

Abstract
Purpose
Autosomal dominant Stargardt-like (STGD3) disease results from mutations in the ELOVL4 gene (elongation of very-long-chain fatty acids). This study was undertaken to characterize a mouse model with a targeted deletion of Elovl4 and to explore the role of this gene in retinal/macular degeneration.
Methods
A construct targeted to exon 2 of the Elovl4 gene was used to suppress expression of the gene. Elovl4 homozygous pups were nonviable and were not available for study. Hence, the analysis was performed on heterozygous Elovl4+/− mice 16 to 22 month of age and littermate wild-type (WT) control mice of the same age. Characterization included examining gene message and protein levels, electroretinogram (ERG), retinal morphology and ultrastructure, and plasma and retinal fatty acid composition.
Results
Although the level of Elovl4 mRNA was reduced in Elovl4+/− retinas, only minimal morphologic abnormalities were found, and the retinal (ERG) function was essentially normal in Elovl4+/− retinas compared with the WT control retinas. Systemic fatty acid profiles of Elovl4+/− mice were unremarkable, although the concentration of several fatty acids was significantly lower in Elovl4+/− retinas, particularly the monounsaturated fatty acids.
Conclusions
The detailed characterization of this animal model provides the first in vivo evidence that Elovl4 haploin-sufficiency is not the underlying key disease mechanism in STGD3. The results are consistent with a dominant negative mechanism for the deletion mutation. The Elovl4 knockout mouse is one of three complementary animal models that will help elucidate the disease mechanism.
Human macular degeneration is generally characterized by a gradual deterioration in central vision accompanied by structural pathologic conditions such as atrophy of the retinal pigment epithelium (RPE), accumulation of lipofuscin in the RPE, and regional loss of photoreceptors.1,2 Stargardt disease 3 (STGD3) is an early-onset autosomal dominant form of atrophic macular degeneration that results from a 5-bp deletion mutation, and possibly other mutations, in the ELOVL4 gene (elongation of very-long-chain fatty acids [LCFAs]), which maps to 6q14.3–6 The deletion causes a frameshift and loss of a 51-amino-acid fragment at the C terminus including a dilysine (KXKXX) targeting signal, creating a premature stop codon and leading to synthesis of an aberrant ELOVL4 peptide.5 The wild-type ELOVL4 protein is found in the endoplasmic reticulum (ER), which is the site of very-LCFA biosynthesis.7 However, the intracellular trafficking of the truncated form is defective and appears to be sequestered in aggresomes in a complex that also includes mislocalized wild-type ELOVL4.5–14
The importance of ELOVL4 protein is implied by its evolutionary conservation. Human ELOVL4 encodes a protein of 314 amino acids with approximately 35% amino acid identity to members of the ELO protein family in yeast,5 and orthologues have been identified in other species, including Caenorhabditis elegans, zebrafish, and chicken.15–18 The cellular function of ELOVL4 is unknown, but homology with yeast proteins suggests that it is an ER-bound transmembrane protein associated with LCFA synthesis.5
LC polyunsaturated fatty acids (PUFAs) from the n-6 and n-3 classes are essential for brain and retinal development and for visual function.19 An important role for docosahexaenoic acid (DHA) within the retina is suggested by its high levels and active conservation in this tissue.5,20,21 ELOVL4 is expressed in rod and cone photoreceptor cells and is probably involved in one of the elongation steps necessary for fatty acid biosynthesis.5,20–23 The essential role of ELOVL4 in polyunsaturated fatty acids (PUFA) synthesis is also suggested by preliminary evidence of improved visual function in human STGD3 disease with supplementation of DHA.24 Another recent study found that phenotypic severity in a family with STGD3 disease correlates with dietary fatty acid intake, and that a high dietary intake of DHA may partially ameliorate the maculopathy.25
We characterized heterozygous animals in a mouse model with a targeted deletion of Elovl4, to further understand the role of this gene in macular degeneration.
Methods
Generation of Elovl4 Heterozygous Knockout Mice
The targeting construct that was used to generate a 62-base-pair deletion in exon 2 of Elovl4 contained a 3.3-kb genomic fragment (5′ arm) cloned 5′ to the SA/IRES/LacZ-PKG-Neo cassette and a 1.5-kb fragment (3′ arm) inserted 3′ to the neomycin cassette (Fig. 1). A linearized vector was used to induce a deletion in the 129/OlaHsd ES cells. Correctly targeted embryonic stem (ES) cell clones were identified by Southern blot hybridization with 199-bp 5′ and 3′ probes. An ES cell line carrying the exon 2 deletion was used to generate chimeric male mice. F1 mice were generated by breeding male chimaeras with C57BL/6 females.
Figure 1.
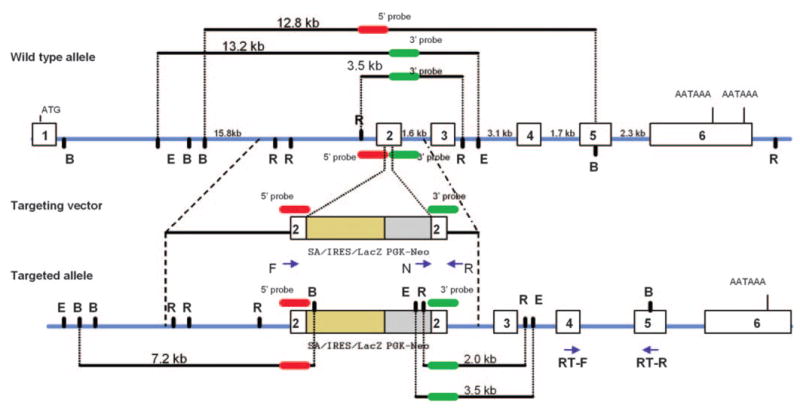
Restriction maps of the wild-type allele, the targeting vector, and the targeted allele of the Elovl4 gene. In the diagram representing the targeted allele of Elovl4 gene, open rectangles are exons and intron sizes are indicated above the solid line. The 199-bp 5′ probe (red) detected 12.8-kb BamHI fragment in the wild-type genomic DNA, whereas the 199-bp 3′ probe (green) detected 13.2-kb EcoRI fragment and 3.5-kb EcoRV fragment in the wild-type genomic DNA. The targeting vector contained 3.3 kb of 5′-flanking sequence and 1.5 kb of 3′-flanking sequence, as well as a splice acceptor site (SA), an internal ribosome entry site (IRES) linked to β-galactosidase (lacZ), and pgk-neo cassette to delete 62 bp in exon 2 in the targeted allele. Arrows: position of forward (F), reverse (R), neo (N), and RT-PCR (RT-F and RT-R) primers that were used to confirm homologous recombination and for expression analysis using qRT-PCR. B, BamHI; E, EcoRI; R, EcoRV.
