

Abstract
PURPOSE
Cone function and survival are compromised in the guanylate cyclase-1 (GC1) knockout mouse. Disruption of the light-driven translocation of cone arrestin is one of the phenotypes of cone cells in this retina: the cone arrestin in these cells is localized to the outer segments and synaptic terminals, regardless of the state of light adaptation. The purpose of this study was to determine whether the expression of GC1 restores cone arrestin translocation in the cone cells of postnatal GC1 knockout mouse retina.
METHODS
Subretinal injections of AAV-GC1 were performed on 3-week-old GC1 KO mice. Electroretinographic and immunohistochemical analyses of treated retinas were carried out 5 weeks after injection. GC1 and cone arrestin antibodies were used to identify photoreceptors transduced by the AAV vector and to localize cone arrestin within cone cells, respectively.
RESULTS
Treatment of GC1 knockout retinas with AAV-GC1 restored the light-driven translocation of cone arrestin in transduced cone cells. Staining patterns for cone arrestin in transduced and wild-type cone cells were indistinguishable after dark and light adaptation. In dark-adapted retinas, cone arrestin was distributed throughout the subcellular compartments of the cone cells. In light-adapted retinas, cone arrestin was concentrated in the cone outer segments. Successful restoration of cone arrestin translocation did not translate to a restoration of cone ERG responses, which remained undetectable in the treated retinas.
CONCLUSIONS
AAV-mediated expression of GC1 in a subpopulation of cone cells in postnatal GC1 knockout retina restores light-driven translocation of cone arrestin in these cells. These findings, which show that fully developed cone cells that have developed in the absence of GC1 can respond to viral-mediated expression of this enzyme, support further analysis of this animal model of Leber congenital amaurosis type 1 (LCA1), a disease that results from null mutations in the gene encoding this enzyme.
The guanylate cyclase 1 (GC1) knockout (KO) mouse1 is a mammalian model of Leber congenital amaurosis 1 (LCA1).2–4 This autosomal recessive disease represents the earliest and most severe form of retinal degeneration. Diagnosis is made at birth or within the first few months of life, when patients display severely impaired vision, extinguished electroretinogram (ERG), and normal fundus. Unlike retinal degeneration in humans with LCA1 that involves rod and cone cells,2 degeneration in the GC1 KO mouse retina is limited to the cone photoreceptors. Rod cells in this retina continue to be able to generate electrical responses to light1 and to exhibit normal light-induced translocation of rod arrestin and rod transducin in the absence of GC1,5 whereas both processes are disrupted in the cone cells.
The ability of mouse rod cells to continue to function in the absence of GC1 suggests that a second GC enzyme is present and functional in these cells. Two variants of retinal GC (GC1 and GC2) have been identified in the vertebrate retina.6–8 GC2 is expressed in photoreceptors and has been colocalized with GC1 in rat rod photoreceptor cells.9 Thus, it is possible that rod function in the GC1 KO mouse is subserved by GC2. The recent observation that rod function is absent in double knockout mice in which both GC1 and GC2 have been disabled supports this possibility (Wolfgang Baehr, personal communication, 2005). The inability of mouse cone cells to function in the absence of GC1 indicates either that these cells do not express a second GC enzyme or that the second enzyme does not support the functional needs of these cells. In either case, restoration of cone function in retinas lacking GC1 is likely to require delivery and expression of a GC1 transgene in these cells.
In the GC1 KO mouse retina, loss of cone function is evident at birth1 and precedes cone cell degeneration, which occurs over the course of the first 6 months of life.10 In considering the possibility of developing effective therapies for LCA1, it is important to determine whether the expression of GC1 can restore any normal function to fully developed cones of the postnatal retina. In this series of experiments, we took advantage of the temporal dissociation of cone function loss and cone degeneration in the GC1 KO mouse to test this hypothesis. AAV serotype 5 vectors were used to deliver functional GC1 transgenes to the retinal photoreceptors of 3-week-old GC1 KO mice. Electrophysiological and immunocytochemical techniques were used to assess the ability of the vectors to restore cone function.
METHODS
Experimental Animals
Homozygous GC1 KO (or GCE KO) mice, originally obtained from the University of Texas Southwestern Medical Center at Dallas (generously provided by David Garbers), were rederived at the University of Florida, as previously described.10 All animals were handled in accordance with the animal use policies of the University of Florida College of Medicine and with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The GC1 KO breeding colony was maintained in an SPF isolator, and treated animals were maintained in SPF housing. A 12-hour light/12-hour dark cycle was used in all housing units, and food and water were available ad libitum.
