposure was measured by staining with fluorescein-conjugated Annexin V. Data presented are the representative of two separate experiments. (B) Caspase activation was measured by staining with the fluorogenic substrate FITC-VAD-FMK and flow cytometry. Data presented are the average of three separate experiments. Significant differences from vector-transfected cells, determined by Student’s t-Test, is indicated as: *, P<0.05. (C) Viability was performed after treatment with diamide at indicated concentrations for 16 h, using the LIVE/DEAD cytotoxicity assay. Duplicate samples were used for each condition. Data presented are the average of three separate experiments (mean ± SEM). Significant differences from vector-transfected cells, determined by Student’s t-Test, are indicated as **, P<0.01.
3.3. Knockdown of TR2 sensitized cells to oxidant-induced cell death
Data from our current and previous studies [15,16] indicate that oxidation of mtTrx is a common consequence to a broad spectrum of environmental toxicities. In order to further investigate the functional consequences of the oxidation, we used siRNA approach to reduce the expression of TR2 in 143B cells. At 48 h after transfection, the amount of TR2 was reduced to 52±7% (mean±SD) of the control level (Fig. 7A). TR2 is essential in maintaining mtTrx in its reduced state via NADPH-dependent redox circuit in the mitochondria. The results suggest that downregulation of TR2 was associated with both decreased expression and oxidation of the mtTrx protein (Fig. 7A, 7B).
Fig. 7.
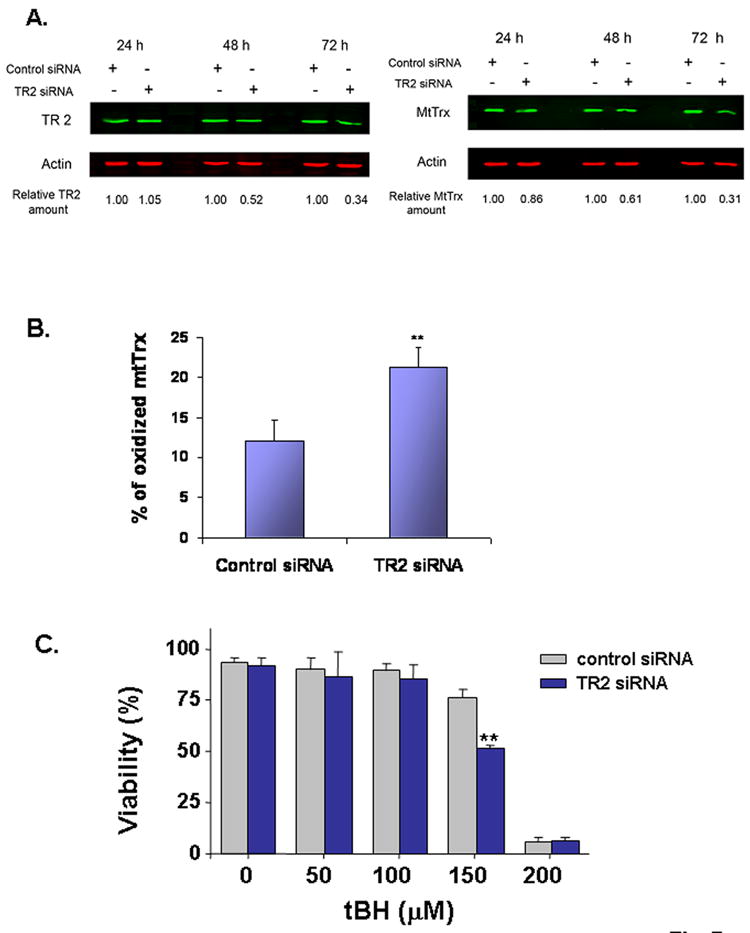
Knockdown TR2 sensitized 143B cells to tBH-induced cell death. Cells transfected with scrambled siRNA or siRNA targeting TR2. (A) The protein levels of TR2 and mtTrx were measured by Western blot analyses and normalized to β-Actin. The ratio between transfection with the scrambled and the TR2-targeting siRNA was indicated on the bottom. Data presented are the representative of three separate experiments. (B) The redox status of mtTrx was measured 48 h after transfection with either control scrambled siRNA or TR2-targeting siRNA. Data presented are the average of three separate experiments (mean± SEM). **, P<0.01, Student’s t-Test. (C) At 36 h after transfection, cells were exposed to different concentrations of tBH for additional 16 h and measured for viability. The data presented are the averages of three separate experiments (mean ± SEM). * P<0.05, Student’s t-Test.