One to three subsequent breedings of progeny were performed with C57BL/6. The number of mice available was limited, as they did not breed well, and the line failed to propagate further. Homozygous Elovl4+/− were nonviable, and only heterozygous Elovl4+/− mice were available for study. Because the retinal degeneration of the human STGD3 disease is progressive, we chose to examine mice at least 16 months of age in the expectation that older mice would exhibit an advanced phenotype. Control mice were Elovl4+/− litter-mates of the same age, unless otherwise indicated. Experiments were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and with protocols approved by the Animal Care and Use Committee of the National Eye Institute of the National Institutes of Health.
Genotyping
Elovl4+/− mice were genotyped by PCR methods using tail DNA as a template, with a set of three oligonucleotide primers: M700KP-WT (5′-CTCCGCAGATAAACGTGTAGCAGAC-3′), M700KP-S (5′-AGAGTGCCGTTAACAAACCTACCTC-3′), and Neo3196 (5′-GGGTGGGATTAGATAAATGCCTGCTCT-3′). The M700KP-WT paired with M700KP-S was used to amplify the wild-type allele; M700KP-WT paired with Neo3196 was used to identify the targeted allele. PCR was performed in a 25-μL reaction volume containing 100 ng of tail DNA, 200 μM deoxynucleotide triphosphates, 250 nM of each primer, 2.5 mM MgCl2, 1.25 units of Taq DNA polymerase, and 2.5 μL of 10× reaction buffer. The PCR conditions were 3 minutes at 95°C for initial denaturation, followed by 35 cycles of 30 seconds at 95°C, 50 seconds at 60°C, 50 seconds at 72°C, and 5 minutes at 72°C for the final extension. The targeted allele gave rise to a 406-bp PCR fragment, and the wild-type allele produced a 217-bp fragment. The PCR products were analyzed by agarose gel electrophoresis.
Electroretinograms
The electroretinogram (ERG) was recorded from six Elovl4+/− 16-month-old mice and four age-matched control littermate wild-type (WT) mice. The mice were anesthetized with an intraperitoneal injection of ketamine (80 mg/kg) and xylazine (4 mg/kg), and body temperature was maintained by placing them on a heating pad. The pupils were fully dilated with topical corneal 0.5% tropicamide and 2.5% phenylephrine HCl, and both eyes were recorded simultaneously using corneal gold-wire loops as the active electrodes with 0.5% proparacaine hydrochloride topical anesthesia. Gold-wire electrodes were placed on the sclera near the limbus, to serve as reference electrodes. A neutral wire was attached to the ear. ERG stimuli were xenon photostrobe 10-μs flashes with 0.6 log cds/m2 maximum intensity in a full-field integrating sphere. Scotopic responses were recorded across a 7-log-unit range of stimulus intensity in 0.5-log-unit steps beginning from below threshold up to maximum intensity, amplified, and filtered (5000 gain, 0.1–1000 Hz). For photopic recordings a constant white background light of 34 cd/m2 was used to suppress rod function.
Dark-adapted and light-adapted a-wave amplitudes were measured from the baseline to the response peak, and b-wave amplitudes were measured from the baseline or from the a-wave trough when the a-wave was present. Data were analyzed by the MIXED procedure (PROC MIXED, SAS for Windows, ver. 9.1; SAS Institute, Inc., Cary, NC), which is used for analysis of repeated-measures data. It fits a variety of mixed linear models to the data and extends the general linear model by allowing a more flexible specification of variance and covariance. The primary ERG outcome variables were log amplitudes of the a- and b-wave responses compared between Elovl4+/− and control eyes. Predictor variables were ERG stimulus intensity and group.
Lipid Extraction and Fatty Acid Analysis
Retinas of 10 Elovl4+/− and 4 control littermate mice, all at 19 to 22 months of age, were removed and stored at −80°C. Retinas were weighed and lipid extraction performed according to the method of Bligh and Dyer.26 To each sample, 50 μg butylated hydroxytoluene (BHT) was added along with the internal standard 22:3n-3 methyl ester (1 μg/mg retina wet weight). Retinas were subsequently transmethylated using the BF3-methanol method of Morrison and Smith27 as modified by Salem et al.28 with the cosolvent hexane. The methyl ester samples were analyzed by gas chromatography as previously described,28 except that the retinas were injected using a splitless method in which 2 μL of the hexane extract was injected, the purge flow rate to the split vent was 14 mL/min, and the valve was opened 0.25 minute after injection. Fatty acid analysis was also performed on plasma samples of all 10 animals. The plasma samples were extracted as previously described by Folch et al.29 and transmethylated as described by Salem et al.28
Tissue Processing for Light and Electron Microscopy
Four Elovl4+/− and two littermate control WT mice, all 17 to 19 months of age, were euthanatized, and the eyes were enucleated. A shallow incision was made through the cornea at the limbus, and the eyes were immersed overnight in primary fixative: 2% formaldehyde (from electron microscopy [EM] grade paraformaldehyde) + 2% glutaraldehyde in 0.086 M sodium phosphate buffer (pH 7.2). The next day, the anterior segment and lens were removed. Eyecups were washed in cold, 0.137 M sodium phosphate buffer and transferred to capped vials containing 1% osmium tetroxide in sodium phosphate buffer for 1 hour. Eyecups were washed in chilled distilled water and serially dehydrated in an ethanol series (30%–100%) on ice. Eyes were transferred from absolute ethanol to propylene oxide for 20 minutes and then infiltrated with a 1:1 mixture of propylene oxide and Araldite 6005 for 24 hours. After infiltration, eyes were embedded in Araldite 6005.
For light microscopy, 1-μm-thick sections were cut with a glass knife through the center of the globe, without regard to orientation about the optical axis of the eye, and were stained with 1% toluidine blue/methylene blue/azure II. Sections were examined with a microscope (BX51; Olympus, Lake Success, NY) and 40× objective and were photographed with a digital camera. Measurements of outer nuclear layer (ONL) width and rod outer segment (ROS) length were made in two peripheral fields approximately 300 μm from each edge, in two midperipheral fields half-way to the central retina and finally in the central field. For ROS length, the maximum and minimum lengths in each field were averaged.
For EM, thin sections (~100 nm) were collected on 200 hex copper grids, stained with 2% aqueous uranyl acetate and Reynold’s lead citrate, and examined by transmission electron microscope (model 1010; JEOL, Tokyo, Japan).
RNA and Protein Isolation and cDNA Preparation
Retinal RNA was isolated (TRIzol reagent; Invitrogen Corp, Carlsbad, CA), and protein was isolated simultaneously from the phenol phase after removal of the aqueous phase containing RNA, both according to the manufacturer’s protocol. Total RNA was treated with RNase-free DNase, to remove the genomic DNA contamination, and purified (RNeasy minikit; Qiagen, Valencia, CA). First-strand cDNA was synthesized for RT-PCR (SuperScript First-Strand Synthesis System; Invitrogen Corp.).