Construction of AAV Vectors
AAV capsid protein serotype type 5 (AAV5) vectors that exhibit a higher transduction efficiency and a faster onset of expression than other AAV serotypes11 were used to deliver bGC1 to the photoreceptor cells. A cell-specific and a ubiquitous promoter were selected to drive expression of bGC1 in our AAV vectors. The cell-specific promoter, mouse opsin promoter (MOP), limits gene expression to photoreceptor cells when used in conjunction with AAV vectors.12 The ubiquitous promoter smCBA is a truncated version of the CBA promoter, which is a fusion of the chicken beta actin promoter and the CMV immediate-early cytomegalovirus enhancer.13 Truncation of the CBA promoter was achieved by removing 780 base pairs of internal sequence and collapsing the hybrid chicken β-actin/rabbit β-globin intron. The expression pattern of the smCBA promoter is similar to that of the full-length CBA promoter in retina (WWH, unpublished data, 2005). The plasmid backbones for AAV vectors were pTR-MOP-GFP12 and pTR-smCBA-GFP. The pTR-MOP-bGC1 and pTR-smCBA-bGC1 plasmids were constructed by replacing the green fluorescence protein (GFP) cDNA and neomycin cassette of the plasmid backbones with cDNA encoding bovine GC1 (bGC1). The cDNA encoding bGC1 was amplified from pSVL-ROS-GC114 using the 5′ fragment (5′-CCA TCG ATA GTT TAA ACG AGC CCC GGA CTT and 5′-GCC CAG CAC TGT TTC) and the 3′ fragment (5′-GGC GAC GTC TTC AGT CT and 5′-CCA TCG ATG ACC CAG CCT CAC TTC C). The resultant 5′ and 3′ bGC1 cDNA fragments were ligated sequentially into a pBSII SK′ vector (Stratagene, La Jolla, CA) from which the SacI site had been removed and joined using the unique SacI site in the sequence to create the full-length cDNA. The bGC1 cDNA was ligated into the AAV vector backbones using NotI and SalI. The integrity of the GC1 coding region was confirmed by sequence analyses. AAV vectors were packaged and purified according to previously reported methods.15
Subretinal Injections
At 21 days of age, mice were anesthetized by intraperitoneal injection (6 μL/g) of a sterile mixture of 100 mg/kg ketamine (Fort Dodge Animal Health, Fort Dodge, IA), 20 mg/kg xylazine (Phoenix Pharmaceutical, St. Joseph, MO), and PBS in a 1:1:5 ratio, respectively. The pupils were dilated with 2.5% phenylephrine (Akorn, Inc., Decatur, IL), and 0.5% proparacaine (Alcon Laboratories Inc., Fort Worth, TX) was applied to the corneas as topical anesthetic. Mydriasis was monitored using a dissecting microscope (Stemi SV6; Zeiss, Oberkochen, Germany). Hypromellose ophthalmic demulcent solution 2.5% (Gonak; Akorn, Inc.) was applied to the corneas to enhance visualization of the ocular surface and fundus during the injection procedure. The deep anesthetic plane was maintained for 20 to 30 minutes, providing ample time for subretinal injections. Subretinal injections of approximately 109 vector genomes in 1 μL were performed using a blunt-tipped 33-gauge needle specifically made to fit a 10-μL Hamilton syringe (Hamilton Co., Reno, NV). Injections were performed according to methods previously described.16 Injections were always performed in the animal’s right eye, leaving the left eye as a contralateral control. Immediately after the injection procedure, an ophthalmic ointment containing bacitracin, neomycin, and polymyxins (Vetropolycin; Pharmaderm, Melville, NY) was applied to the cornea to prevent corneal opacification and infection.
Electroretinograms
Mice were dark adapted overnight (more than 12 hours), and all subsequent procedures were carried out under dim red light (more than 650 nm). Immediately before the recording session, mice were anesthetized and eyes were dilated as described for the subretinal injection procedure. Hydroxypropyl methylcellulose 2.5% (Gonak; Akorn, Inc.) was applied to each eye to prevent corneal dehydration and to allow for optimal electrical conductivity. When they were fully sedated, mice were placed prostrate onto the electroretinogram (ERG) platform that was positioned so that the entire head of the mouse was inside the Ganzfeld stimulus dome. ERGs were recorded simultaneously from the left and right eyes of the animal using custom, gold loop cornea electrodes (2-mm diameter). Aluminum hub needles (27 gauge, 0.5 inch; Monoject; Kendall, Mansfield, MA) placed subcutaneously between the eyes and in a hind leg were used for the reference and ground electrodes, respectively. ERGs were recorded using a PC-based control and recording unit (Toennies Multiliner Vision; Jaeger/Toennies Höchberg, Germany) equipped with a Ganzfeld-stimulator. Dark-adapted (scotopic) ERGs were recorded first, and then light-adapted (photopic) ERGs were recorded. Scotopic rod ERG luminance-response functions were elicited through a series of white flashes of seven increasing intensities (−5.0 to 0.7 log cd · s/m2; 1.0 log unit steps). The interstimulus interval for the low-intensity stimuli was 1.1 second. Interstimulus intervals for the three highest intensity stimuli (−1.0 to 0.7 log cd · s/m2) were 2.5, 5.0, and 20.0 seconds, respectively. Ten responses were recorded and averaged at each of these stimulus intensities. On completion of the scotopic series, the animals were light adapted to a 100 cd · s/m2 white background for 5 minutes. Photopic cone responses were examined using a stimulus series consisting of seven increasing intensities (−3.0 to 1.08 log cd · s/m2). Fifty responses were recorded and averaged at each of these stimulus intensities. All stimuli were presented in the presence of the 100 cd · s/m2 white background. Vetropolycin ointment (Bacitracin-Neomycin-Polymyxin B; PharmaDerm, Duluth, GA) was applied to each eye at the end of the recording session. Amplitudes of the ERG waveforms were measured conventionally: a-waves were measured from the baseline to the trough; b-waves were measured from the a-wave trough to the positive peak.