Transfection with siRNA did not induce apparent cytotoxic effects under the experimental conditions (Fig. 7C). However, when these cells were further exposed to tBH, there was an increased sensitivity to cell death. At 150 μM, tBH induced minimal cell death in cells transfected with control scrambled siRNA, but caused significant toxicity in cells transfected with the siRNA against TR2. Thus, the proper function of the mtTrx/TR2 system controls the sensitivity to the oxidant-induced cell death in the 143B cells.
4. DISCUSSION
In the present study, we evaluated the oxidative stress-induced changes in the redox status of human mtTrx and Trx 1, two of the mammalian Trx family members with pleiotropic functions but with distinct subcellular localizations. We demonstrated that both mtTrx and Trx 1 were quickly oxidized by peroxides, but to different levels. Although mtTrx and Trx 1 belong to the same protein family, contain the conserved invariable active center, show similar in vitro insulin reduction activities and function as critical components of local antioxidant networks in individual subcellular compartments [7–9], they showed different sensitivities to oxidative stress. Compared to the cytosolic Trx 1, mtTrx was oxidized much more extensively and persistently (Fig. 2 and Fig. 3). Although the initial oxidation induced by diamide was similar between Trx 1 and mtTrx, the latter recovered with a slower rate (Fig. 4). Results from our current and previous studies [16] indicate that mtTrx is likely to be one of the mitochondrial proteins, including adenine nucleotide translocase [17,18] and aconitase [19], that are particularly sensitive to oxidative damage.
The redox status of Trxs reflects the balance between the local oxidizing power and the reducing capacity of the TRs. There are two possible mechanisms account for the observed higher susceptibility of mtTrx to oxidation. First, it may result from a higher level of local oxidative stress, which is exemplified by the tBH treatment. In cells exposed to tBH, oxidation of mtTrx lasted much longer than that of Trx 1. As demonstrated by other studies [13], tBH targets mitochondria and the greater extent of oxidation of mtTrx may be partially resulted from the local reactive intermediates production. Alternatively, the disparity between mtTrx and Trx1 may be caused by a lower activity of the mitochondrial TR system. Spyrou et al showed that in the presence of TR1, oxidized Trx 1 had lower insulin reduction activity than oxidized mtTrx [7]. Their data may suggest that mtTrx is less prone to oxidation in vitro. Interestingly, however, data from Rhee’s group suggested that it may not be the case because TR2 has similar, if not greater, catalytic activity compared to TR1 in vitro [8]. Despite the controversy, our measurements using intact cells clearly indicate that mtTrx is more prone to oxidation than Trx1 after exposure to peroxides and diamide.
Although Trx1 and mtTrx are functional homologues, they are different proteins encoded by different genes. There are potentially various factors that can influence their redox status. In diamide treatment (Fig. 4 and 5), the initial oxidation was similar between Trx1 and mtTrx; however, the recovery rate was distinct between the two proteins. The data suggest that there are differences in the kinetics and/or the capability of reducing oxidized mtTrx and Trx1 in different subcellular compartments. Compared to the in vitro reconstitution system, Trxs in vivo are exposed to a more diverse and dynamic microenvironment and, consequently, interact with a variety of proteins. The protein interactions will likely influence the function by which TRs control the redox status of Trxs.