Real-Time Quantitative RT-PCR
All real-time PCR (qRT-PCR) primer pairs were designed so that one primer was located in one exon, and the second primer was in a different exon, resulting in an intron-spanning amplification event that could distinguish amplification of reverse-transcribed RNA from genomic DNA21 (Fig. 1). Quantitative PCR and melting curve analyses were performed (iQ SYBR Green Supermix and iCycler; Bio-Rad, Hercules, CA). Expression of four housekeeping genes—Gapdh, Hgprt, Actin-b, and RpL19—was used as the control, to normalize and validate the expression data. The relative quantity of expression was calculated by the comparative Ct (threshold cycle) method for each of the house-keeping genes, as described previously.21 The expression data are presented as a percentage of the relative value of the age-matched controls.30 Samples from four Elovl4+/− mice and three C57BL/6 control animals, all at 19 to 22 months of age, were analyzed.
Western Blot Analysis
Protein isolation and Western blot analysis were performed as previously described18 with the same four Elovl4+/− and three C57BL/6 control retinas used for qRT-PCR. Total protein quantity was measured with the bicinchoninic acid (BCA) reagent (Pierce, Rockford, IL), and equal quantities were resolved on 10% SDS-PAGE. Western blot analysis was performed using a rabbit polyclonal anti-ELOVL4 antibody generated against the synthetic peptide corresponding to internal sequence amino acids (107–121) in exon 3 of human ELVOL4. This antibody, available through Abcam (Cambridge, MA), has been tested and found to cross-react with ELVOL4 from mouse tissues (1:500 dilution). Signal was detected with chemiluminescence (ECL Kit; Amersham Biosciences, Piscataway, NJ), according to the manufacturer’s instructions.
Results
Litters were genotyped at the age of 3 to 4 weeks, at which time no homozygous Elovl4+/− mice were found. Dead pups found within 2 to 3 days of birth were genotyped and were identified as either heterozygous Elovl4+/− or wild type. Therefore, we have no indication that any Elovl4+/− mice were born.
The Elovl4+/− mice were not different from littermate control WT mice in weight, apparent behavior, or longevity. Semiquantitative (q)RT-PCR analysis of retinal cDNA from Elovl4+/− mice with primers designed to amplify Elovl4 mRNA confirmed the presence of message from the WT Elovl4 gene. However, the WT Elovl4 message level was reduced to 10% to 25% of the expression in age-matched C57BL/6 mice (Fig. 2A). On a Western blot, the anti-ELOVL4 antibody detected a single band of the expected size (37 kDa) in both Elovl4+/− and C57BL/6 control retina, indicating expression of the WT ELOVL4 protein in the Elovl4+/− retina (Fig. 2C). Despite the considerable reduction in Elovl4 message, the morphologic appearance of the retina in the Elovl4+/− mice was minimally different from age-matched littermate WT mice (Fig. 3A). The photoreceptor outer nuclear layer (ONL) was at most only 1 to 2 cells thinner in some areas of the retina in the Elovl4+/− mice, and measurements of ONL width indicated only minimal thinning of 10% ± 7.9% (mean ± SD, n = 5), with no statistical significance at any region (Fig. 3B). The ROS of Elovl4+/− mice were shorter than the WT by an average of 20% ± 18% (mean ± SD, n = 5, P < 0.01 in the PROCMIXED procedure) across the retina (Fig. 3B). Rhodopsin message level was 33% lower in four Elovl4+/− retinas relative to aged-matched C57BL/6 mice (Fig. 2B), but was not statistically significant (P = 0.36) indicating that, overall, there was no detectable loss of photoreceptors at this advanced age by this method. The Elovl4+/− retinas showed scattered regions in which apoptotic photoreceptor nuclei were present and rod outer segments were less densely packed.
Figure 2.
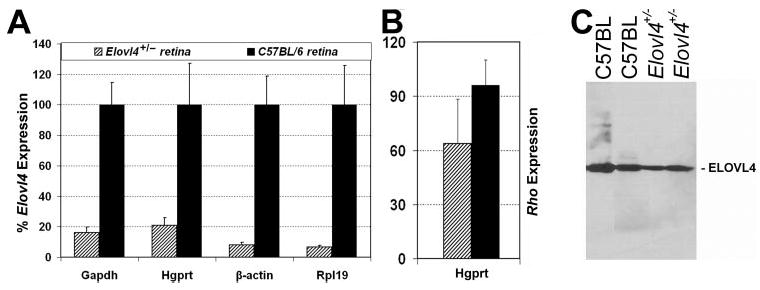
(A) Comparison of expression of the Elovl4 gene in Elovl4+/− (n = 4) and age-matched C57BL/6 (n = 3) retinas. Expression level in Elovl4+/− retinas is presented relative to the expression level in age-matched C57BL/6 retinas. The relative quantity of expression was calculated by the comparative threshold cycle (Ct) method for the four housekeeping genes indicated on the x-axis. (B) Comparison of the expression of the rhodopsin gene in Elovl4+/− retinas (n = 4) and age-matched C57BL/6 (n = 3, black) retinas. qRT-PCR data were analyzed by using the comparative Ct method and presented relative to the expression of the housekeeping gene Hgprt. (C) Immunoblot assay: Retinal protein extracts (40 μg) from Elovl4+/− (n = 4) mice and C57BL/6 wild-type mice (n = 3) were analyzed by SDS-PAGE, followed by immunoblot analysis with the anti-ELOVL4 antibody.
Figure 3.
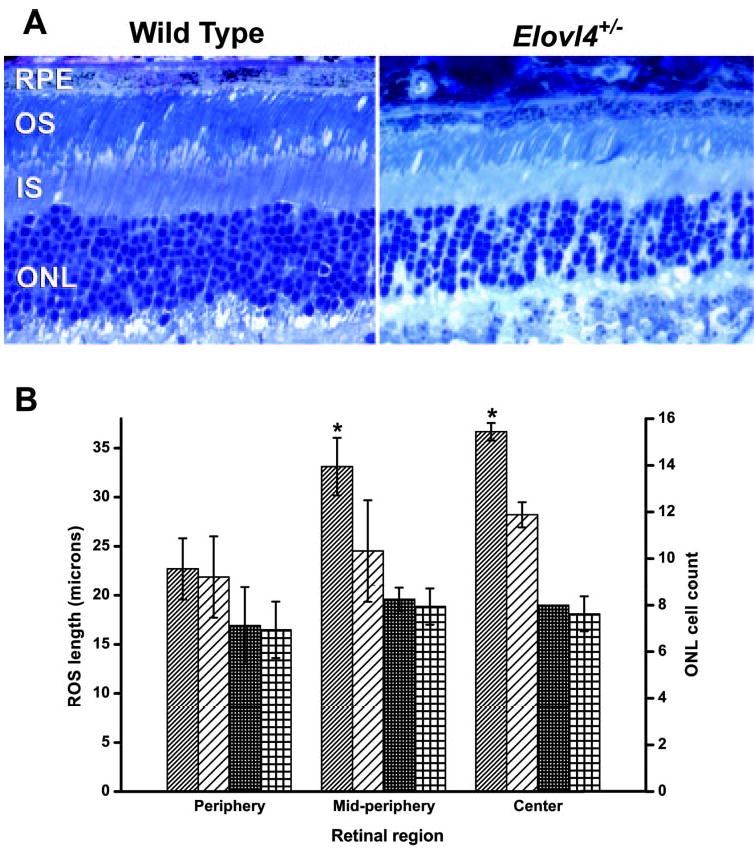
Shortening of outer segments and a minor loss of photoreceptor cells was seen in the retinas of Elovl4+/− mice. (A) Representative midperipheral retinal sections of WT and Elovl4+/− mice. Magnification, ×40. ONL, outer nuclear layer; IS, inner segments; OS, outer segments; RPE, retinal pigment epithelium. (B) Average ROS length (diagonal stripes) and ONL cell count (squares) of Elovl4+/− (n = 4, sparse pattern) and age-matched littermate WT (n = 2, dense pattern) retinas. Error bars: standard deviation. *Significant differences between Elovl4+/− and WT, Student’s t-test, P < 0.02.