Tissue Preparation
Five weeks after subretinal injections, GC1 KO and age-matched wild-type (WT) mice were dark adapted for 2 hours. Immediately after dark adaptation, enucleation was performed under dim red light–emitting diode. Injected and uninjected eyes were fixed in 4% paraformaldehyde overnight at 4°C. Subsequently, the cornea, lens, and vitreous were removed from each eye without disturbance to the retina. Eye-cups were placed in 30% sucrose in PBS for at least 1 hour at 4°C. Then they were then embedded in cryostat compound (Tissue-Tek OCT 4583; Sakura Finetek USA, Inc., Torrance, CA), quick frozen in a bath of liquid nitrogen, and serially sectioned at 12 μm with the use of a cryostat (Microtome HM550; Walldorf, Germany). Sections designated for immediate analysis were allowed to dry overnight at room temperature. Remaining sections were stored at −20°C until use.
Production of IS4 Monoclonal Antibody
Monoclonal anti–GC1 antibody (IS4) was generated against the 15-amino acid C-terminal peptide of bovine GC1 (RQKLEKARPGQFSGK; accession no. AAB86385). The peptide (2 mg) was mixed with 4 mg keyhole limpet hemocyanin in 5 mL of 10 mM sodium phosphate buffer, pH 7.4, containing 150 mM NaCl. A total volume of 100 μL glutaraldehyde (2.5%) was added to the sample in 20-μL aliquots. Samples were then mixed by rotation at room temperature for 3 hours and dialyzed overnight against 10 mM sodium phosphate and 150 mM NaC1, pH 7.4, using 1000 Da molecular weight cutoff (MWCO) dialysis tubing. Antisera were raised in BALB/c mice by subcutaneous immunization with approximately 10 μL of the peptide–keyhole limpet hemocyanin solution mixed with an equal volume of complete Freund adjuvant. Animals were boosted at 1- to 2-week intervals with the peptide solution mixed with incomplete adjuvant. Monoclonal IS4 antibody was prepared by a standard procedure.17 Hybridoma cells producing antibody were screened using immunoblotting against GC1 purified from bovine rod outer segment (ROS) and by immunocytochemical analyses of WT and GC1 KO mouse retinal sections.
Immunohistochemistry and Microscopy
Retinal sections were rinsed in 1 × PBS and were incubated in blocking solution (10% goat serum in PBS) for 30 minutes at room temperature. Sections were then incubated in primary dilution buffer (0.3% Triton X-100 and 1% BSA in PBS) containing the primary antibody overnight at 4°C. Bovine GC1 was detected with monoclonal antibody IS4 (1: 1000 dilution). Cone arrestin was detected with the polyclonal antibody LUMIj (Luminaire junior)18 at 1:1000 dilution. GFP was detected with monoclonal antibody MAB3580 (1:500 dilution; Chemicon, Temecula, CA). After incubation in primary antibodies, sections were rinsed in PBS and incubated for 1 hour at room temperature with IgG secondary antibodies tagged with either Alexa-594 or Alexa-488 fluorophore (Molecular Probes, Eugene, OR) diluted 1:500 in PBS. After incubation with secondary antibodies, sections were rinsed in PBS. Control sections were incubated in secondary antibodies alone. After a final rinse step, the sections were counterstained with 4′, 6′-diamino-2-phenylindole (DAPI) for 5 minutes at room temperature. After a rinse with PBS and water, the sections were mounted in an aqueous-based medium (Gel Mount; Biomedia, Foster City, CA) and coverslipped. Sections were examined with a microscope (Axiostop 2 Plus; Zeiss) fitted with a camera (RT color Spot, model 2.2.1; Diagnostic Instruments, Sterling Heights, MI). Retinal images were acquired with software (Spot version 3.4.5; Diagnostic Instruments). All fluorescent images were acquired using identical camera gain-and-exposure settings and were saved as .tiff files.
Confocal microscopy was used to obtain images of selected sections. Images were acquired (1024ES instrument; Bio-Rad, Hemel Hempstead, UK) using excitation lasers and emission filters optimized for FITC and Texas red. Images were attained with a 40 × objective supplemented with 1.5 × magnification. Laser settings were optimized so that signals arising from GC1, cone arrestin, or GFP expression were not saturated and autofluorescence was undetectable. Noise reduction was achieved using four to five Kalman reduction algorithms. Software (Lasersharp 3.0; Bio-Rad) was used to acquire a Z-series consisting of approximately 20 images spaced at 0.5-μm intervals. These images were combined to create a two-dimensional montage allowing visualization of entire cells.
Analysis of Fluorescent Images
Red-, green-, and blue-filtered .tiff images were overlaid (Adobe Photoshop version 8.0). With the use of the histogram function in the image program, measurements of the fluorescence intensity of each secondary antibody’s tagged fluorophore were obtained. Fluorescence intensity values for single cells were measured within the specific subcompartments (outer segments, inner segments) of the cell. Values obtained for treated, untreated, and WT cells were compared using one-way ANOVA (SigmaStat version 2.03; Systat Software, Inc., Point Richmond, CA). Fluorescence values were also measured across entire retinal cross-sections. These results represented estimates of the levels of GC1, cone arrestin, and GFP expression within the subcellular compartments of the photoreceptor cells. These measurements were corrected for background staining by subtracting fluorescence values obtained from control sections stained with secondary antibody alone. Intensity values were plotted and graphed (SigmaPlot version 8.0; Systat Software, Inc.).