Although oxidation of mtTrx has been reported, the implications in the redox regulated functions have not been well characterized. Our data indicate that the initial oxidation of mtTrx was caused by chemical exposure and may not always be associated to perturbation of the cellular function. For instance, both 50 μM tBH and 100 μM H2O2 caused nearly complete oxidation of mtTrx (Fig. 2). However, neither condition was lethal. Cells recovered quickly after the initial exposure. In fact if the peroxides were removed from the media, the redox status of mtTrx returned to basal level within minutes (data not shown). Similar observations were obtained with diamide treatment, which at 200 μM concentration induced nearly maximal oxidation of mtTrx (Fig. 4A) but was proved to be not lethal in the 143B osteosarcoma cells (Fig. 6C).
In contrast to the initial reversible oxidation, prolonged oxidation of mtTrx is associated with severe cytotoxicity and cell death. At 300 μM, tBH induced persistent oxidation of mtTrx (Fig. 3A), mitochondrial depolarization (Fig. 3C) and cell death [10]. H2O2 at the same concentration was not lethal and, accordingly, the oxidation of mtTrx lasted only less than 60 minutes (Fig. 3B) and was not associated with mitochondrial damage (Fig. 3C). Diamide at 300 μM also induced oxidation longer than 2 h (Fig. 5B) and was associated with mitochondrial depolarization (Fig. 5C), caspase activation (Fig. 6B) and cell death (Fig. 6C). Overexpression of mtTrx facilitated the recovery from oxidation (Fig. 5B) and protected against the cell death (Fig. 6C). Oxidation and downregulation of mtTrx by the siRNA approach potentiated the tBH-induced cell death (Fig. 7C). Taken together, these data indicate that the persistent oxidation of mtTrx is a sign of mitochondrial dysfunction. The redox status of mtTrx is a modulatory factor of sensitivity to oxidant-induced cell death.
Several redox sensitive components of the mitochondrial pathway of apoptosis have been identified. ASK-1 was recently shown to have a mitochondrial localization and may directly interact with mtTrx [20]. The adenine nucleotide translocator (ANT) is known to have critical thiols that modulate the permeability transition (17). Whether the effects of mtTrx were mediated through these proteins or whether other unknown proteins are involved remain to be determined.
The sensitivity of mtTrx to oxidative stress may have important implications in aging and age-related diseases. Oxidative damages in mitochondria DNA and lipids have been observed during the aging process [21,22]. In aging tissues, mitochondrial protein damage has also been reported, such as aconitase and ANT [23]. Our data suggest that mtTrx is another important target. Like its cytosolic isoform Trx 1, mtTrx is an essential component of antioxidant systems. An oxidation of the mtTrx pool may trigger a chain reaction that dramatically alter the posttranslational modifications of a variety of mitochondrial proteins and lead to catastrophic damages to mitochondrial structure and function.
Acknowledgments
This research was supported by grants from the NIH (ES09047, EY07892, ES014668); Carl M. Reeves and Mildred A. Reeves Foundation; International Retinal Research Foundation, Inc.; a departmental Challenge Grant and Sybil Harrington Award (to JC) from Research to Prevent Blindness, Inc.
References
- 1.Finkel T. Reactive oxygen species and signal transduction. IUBMB Life. 2001;52:3–6. doi: 10.1080/15216540252774694. [DOI] [PubMed] [Google Scholar]
- 2.Chiarugi P. Reactive oxygen species as mediators of cell adhesion. Italian J Biochem. 2003;52:28–32. [PubMed] [Google Scholar]
- 3.Xu D, Rovira II, Finkel T. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev Cell. 2002;2:251–252. doi: 10.1016/s1534-5807(02)00132-6. [DOI] [PubMed] [Google Scholar]