As membrane structure may be affected by changes in fatty acid composition,31 we examined disc morphology in the outer segments by EM. The ROS were disrupted in the Elovl4+/− retinas, and irregular gaps were seen between stacks of discs. Some disc stacks were disoriented, and ROS membranous whorls were seen in some areas (Fig. 4). These changes were scattered intermittently amid normal-looking areas. Comparable abnormalities were found in age-matched littermate WT retinas although to a lesser extent and severity, indicating that some of these changes may result from the advanced age of the mice32 and suggesting Elovl4+/− retinas undergo an accelerated aging process. Unfortunately, the Elovl4+/− mice bred poorly, and younger mice were not available for study, despite considerable breeding efforts by a proficient animal resource facility over a 2-year period.
Figure 4.
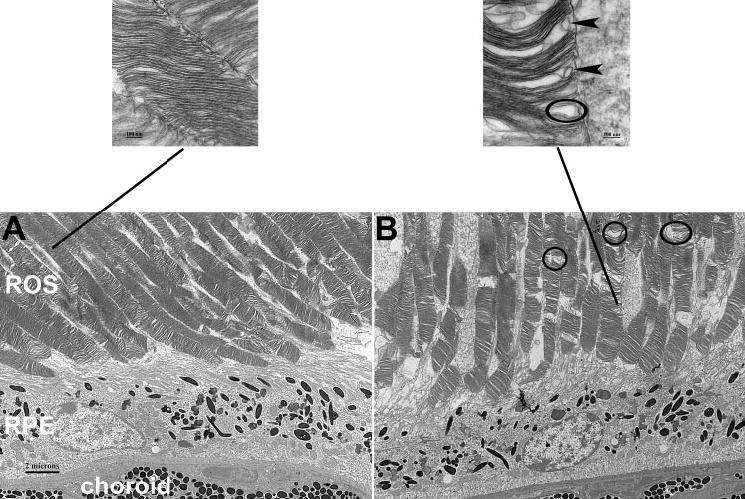
Electron micrographs. Compared with the wild-type (A), the Elovl4+/− (B) retina shows disrupted ROS disc structure. Circles: examples of unusual gaps in the ROS. Insets: OS discs at a higher magnification illustrating gaps (circle) and membranous whorls (arrow-heads) in the Elovl4+/− retina. Magnification: (A, B) × 4,000; insets ×100,000.
A few focal retinal pigment epithelium (RPE) irregularities were also noted in Elovl4+/− mice, including a wavy appearance of the basal RPE-Bruch’s membrane interface and, occasionally, the presence of more than one layer of RPE cells. Although these changes may be indicative of disease, they were observed only in very short stretches of the RPE (1–3 cells long), and the overall appearance of the RPE was otherwise unremarkable. The RPE of both WT and Elovl4+/− mice showed pigment granules at the basal side of the cells in some areas, which may be attributed to aging of the retina.33
ERGs were recorded from 16-month-old Elovl4+/− mice and littermate WT control animals to evaluate retinal function (Fig. 5). Dark-adapted a- and b-wave amplitudes were both consistently larger in the Elovl4+/− mice than in the WT mice, although the difference was statistically significant at only one stimulus intensity (Figs. 5A, 5B). When all points of the entire intensity response curve were analyzed together by the “mixed procedure” method, no significant difference was found between the groups. Similarly, the b-wave amplitudes of the light-adapted responses were consistently larger in the Elovl4+/− mice for all stimulus intensities, but the results were not statistically different from WT (Figs. 5A, 5C). No differences in wave shape or implicit time were observed. Hence, one can conclude that the Elovl4+/− responses were definitively not reduced compared with WT, despite the slight decrease in ROS length. We also examined the fundus of five 17-to 19-month-old Elovl4+/− mice by ophthalmoscopy, and the gross appearance was unremarkable and showed no change of RPE pigmentation or accumulations (data not shown).
Figure 5.
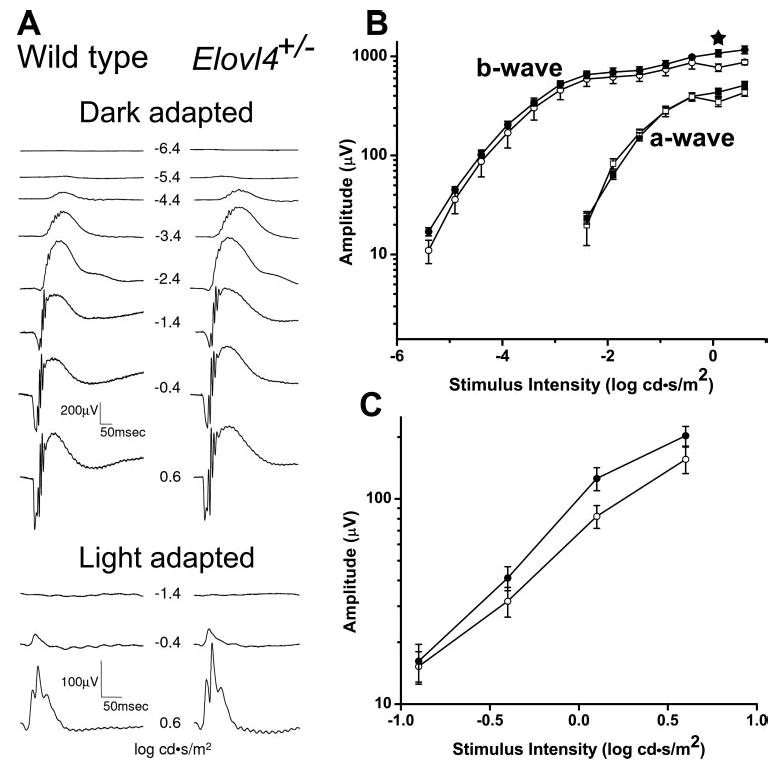
Electroretinogram responses of Elovl4+/− and wild-type mice were similar. (A) Representative waveforms of dark- and light-adapted flash ERG responses of wild-type and Elovl4+/− mice. (B) The a-wave (squares) and b-wave (circles) intensity–response curves of the dark-adapted ERGs of Elovl4+/− (n = 6, full symbols) and age-matched littermate wild-type (n = 4, empty symbols) mice. Averages and SE bars are shown. A significant difference between the groups was found at only one stimulus intensity (★). (C) Intensity-response curves of the light-adapted ERGs.