RESULTS
Monoclonal Antibody IS4 Specific for GC1
The kinase-like and catalytic domains of all vertebrate GCs are highly conserved. To generate a specific antibody against GC1, we selected the C-terminal region of GC1 that is conserved among species (Fig. 1A) but is distinguished from the closely related guanylate cyclase, GC2 (Fig. 1B). The C-terminal peptide was coupled to keyhole limpet hemocyanin, and 10 mice were immunized. One mouse responded strongly to the expressed GC1 and was used to generate the monoclonal antibody. Using bovine ROS extract for screening, IS4 was isolated (Fig. 1C), and its specificity was tested immunohistochemically with WT and GC1 knockout mouse retina. In WT retina, immunostaining was highest in the photoreceptor outer segment layer, with lighter staining in the photoreceptor inner segments (Fig. 1D, left). No staining was observed in the photoreceptors of the GC1 KO mouse retina (Fig. 1D, right). RPE staining in this section was background staining resulting from autofluorescence of the layer.
FIGURE 1.
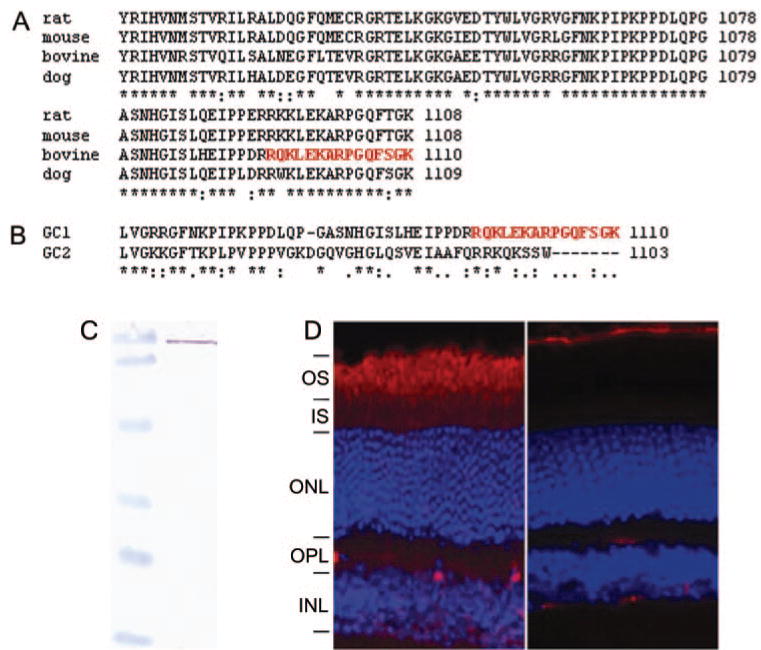
Production and characterization of anti–GC1 monoclonal antibody. (A) Alignment of the C-terminal regions of selected mammalian GC1 amino acid sequences. (B) Alignment of the C-terminal region of bovine GC1 and GC2. (C) Immuno-blotting of bovine ROS (10 μg) with IS4 antibody. Molecular weight standards (left) are 120, 86, 47, 34, 26, and 20 kDa. A single reactive band was observed in ROS. (D) Localization of GC1 in WT (left) and GC KO (right) mouse retinas. Frozen sections of 8-week-old mice were probed with the monoclonal antibody IS4.
MOPS and smCBA rAAV Vector Expression in Cone Photoreceptors
Cone photoreceptors represent 3% to 5% of the total population of photoreceptors in mouse retina. Because these cells were the focus of this study, it was important to ensure that the promoters we selected to drive expression of the bGC1 transgene were active in cone cells. The ability of cell-specific (MOP) and ubiquitous (smCBA) promoters to drive transgene expression in these cells was determined by examining the retinas of GC1 KO mice that had been injected with pTR-MOP-bGC1 or pTR-smCBA-bGC1 AAV vectors at 3 weeks of age. Examination of the retinas of these animals at 5 weeks after injection showed that both the cell-specific MOP promoter (Fig. 2A) and the ubiquitous smCBA promoter (data not shown) efficiently drove GC1 expression in rod and cone photoreceptor cells. Transduction efficiency and cell specificity of pTR-MOP-bGC1 and pTR-smCBA-bGC1 were identical. Subretinal injections typically cause detachment of approximately 80% of the retina. Based on histologic observation, we estimate that approximately 50% of cones within this area were transduced with therapeutic virus. Retinas of GC1 KO mice injected with either pTR-smCBA-bGC1 (Fig. 2B) or pTR-MOP-bGC1 (data not shown) were stained for GC1 and cone transducin alpha (CTα). Examination of these retinas using confocal microscopy revealed that several of the cone cells had been successfully transduced by these vectors and expressed GC1.
FIGURE 2.