- 4.Hoshi T, Heinemann S. Regulation of cell function by methionine oxidation and reduction. J Physiol. 2001;531(Pt 1):1–11. doi: 10.1111/j.1469-7793.2001.0001j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xing K, Lou M. The possible physiological function of thioltransferase in cells. FASEB J. 2003;17:2088–2090. doi: 10.1096/fj.02-1164fje. [DOI] [PubMed] [Google Scholar]
- 6.Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Pharmcol Toxicol. 2001;41:261–295. doi: 10.1146/annurev.pharmtox.41.1.261. [DOI] [PubMed] [Google Scholar]
- 7.Spyrou G, Enmark E, Miranda-Vizuete A, Gustafsson J. Cloning and expression of a novel mammalian thioredoxin. J Biol Chem. 1997;272:2936–2941. doi: 10.1074/jbc.272.5.2936. [DOI] [PubMed] [Google Scholar]
- 8.Lee SR, Kim JR, Kwon KS, Yoon HW, Levine RL, Ginsburg A, Rhee SG. Molecular cloning and characterization of a mitochondrial selenocysteine-containing thioredoxin reductase from rat liver. J Biol Chem. 1999;274:4722–4734. doi: 10.1074/jbc.274.8.4722. [DOI] [PubMed] [Google Scholar]
- 9.Miranda-Vizuete A, Damdimopoulos AE, Pedrajas JR, Gustafsson J, Spyrou G. Human mitochondrial thioredoxin reductase cDNA cloning, expression and genomic organization. Eur J Biochem. 1999;261:405–412. doi: 10.1046/j.1432-1327.1999.00286.x. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Cai J, Murphy TJ, Jones DP. Overexpressed human mitochondrial thioredoxin confers resistance to oxidant-induced apoptosis in human osteosarcoma cells. J Biol Chem. 2002;277:33242–33248. doi: 10.1074/jbc.M202026200. [DOI] [PubMed] [Google Scholar]
- 11.Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 2001;20:6288–6296. doi: 10.1093/emboj/20.22.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritz D, Beckwith J. Redox state of cytoplasmic thioredoxin. Methods Enzymol. 2002;347:360–370. doi: 10.1016/s0076-6879(02)47036-x. [DOI] [PubMed] [Google Scholar]
- 13.Moore GA, Jewell SA, Bellomo G, Orrenius S. On the relationship between Ca2+ efflux and membrane damage during t-butylhydroperoxide metabolism by liver mitochondria. FEBS Lett. 1983;153:289–292. doi: 10.1016/0014-5793(83)80626-7. [DOI] [PubMed] [Google Scholar]
- 14.Pias EK, Aw TY. Apoptosis in mitotic competent undifferentiated cells is induced by cellular redox imbalance independent of reactive oxygen species production. FASEB J. 2002;16:781–790. doi: 10.1096/fj.01-0784com. [DOI] [PubMed] [Google Scholar]
- 15.Hansen JM, Zhang H, Jones DP. Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions. Free Radic Biol Med. 2006;40:138–145. doi: 10.1016/j.freeradbiomed.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Yu M, Jones DP, Greenamyre JT, Cai J. Protection against oxidant-induced apoptosis by mitochondrial thioredoxin in sh-sy5y neuroblastoma cells. Toxicol Appl Pharmacol. 2006;216:256–262. doi: 10.1016/j.taap.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 17.McStay GP, Clarke SJ, Halestrap AP. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem J. 2002;367:541–548. doi: 10.1042/BJ20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imai H, Koumura T, Nakajima R, Nomura K, Nakagawa Y. Protection from inactivation of the adenine nucleotide translocator during hypoglycaemia-induced apoptosis by mitochondrial phospholipid hydroperoxide glutathione peroxidase. Biochem J. 2003;371:799–809. doi: 10.1042/BJ20021342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yarian CS, Toroser D, Shoal RS. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice Mech. Ageing Dev. 2006;127:79–84. doi: 10.1016/j.mad.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Thioredoxin-2 inhibits mitochondri-located ASK 1-mediated apoptosis in a JNK-independent manner. Cir Res. 2004;94:1483–1491. doi: 10.1161/01.RES.0000130525.37646.a7. [DOI] [PubMed] [Google Scholar]
- 21.Lenaz G. Role of mitochondria in oxidative stress and ageing. Biochim Biophys Acta. 1998;1366:53–67. doi: 10.1016/s0005-2728(98)00120-0. [DOI] [PubMed] [Google Scholar]
- 22.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 23.Levine RL, Stadtman ER. Oxidative modification of proteins during aging. Exp Gerontol. 2001;36:1495–1502. doi: 10.1016/s0531-5565(01)00135-8. [DOI] [PubMed] [Google Scholar]