Because ELVOL4 is assumed to be involved in fatty acid metabolism, we examined the systemic and retinal fatty acid profiles (summarized in Tables 1, 2). No major differences were noted in the fatty acid profile of the plasma between the groups, although several fatty acids had a significantly lower concentration in the Elovl4+/− retinas. After we applied the Bonferroni correction for multiple comparisons made simultaneously on the same data set, we found no significant differences in the fatty acid profile between Elovl4+/− and WT mice. However, it is noteworthy that five of the nine fatty acids that showed a significant difference at the P < 0.05 level were of the monounsaturated group.
Table 1.
Plasma Concentration of Fatty Acids
| Fatty Acid | WT | Elovl4+/− |
|---|---|---|
| 14:0 | 4 ± 1 | 4.8 ± 2.5 |
| 16:0 | 300 ± 143 | 336 ± 156 |
| 18:0 | 128 ± 56 | 195 ± 91 |
| 20:0 | 3.3 ± 2.3 | 2.6 ± 1.6 |
| 22:0 | 3.5 ± 1.7 | 2.8 ± 1.5 |
| 24:0 | 2.4 ± 0.6 | 2.5 ± 0.9 |
| Total saturated | 441 ± 201 | 542 ± 240 |
| 14:1 | 5.1 ± 1.8 | 3.9 ± 0.8 |
| 16:1 | 38 ± 20 | 33 ± 18 |
| 18:1n9 | 218 ± 72 | 204 ± 97 |
| 18:1n7 | 38 ± 18 | 40 ± 25 |
| 20:1n9 | 4.5 ± 1.9 | 6.9 ± 4.8 |
| 24:1n9 | 5.9 ± 0.5 | 8.3 ± 2.8 |
| Total monounsaturated | 309 ± 110 | 296 ± 137 |
| 20:3n9 | 3 ± 1.7 | 3.5 ± 2.1 |
| 18:2n6 | 347 ± 149 | 352 ± 154 |
| 18:3n6 | 2.8 ± 1.2 | 3.5 ± 1.9 |
| 20:2n6 | 4.5 ± 1.8 | 4.3 ± 2.4 |
| 20:3n6 | 26 ± 15 | 30 ± 24 |
| 20:4n6 (AA) | 159 ± 58 | 209 ± 125 |
| 22:4n6 | 2.4 ± 0.2 | 2.1 ± 1.2 |
| 22:5n6 | 2.5 ± 1 | 2.2 ± 1.4 |
| Total n6 | 544 ± 210 | 603 ± 290 |
| 18:3n3 | 7 ± 3.1 | 6.2 ± 4.2 |
| 20:5n3 | 21.3 ± 9.5 | 29.9 ± 13.3 |
| 22:5n3 | 10.2 ± 3.9 | 8.6 ± 3.7 |
| 22:6n3 (DHA) | 114 ± 31 | 144 ± 73 |
| Total n3 | 153 ± 45 | 188 ± 84 |
| n6/n3 ratio | 3.5 ± 0.4 | 3.2 ± 0.6 |
| AA/DHA ratio | 1.4 ± 0.3 | 1.4 ± 0.5 |
Data are average micrograms/milliliters ± standard deviations. WT (n = 4), Elovl4+/− (n = 13). AA, arachidonic acid; DHA, docosahexaenoic acid.
Table 2.
Retinal Concentration of Fatty Acids
| Fatty Acid | WT | Elovl4+/− |
|---|---|---|
| 14:0* | 0.2 ± 0.2 | 0.07 ± 0.03 |
| 16:0DMA | 0.3 ± 0.9 | 0.26 ± 0.1 |
| 16:0 | 4.1 ± 1.9 | 3.1 ± 1 |
| 18:0DMA | 0.3 ± 0.1 | 0.3 ± 0.1 |
| 18:0 | 2.90 ± 0.64 | 3.1 ± 1.1 |
| 20:0 | 0.07 ± 0.03 | 0.06 ± 0.01 |
| 22:0 | 0.06 ± 0.04 | 0.05 ± 0.01 |
| 24:0* | 0.06 ± 0.04 | 0.04 ± 0.01 |
| Total saturated | 8 ± 2.91 | 7 ± 2.2 |
| 16:1* | 0.4 ± 0.3 | 0.1 ± 0.1 |
| 18:1DMA | 0.05 ± 0.002 | 0.08 ± 0.1 |
| 18:1n9* | 2.9 ± 1.5 | 1.7 ± 0.5 |
| 18:1n7* | 0.5 ± 0.2 | 0.3 ± 0.1 |
| 20:1n9 | 0.04 ± 0.02 | 0.04 ± 0.01 |
| 24:1n9* | 0.05 ± 0.01 | 0.04 ± 0.01 |
| Total monounsaturated* | 3.9 ± 2 | 2.2 ± 0.6 |
| 20:3n9* | 0.03 ± 0.02 | 0.02 ± 0.003 |
| 18:2n6* | 0.4 ± 0.3 | 0.2 ± 0.05 |
| 20:2n6 | 0.04 ± 0.02 | 0.03 ± 0.01 |
| 20:3n6 | 0.09 ± 0.03 | 0.08 ± 0.03 |
| 20:4n6 | 1.3 ± 0.3 | 1.3 ± 0.4 |
| 22:4n6 | 0.09 ± 0.07 | 0.11 ± 0.05 |
| 22:5n6 | 0.03 ± 0.02 | 0.02 ± 0.01 |
| 24:4n6 | 0.02 ± 0.01 | 0.02 ± 0.03 |
| 24:5n6 | 0.01 ± 0.002 | 0.005 ± 0.002 |
| Total n6 | 2 ± 0.8 | 1.7 ± 0.5 |
| 20:5n3 | 0.07 ± 0.03 | 0.06 ± 0.02 |
| 22:5n3 | 0.14 ± 0.03 | 0.14 ± 0.04 |
| 22:6n3 | 1.1 ± 1.8 | 3.2 ± 2.5 |
| 24:5n3 | 2.7 ± 2.3 | 1.3 ± 2.3 |
| 24:6n3 | 0.13 ± 0.08 | 0.09 ± 0.04 |
| Total n3 | 4.1 ± 0.8 | 4.9 ± 1.5 |
| Total fatty acids | 20.6 ± 8 | 18.2 ± 6.7 |
| n6/n3 ratio | 0.5 ± 0.11 | 0.4 ± 0.1 |
| AA/DHA ratio | 12.1 ± 5.2 | 15.2 ± 3.5 |
Data are average micrograms/milligram ± standard deviations. WT (n = 3), Elovl4+/− (n = 10).