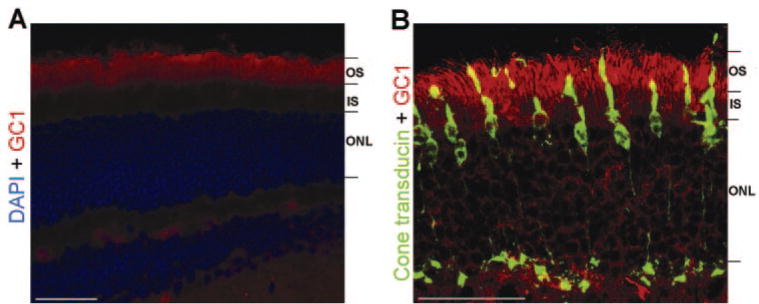
GC1 and cone Tα immunostaining of a GC1 KO mouse retina 5 weeks after subretinal injection of therapeutic AAV vector. (A) GC1 KO retina treated with pTR-MOP-bGC1. GC1 staining (red) was present in rod and cone photoreceptors and was localized to the outer segment regions of these cells. Sections were counterstained with DAPI. (B) Confocal image of GC1 KO retina treated with pTR-smCBA-GC1 stained with antibodies against GC1 (red) and cone transducin alpha (green). Cone Tα was detected throughout the cone cells in these dark-adapted retinas. GC1 and cone Tα were coexpressed in the outer segments of several of the cone cells, as evidenced by the yellow signal in these regions. Scale bars: 50 αm. OS, outer segment; IS, inner segment; ONL, outer nuclear layer.
Cone ERG Responses in AAV-GC1–Treated Retinas
Cone cells in the GC1 KO retina did not translocate cone arrestin or generate ERGs when stimulated by light. In this series of experiments, we recorded ERG responses from treated GC1 KO mouse retinas under photopic and scotopic recording conditions to determine whether AAV-GC1 treatment had any impact on the ability of the transduced cells to generate ERGs.
At 5 weeks after AAV-bGC1 treatment (postnatal day 56), the full-field photopic responses recorded from treated eyes of GC1 KO mice could not be distinguished from background levels. At the three highest stimulus intensities, the maximum photopic b-wave amplitudes recorded from WT P56 mice were significantly greater than those recorded from age-matched GC1 KO, pTR-MOP-bGC1-, and pTR-smCBA-bGC1- treated mice (Fig. 3A). No significant differences were detected between these groups at the four lowest intensities. One-way ANOVA was used to identify the main effects of treatment. Fisher PLSD post hoc tests (P = 0.05) were used to localize significant differences between groups. Significant treatment effects were observed at 0.7 log cd · s/m2 [F(3,51) = 15.03; P = 0.0001], 1.0 log cd · s/m2 [F(3,51) = 30.5; P < 0.0001], and 1.08 log cd · s/m2 [F(3,51) = 41.6; P < 0.0001]. At each of these intensities, the amplitudes of the responses produced by WT animals were larger than those observed in all other groups. No differences were observed in the b-waves recorded from pTR-MOP-bGC1 and pTR-smCBA-bGC1–treated eyes in response to the photopic stimulus series.
FIGURE 3.
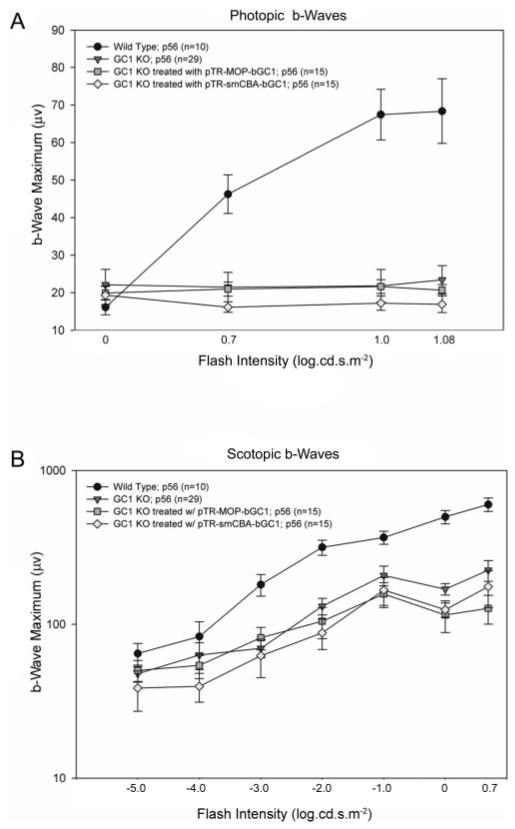
Electrophysiological analyses of AAV-GC1 retinas. (A) Comparisons of cone responses in age-matched (P56) WT (n = 10), GC1 KO (n = 29), pTR-MOP-bGC1– (n = 15) and pTR-smCBA-GC1–(n = 15) treated mice 5 weeks after treatment. Photopic responses were elicited using seven stimulus intensities (−3.0 to 1.08 log cd · s/m2), and the amplitudes of the b-waves of the ERG were measured. No responses were obtained at the three lowest flash intensities and are not plotted. Each point represents the mean ± SEM of the amplitudes of the b-waves recorded for each group at the indicated flash intensity. (B) Comparisons of rod responses in the same age-matched WT, GC1 KO, pTR-MOP-bGC1–, and pTR-smCBA-GC1–treated mice. Scotopic responses were elicited using seven stimulus intensities (−5.0 to 0.7 log cd · s/m2), and the amplitudes of the b-waves of the ERG were measured. Each point represents the mean ± SEM of the amplitudes of the b-waves recorded for each group at the indicated flash intensity. Scotopic responses in AAV-bGC1–treated animals were indistinguishable from those of untreated controls at all flash intensities.