Fatty acids with significantly lower concentrations in the retinas of Elovl4+/− mice (Student’s t-test, P < 0.05). DMA indicates the dimethylacetal derivatives. AA, arachidonic acid; DHA, docosahexaenoic acid.
Discussion
Total knockout of Elovl4 is lethal, and no adult homozygous knockout mice were produced, indicating that the gene and protein are essential at least during development. Mutations in the human ELOVL4 gene result in STGD3, causing vision loss from disruption of the photoreceptor-RPE complex as an autosomal dominant trait, and the Elovl4+/− retina of mouse showed subtle yet consistent thinning of the photoreceptor outer segment layer. RPE changes including atrophy and lipofuscin accumulation are believed to contribute to photoreceptor loss in patients with STGD1,34 and similar changes have been found in Elovl4 transgenic STGD3 mice.12,34 However, in this gene-knockout study, we observed only sporadic RPE abnormalities in the Elovl4+/− mice, and it is reasonable to propose that photoreceptor loss in the mice may be primary rather than secondary to RPE failure. This is consistent with the known expression of ELOVL4 protein in rod and cone inner segments (IS).5
Elovl4 mRNA levels in Elovl4+/− retina were significantly lower than those in age-matched C57BL/6 mice. The rhodopsin mRNA level was, on average, 33% lower in Elovl4+/− retinas. Although this difference was not statistically significant, it could reflect a decreased requirement for rhodopsin brought about by the slightly reduced number of rods and reduced ROS length.
Despite the 20% decrease in ROS length and the approximately 10% loss of photoreceptor cells, retinal ERG responses of the Elovl4+/− mice were not significantly different from the littermate control mice and, if anything, the responses tended to be larger than WT. ERG responses in ELOVL4+/− patients are generally only slightly smaller than normal, consistent with involvement of only macular function rather than having fullfield retinal involvement.3,4 This finding is dissimilar to that in autosomal recessive STGD1 patients, who, because of ABCR gene mutations, can exhibit severe functional deficits across the entire retina.35,36
The amino acid sequence of ELOVL4 is well conserved evolutionarily from yeast to human.18 Members of the ELO family are believed to be involved in fatty acid elongation.37,38 ELOVL4 is expressed in the photoreceptor cells in a number of species.18 In the mouse the retina, brain, testis, and skin express high amounts of Elovl4, and these tissues are known to have high requirements for LCFAs in their cellular membranes.21 Elovl4 expression increases in the retina during the period that photoreceptor outer segments are developing, and a high level of ELOVL4 is maintained throughout life, perhaps because of the requirement for LCFAs in the retina, owing to turnover of photoreceptor outer segment membrane as they undergo repetitive, daily shedding and renewal.39 Very-long-chain polyunsaturated fatty acids of phosphatidylcholine are synthesized in photoreceptors.40 Moreover, lower blood levels of DHA and other long-chain fatty acids have been noted in patients with several different genetic forms of photoreceptor degeneration.41,42 We evaluated but did not find any abnormalities in blood fatty acid composition of the Elovl4+/− mice, despite reduced concentration of several fatty acids in the retina. Although we could discern no obvious pattern in the reduced fatty acids that would place the ELOVL4 enzyme along the fatty acid metabolic pathway in the retina, two LCFAs of 24 carbon atoms were diminished, suggesting that the ELOVL4 protein may be necessary for their synthesis. It was intriguing to note that retinal levels of several shorter fatty acids were also decreased in the Elovl4+/− retinas. LCFAs are broken down and recycled into short-chain nonessential fatty acids, and it is possible that a decrease in long- or very-LCFAs results in lower amounts of short-chain fatty acids. Shortening of the outer segments and photoreceptor loss would reduce the proportion of membrane-rich tissue in the Elovl4+/− retinas relative to the WT and may therefore account for some of the reduced fatty acid content in the Elovl4+/− retinas. However, in such case we would expect the content of DHA, which is the prominent fatty acid in the disc membranes, to be reduced. Because DHA levels were similar in Elovl4+/− and in WT retinas, we suggest that most of the difference in fatty acid content reflects an actual change in fatty acid composition and is not an artifact of different membrane contents in the retinal tissues.
Lower concentrations of monounsaturated fatty acids were noted in the retina of the Elovl4+/− mice. Monounsaturated fatty acids, including specifically oleic acid (18:1n9), have a high affinity for the interphotoreceptor retinoid-binding protein (IRBP) and may be involved in regulating the binding of all-trans-retinol to IRBP.43 Hence, a defect in ELOVL4 activity may have both regulatory and structural consequences. Further work is needed to establish the role of ELOVL4 in fatty acid metabolism in the retina.
Two other Elovl4 mutant mouse models have been produced, both of which are different from this gene-knockout model. One was a transgenic model in which one or more gene copies of mutant Elovl4 with a 5-bp deletion were added to the complement of two endogenous normal gene alleles, causing expression of three and in some cases more copies.12 A second model is a gene-knockin model in which a mutant Elovl4 gene harboring a 5-bp deletion identical with the mutation identified in humans with autosomal dominant STGD3 maculopathy replaces one normal WT allele. The third model that we present here, the Elovl4 knockout, provides unique information as to the possible disease mechanism.
Autosomal dominant human STGD3 disease could hypothetically lead to photoreceptor degeneration through several mechanisms. Haploinsufficiency may result for conditions in which expression of both gene copies is necessary to produce a sufficient quantity of protein to maintain normal function. Because the metabolic requirement for ELOVL4 due to the high level of photoreceptor turnover in the retina may be greater than in other tissues, the loss of one functional allele may make the retinal tissue more susceptible to damage and degeneration than it does other tissues with lower metabolic requirements. One example of haploinsufficiency is the heterozygous rhodopsin knockout rho+/− mouse in which an age-dependent loss of photoreceptors and shortening of rod outer segments are observed as early as 3 to 4 months of age.44,45 By 1 year of age, ONL width and outer segment length in rho+/− mice were reduced to only 50% of wild-type animals, and the ERG b-wave was 80% reduced (Bush R, unpublished observation, 1999). Another example is haploinsufficiency of the rds/peripherin photoreceptor protein that results in abnormal photoreceptor structure and progressive degeneration in the rds+/− mouse.46,47 However, if haploinsufficiency were the underlying mechanism of STGD3 disease, our heterozygous Elovl4+/− mice that expressed greatly reduced Elovl4 message levels would be likely to manifest overt retinal disease. Further, they would have the same phenotype as heterozygous mice harboring the 5-bp deletion human STGD3 mutation. This was not the case, however, as the 5-bp mutation knockin heterozygous mouse is reported to show progressive photoreceptor loss beginning as early as 2 months (Ayyagari R, personal communication, 2005). By contrast, our Elovl4+/− animals showed only subtle structural photoreceptor abnormalities and exhibited no functional ERG abnormality even at the advanced age of 16 months and older, despite the considerable reduction of the normal ELOVL4 message quantity. This is a fundamental difference between the phenotypes of these two models that indicates that retinal degeneration in the STGD3 in mouse does not result primarily from haploinsufficiency.