At 5 weeks after AAV-bGC1 treatment, the full-field scotopic responses recorded from treated eyes of the GC1 KO mouse showed no improvement over untreated controls (Fig. 3B). In both the treated and untreated retinas, the amplitudes of the b-waves generated were approximately 20% of the WT responses. One-way ANOVA was used to identify the main effects of treatment in the scotopic series. Fisher PLSD post hoc tests (P = 0.05) were used to identify significant differences between groups. Significant treatment effects were observed at −3.0 log cd · s/m2 ([F(3,59) = 7.3; P < 0.0003]), −2.0 log cd · s/m2 ([F(3,59) = 9.4; P < 0.0001]), −1.0 log cd · s/m2 ([F(3,59) = 3.4; P < 0.0246]), 0.0 log cd · s/m2 ([F(3,59) = 24.9; P < 0.0001]), and 0.7 log cd · s/m2 ([F(3,59) = 11.9; P < 0.0001]). At each of these intensities, WT animals produced significantly larger responses than all other groups. At the two highest stimulus intensities, 0.0 log cd · s/m2 and 0.7 log cd · s/m2, the amplitudes of the b-waves recorded from the untreated GC1 KO mice were significantly higher than those of pTR-MOP-bGC1–treated mice ([F(3,59) = 24.5; P < 0.0341] and [F(3,59) = 24.5; P < 0.0307], respectively). This result prompted reexamination of the electrophysiological data obtained from the pTR-MOP-bGC1–treated mice at these stimulus intensities. Examination of these data revealed that the amplitudes of the b-wave responses of 4 of the 15 treated mice were >4 SD below the group mean b-wave amplitude. Histologic examination of the retinas of these animals, which revealed injection-induced retinal damage, suggested that the abnormally small ERG responses recorded from these animals reflected mechanical rather than vector-induced damage to the retinas. No significant differences were observed in the amplitudes of the b-waves recorded from pTR-MOP-bGC1– and pTR-smCBA-bGC1–treated eyes in response to the stimulus intensities examined.
Translocation of Cone Arrestin in rAAV-Transduced GC1 KO Cone Cells
We have previously shown that the light-driven movement of cone arrestin is disrupted in cone photoreceptors of GC1 KO mice.5 In dark-adapted WT retina, cone arrestin is distributed throughout the subcellular compartments of the cone photoreceptors; however, in the dark-adapted GC1 KO retina, cone arrestin is localized primarily to the outer segments and synaptic terminals of the cone cells (Fig. 4A). The distribution of cone arrestin within the dark-adapted cone cells of retinas treated with either pTR-MOP-bGC1 or pTR-smCBA-bGC1 was indistinguishable from that observed in dark-adapted WT retina (Fig. 4A). In dark-adapted, WT, and treated cone cells, arrestin immunostaining was approximately equal in the outer and inner segments. Quantitative analyses of cone arrestin immunostaining in these retinas confirmed this result. Cone arrestin staining was present in the outer and inner segments of the WT (Fig. 4B), pTR-MOP-bGC1–treated (Fig. 4D), and pTR-smCBA-bGC1–treated (data not shown) cone cells. The distribution of cone arrestin fluorescence in pTR-MOP-bGC1–treated cone cells (Fig. 4D) was representative of the distribution seen in retinas of mice treated with pTR-smCBA-b-GC1. In dark-adapted, untreated GC1 KO retina, cone arrestin staining was significantly higher in the outer segments compared with the inner segments of the cone cells (Fig. 4C). A comparison of these analyses (Fig. 4E) confirms that the largest change in the distribution of cone arrestin in the dark-adapted, AAV-treated cones occurred in the outer segments of these cells. Staining within individual cone photoreceptors was also analyzed. Differences in cone arrestin fluorescent staining between outer and inner segments of WT, GC1 KO, and pTR-MOP-bGC1–treated cone cells were calculated (n = 10 per group) and analyzed using one-way ANOVA (Fig. 4F). Results of these analyses showed that there was a significant treatment effect ([F(2,29) = 54.720; P < 0.001]). Post hoc Tukey tests (P = 0.05) revealed that there was not a significant difference between the distribution of cone arrestin in WT and AAV-treated cells; however, the distribution of cone arrestin in WT and AAV-treated cells was significantly different from that observed in untreated GC1 KO cone cells.
FIGURE 4.