An alternative mechanism for the autosomal dominant mutation underlying STGD3 is gain of function. A subset of gain-of-function mutations is the dominant negative mechanism, in which the abnormal protein from the mutant allele antagonizes the function of the wild-type protein. Some dominant negative mutations occur in multimeric proteins, in which a single mutant subunit can eliminate normal activity of the entire protein complex.11 In dominant negative disease the heterozygous knockout would have no or a very mild phenotype, as we found in the present Elovl4+/− model.
Elimination or decrease in enzyme activity can lead to abnormal accumulation of a substrate, as occurs in several neurologic disorders involving lysosomal or peroxisomal defects.48 In the case of ELOVL4 disease, a deleterious effect could arise from abnormal accumulation of a precursor fatty acid that is not metabolized appropriately due to insufficient enzymatic activity. However, we did not find evidence toward this in the fatty acid profiles of the Elovl4+/− retinas. It is more likely that the detrimental effect of the mutation involves the lack of some enzymatic product, which would be consistent with the reduced amounts of some fatty acids that we found in the Elovl4+/− retinas.
The present work shows for the first time that haploinsufficiency does not play a key role in the Elovl4 disease mechanism in vivo. Though haploinsufficiency and dominant negative mechanisms are not mutually exclusive, the results of the present study further suggest that the disease observed for the 5-bp deletion ELOVL4 mutation in STGD3 results from a dominant negative mechanism. This mechanism has also been suggested based on in vitro cell line studies and in vivo studies.5–14 The evidence that is accumulating from the three different Elovl4 mouse models begins to elucidate the disease mechanism in STGD3 and opens the way to exploring treatment strategies.
Finding that haploinsufficiency is not the key mechanism of pathogenesis in STGD3 ELOVL4 disease indicates that decreasing the quantity of mutant protein could be a useful therapeutic strategy. This approach was successful in reducing retinal degeneration in the P23H rhodopsin transgenic rat using ribozyme knockdown of mutant RNA.49 As the Elovl4 transgenic mouse model indicated that disease manifestation worsened as the load of mutant protein increased,12 a strategy of decreasing the ratio of mutant to normal protein should be therapeutically beneficial.
Acknowledgments
The authors thank Arvydas Maminishkis and Robert Mullins for help with fundus images, Yong Zeng for genotyping, Mary A. Crawford for EM imaging, Terry Cox for helpful advice on statistical analysis, and Tricia Barr-Hughes (Taconic Farms, Germantown, NY) for animal husbandry information.
Footnotes
Disclosure: D. Raz-Prag, None; R. Ayyagari, None; R.N. Fariss, None; M.N.A. Mandal, None; V. Vasireddy, None; S. Majchrzak, None; A.L. Webber, Merck Research Laboratories (E); R.A. Bush, None; N. Salem, Jr, None; K. Petrukhin, Merck Research Laboratories (E); P.A. Sieving, None
Supported by the Intramural Research Program of the NIH, NIDCD, NIAAA and NEI.
References
- 1.Tezel TH, Bora NS, Kaplan HJ. Pathogenesis of age-related macular degeneration. Trends Mol Med. 2004;10:417–420. doi: 10.1016/j.molmed.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Shroyer NF, Lewis RA, Allikmets R, et al. The rod photoreceptor ATP-binding cassette transporter gene, ABCR, and retinal disease: from monogenic to multifactorial. Vision Res. 1999;39:2537–2544. doi: 10.1016/s0042-6989(99)00037-1. [DOI] [PubMed] [Google Scholar]
- 3.Stone EM, Nichols BE, Kimura AE, et al. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch Ophthalmol. 1994;112:765–772. doi: 10.1001/archopht.1994.01090180063036. [DOI] [PubMed] [Google Scholar]
- 4.Griesinger IB, Sieving PA, Ayyagari R. Autosomal dominant macular atrophy at 6q14 excludes CORD7 and MCDR1/PBCRA loci. Invest Ophthalmol Vis Sci. 2000;41:248–255. [PubMed] [Google Scholar]
- 5.Zhang K, Kniazeva M, Han M, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein PS, Tammur J, Singh N, et al. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest Ophthalmol Vis Sci. 2001;42:3331–3336. [PubMed] [Google Scholar]
- 7.Ambasudhan R, Wang X, Jablonski MM, et al. Atrophic macular degeneration mutations in ELOVL4 result in the intracellular misrouting of the protein. Genomics. 2004;83:615–625. doi: 10.1016/j.ygeno.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Karan G, Yang Z, Zhang K. Expression of wild type and mutant ELOVL4 in cell culture: subcellular localization and cell viability. Mol Vis. 2004;10:248–253. [PubMed] [Google Scholar]
- 9.Maugeri A, Meire F, Hoyng CB, et al. A novel mutation in the ELOVL4 gene causes autosomal dominant Stargardt-like macular dystrophy. Invest Ophthalmol Vis Sci. 2004;45:4263–4267. doi: 10.1167/iovs.04-0078. [DOI] [PubMed] [Google Scholar]
- 10.Jackson MR, Nilsson T, Peterson PA. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J. 1990;9:3153–3162. doi: 10.1002/j.1460-2075.1990.tb07513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grayson C, Molday RS. Dominant-negative mechanism underlies autosomal dominant stargardt-like macular dystrophy linked to mutations in elovl4. J Biol Chem. 2005 doi: 10.1074/jbc.M503411200. [DOI] [PubMed] [Google Scholar]
- 12.Karan G, Lillo C, Yang Z, et al. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: a model for macular degeneration. Proc Natl Acad Sci USA. 2005;102:4164–4169. doi: 10.1073/pnas.0407698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karan G, Yang Z, Howes K, et al. Loss of ER retention and sequestration of the wild-type ELOVL4 by Stargardt disease dominant negative mutants. Mol Vis. 2005;11:657–664. [PubMed] [Google Scholar]
- 14.Vasireddy V, Vijayasarathy C, Huang J, et al. Stargardt-like macular dystrophy protein ELOVL4 exerts a dominant negative effect by recruiting wild-type protein into aggresomes. Mol Vis. 2005;11:665–676. [PubMed] [Google Scholar]
- 15.Zhang XM, Yang Z, Karan G, et al. Elovl4 mRNA distribution in the developing mouse retina and phylogenetic conservation of Elovl4 genes. Mol Vis. 2003;9:301–307. [PMC free article] [PubMed] [Google Scholar]
- 16.Leonard AE, Bobik EG, Dorado J, et al. Cloning of a human cDNA encoding a novel enzyme involved in the elongation of long-chain polyunsaturated fatty acids. Biochem J. 2000;350:765–770. [PMC free article] [PubMed] [Google Scholar]