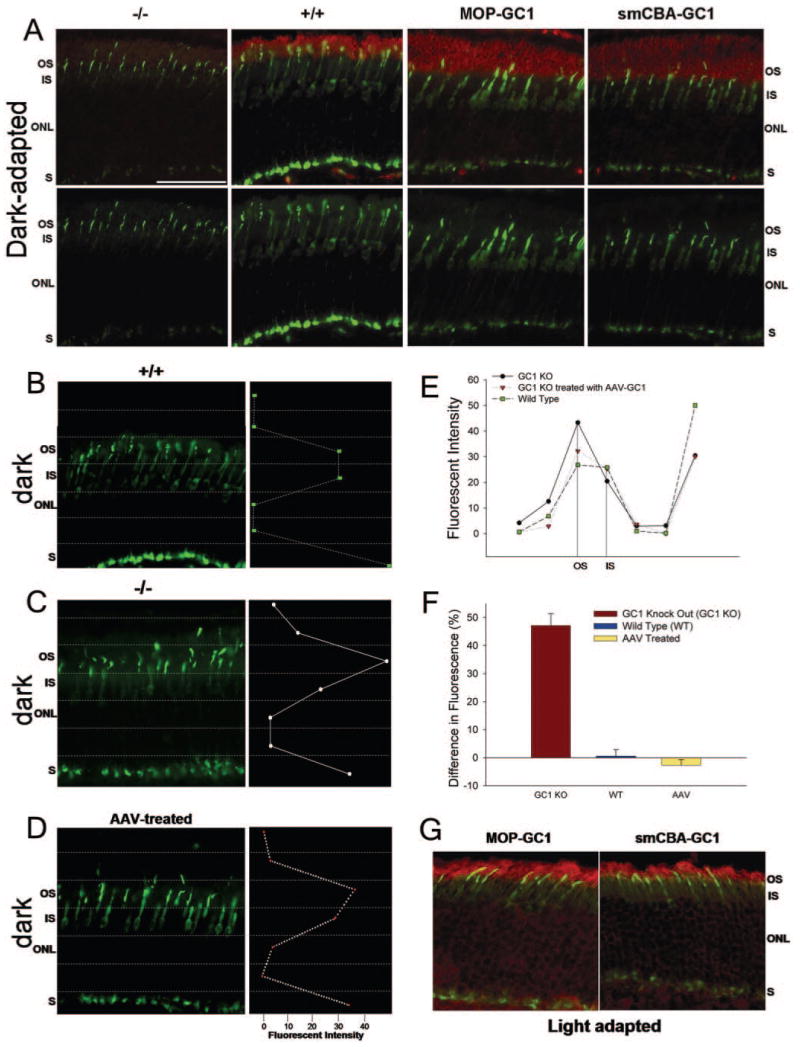
Distribution of cone arrestin in GC1 KO, WT, and AAV-treated mouse retinas in dark- and light-adapted conditions. (A) Retinas of dark-adapted mice were stained for GC1 (red) and cone arrestin (green). Examples of the staining observed in GC1 KO (−/ −), WT (+/+), and GC1 KO retinas treated with either pTR-MOP-bCG1 or pTR-smCBA-bGC1 are shown from left to right. The upper panel shows GC1 and cone arrestin staining. The lower panel shows only cone arrestin staining. (B–D) Quantitative analyses of the intensity of cone arrestin staining within the subcellular compartments of dark-adapted photoreceptor cells in wild type (+/+) (B), GC1 KO (−/ −) (C), and pTR-MOP-bGC1–treated (D) retinas. The distribution of cone arrestin staining was identical in cone cells treated with either pTR-MOP-bGC1 or pTR-smCBA-bGC1. (E) Comparison of the quantitative data shown in panels B to D. (F) Analyses of the differences in cone arrestin fluorescence staining of the outer and inner segments of GC1 KO (n = 10), WT (n = 10), and AAV-treated (n = 10) cone photoreceptors. Differences in cone arrestin fluorescence intensity values between outer and inner segments were calculated for each cell within each treatment group. The average of these differences for each respective group is plotted. (G) Distribution of cone arrestin in light-adapted retinas of mice treated with either pTR-MOP-bGC1 (left) or pTR-smCBA-bGC1 (right). Scale bar (A): 50 μm. S, synaptic terminal.
The distribution of cone arrestin within the cone cells of light-adapted GC1 KO mice treated with pTR-MOP-bGC1 (Fig. 4G, left) or pTR-smCBA-bGC1 (Fig. 4G, right) was similar to that observed in cone cells of light-adapted WT mice.5 Cone arrestin was localized to the cone outer segments and synaptic termini. Quantitative analysis of the cone arrestin immunostaining confirmed these results. No significant differences were found in cone arrestin fluorescent staining between the outer and inner segments of WT and AAV-treated cone cells of light-adapted mice (n = 10 per group).
The cone arrestin staining pattern in retinas of dark-adapted GC1 KO mice injected with control vector, either pTR-MOP-GFP or pTR-smCBA-GFP, was identical with that observed in untreated GC1 KO retinas. Unlike the redistribution of cone arrestin observed in cone cells expressing GC1 (Fig. 5A), cone arrestin in cone cells treated with control vector was localized primarily to the outer segments of transduced cells (Fig. 5B). Quantitative analyses of single cells transduced with the pTR-MOP-GFP control vector confirmed that the intensity of cone arrestin staining was greatest in the outer segments of the transduced cone cells expressing GFP (Fig. 5C).
FIGURE 5.