- 17.Oh CS, Toke DA, Mandala S, et al. ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J Biol Chem. 1997;272:17376–17384. doi: 10.1074/jbc.272.28.17376. [DOI] [PubMed] [Google Scholar]
- 18.Lagali PS, Liu J, Ambasudhan R, et al. Evolutionarily conserved ELOVL4 gene expression in the vertebrate retina. Invest Ophthalmol Vis Sci. 2003;44:2841–2850. doi: 10.1167/iovs.02-0991. [DOI] [PubMed] [Google Scholar]
- 19.Uauy R, Hoffman DR, Peirano P, et al. Essential fatty acids in visual and brain development. Lipids. 2001;36:885–895. doi: 10.1007/s11745-001-0798-1. [DOI] [PubMed] [Google Scholar]
- 20.Jeffrey BG, Weisinger HS, Neuringer M, et al. The role of docosahexaenoic acid in retinal function. Lipids. 2001;36:859–871. doi: 10.1007/s11745-001-0796-3. [DOI] [PubMed] [Google Scholar]
- 21.Mandal MN, Ambasudhan R, Wong PW, et al. Characterization of mouse orthologue of ELOVL4: genomic organization and spatial and temporal expression. Genomics. 2004;83:626–635. doi: 10.1016/j.ygeno.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Sprecher H, Luthria DL, Mohammed BS, et al. Reevaluation of the pathways for the biosynthesis of polyunsaturated fatty acids. J Lipid Res. 1995;36:2471–2477. [PubMed] [Google Scholar]
- 23.Luthria DL, Mohammed BS, Sprecher H. Regulation of the biosynthesis of 4,7,10,13,16,19-docosahexaenoic acid. J Biol Chem. 1996;271:16020–16025. doi: 10.1074/jbc.271.27.16020. [DOI] [PubMed] [Google Scholar]
- 24.MacDonald IM, Hebert M, Yau RJ, et al. Effect of docosahexaenoic acid supplementation on retinal function in a patient with autosomal dominant Stargardt-like retinal dystrophy. Br J Ophthalmol. 2004;88:305–306. doi: 10.1136/bjo.2003.024299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hubbard AF, Askew EW, Singh N, et al. Association of adipose and red blood cell lipids with severity of dominant Stargardt macular dystrophy (STGD3) secondary to an ELOVL4 mutation. Arch Ophthalmol. 2006;124:257–263. doi: 10.1001/archopht.124.2.257. [DOI] [PubMed] [Google Scholar]
- 26.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 27.Morrison WR, Smith LM. Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. J Lipid Res. 1964;53:600–608. [PubMed] [Google Scholar]
- 28.Salem N, Jr, Reyzer M, Karanian J. Losses of arachidonic acid in rat liver after alcohol inhalation. Lipids. 1996;31:S153–S156. doi: 10.1007/BF02637068. [DOI] [PubMed] [Google Scholar]
- 29.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 30.Znoiko SL, Rohrer B, Lu K, et al. Downregulation of cone-specific gene expression and degeneration of cone photoreceptors in the Rpe65−/− mouse at early ages. Invest Ophthalmol Vis Sci. 2005;46:1473–1479. doi: 10.1167/iovs.04-0653. [DOI] [PubMed] [Google Scholar]
- 31.Haag M. Essential fatty acids and the brain. Can J Psychiatry. 2003;48:195–203. doi: 10.1177/070674370304800308. [DOI] [PubMed] [Google Scholar]
- 32.Gartner S, Henkind P. Aging and degeneration of the human macula. 1. Outer nuclear layer and photoreceptors. Br J Ophthalmol. 1981;65:23–28. doi: 10.1136/bjo.65.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delori FC, Goger DG, Dorey CK. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci. 2001;42:1855–1866. [PubMed] [Google Scholar]
- 34.Cideciyan AV, Aleman TS, Swider M, et al. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet. 2004;13:525–534. doi: 10.1093/hmg/ddh048. [DOI] [PubMed] [Google Scholar]
- 35.Klevering BJ, Deutman AF, Maugeri A, et al. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol. 2005;243:90–100. doi: 10.1007/s00417-004-1079-4. [DOI] [PubMed] [Google Scholar]
- 36.Lois N, Halfyard AS, Bird AC, et al. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am J Ophthalmol. 2004;138:55–63. doi: 10.1016/j.ajo.2004.02.056. [DOI] [PubMed] [Google Scholar]
- 37.Cinti DL, Cook L, Nagi MN, et al. The fatty acid chain elongation system of mammalian endoplasmic reticulum. Prog Lipid Res. 1992;31:1–51. doi: 10.1016/0163-7827(92)90014-a. [DOI] [PubMed] [Google Scholar]
- 38.Tvrdik P, Westerberg R, Silve S, et al. Role of a new mammalian gene family in the biosynthesis of very long chain fatty acids and sphingolipids. J Cell Biol. 2000;149:707–718. doi: 10.1083/jcb.149.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young RW. The renewal of photoreceptor cell outer segments. J Cell Biol. 1967;33:61–72. doi: 10.1083/jcb.33.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rotstein NP, Aveldano MI. Synthesis of very long chain (up to 36 carbon) tetra, penta and hexaenoic fatty acids in retina. Biochem J. 1988;249:191–200. doi: 10.1042/bj2490191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Converse CA, Hammer HM, Packard CJ, et al. Plasma lipid abnormalities in retinitis pigmentosa and related conditions. Trans Ophthalmol Soc U K. 1983;103:508–512. [PubMed] [Google Scholar]
- 42.Hoffman DR, Birch DG. Docosahexaenoic acid in red blood cells of patients with X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1995;36:1009–1018. [PubMed] [Google Scholar]
- 43.Semenova EM, Converse CA. Comparison between oleic acid and docosahexaenoic acid binding to interphotoreceptor retinoid-binding protein. Vision Res. 2003;43:3063–3067. doi: 10.1016/j.visres.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 44.Humphries MM, Rancourt D, Farrar GJ, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- 45.Lem J, Krasnoperova NV, Calvert PD, et al. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci USA. 1999;96:736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hawkins RK, Jansen HG, Sanyal S. Development and degeneration of retina in rds mutant mice: photoreceptor abnormalities in the heterozygotes. Exp Eye Res. 1985;41:701–720. doi: 10.1016/0014-4835(85)90179-4. [DOI] [PubMed] [Google Scholar]
- 47.Sanyal S, Dees C, Zeilmaker GH. Development and degeneration of retina in rds mutant mice: observations in chimaeras of heterozygous mutant and normal genotype. J Embryol Exp Morphol. 1986;98:111–121. [PubMed] [Google Scholar]
- 48.Wanders RJ, Waterham HR. Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin Genet. 2004;67:107–133. doi: 10.1111/j.1399-0004.2004.00329.x. [DOI] [PubMed] [Google Scholar]
- 49.Lewin AS, Drenser KA, Hauswirth WW, et al. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med. 1998;4:967–971. doi: 10.1038/nm0898-967. [DOI] [PubMed] [Google Scholar]