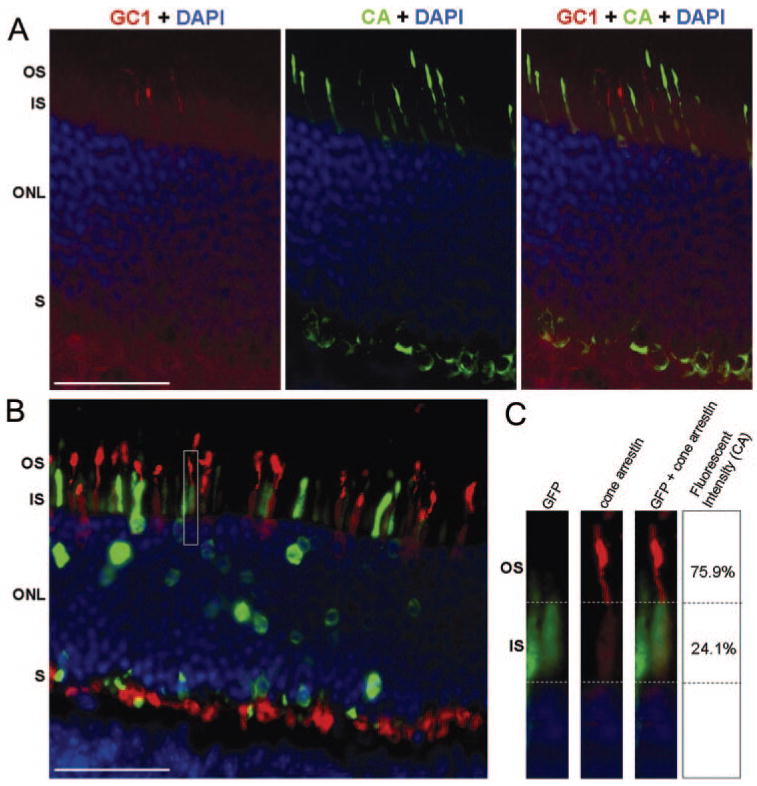
Comparison of the distribution of cone arrestin in retinas treated with the therapeutic vector, pTR-MOP-bGC1, or the control vector, pTR-MOP-GFP. (A) Retina of dark-adapted GC1 KO mouse treated with pTR-MOP-GC1. Outer segments of three cells expressing GC1 (red) are visible in the left panel. Cone arrestin staining (green) of this same section is shown in the middle panel. Overlay of the GC1 and cone arrestin images (right panel) shows that arrestin staining is reduced in the outer segments of cone cells expressing GC1. (B) Retina of dark-adapted GC1 KO retina treated with pTR-MOP-GFP showing cone arrestin (red) and GFP (green) immunostaining. (C) Analyses of cone arrestin staining in a single representative cell shown in boxed region of B. Left: GFP (green) staining in cone cell indicating expression of the MOP-GFP transgene. Middle: Cone arrestin (red) staining of cells shown in left panel. Right: Overlay of GFP (green) and cone arrestin (red) staining. Staining intensity for cone arrestin within the OS and IS of these cells is shown to the right. These values represent percentage fluorescence over background. Scale bars (A, B): 50 μm.
DISCUSSION
The goal of this study was to examine the feasibility of restoring function to cone photoreceptors in postnatal retinas that develop in the absence of GC1. The results of this study show that the expression of GC1 in fully developed cone cells restores some function to these cells, as evidenced by light-dependent cone arrestin translocation. Examination of the treated retinas of GC1 KO mice showed that both the cell-specific MOP promoter and the ubiquitous smCBA promoter effectively drove GC1 expression in rod and cone photoreceptors cells. Approximately equal levels of transgene expression were observed in treated retinas up to 6 months after injection with either therapeutic vector. Using a similar strategy, we recently restored vision to the GUCY1*B chicken model of LCA1, an animal blind from hatching if left untreated.19 In that study, visual behavior and dark- and light-adapted ERG responses were present in animals that had been treated with lentiviral vectors carrying transgenes encoding bovine GC1. Dark- and light-adapted ERG responses recorded from the treated animals were similar in shape to those recorded from the WT animals but had lower amplitudes. Amplitudes of the dark-adapted b-waves recorded from the treated animals were approximately 9% of the WT response at the highest flash intensity tested. Visual behaviors of the treated animals, which were assessed using optokinetic and volitional visual tests, were nearly identical with WT behaviors.19 These results, which are consistent with clinical observations that show patients can have serviceable vision while exhibiting major ERG deficits,20–22 demonstrate that ERG responses, while informative, do not necessarily correlate with other measures of photoreceptor function. In the present study, we attempted to obtain electrophysiological evidence of cone function in the AAV-GC1–treated retinas but were unable to resolve cone signals from background noise. In mice, cones constitute approximately 3% of the total photoreceptor population and are distributed uniformly across the entire extent of the retina. It is unclear why the cone responses in the GC1 KO retina did not improve with treatment. The relatively low numbers of these cells in mouse retina and their distribution might have contributed to our inability to record signals from the transduced cone cells using a full-field recording paradigm.
Based on studies of the GUCY1*B chicken, which also carries a null mutation in GC1,23 the levels of cGMP in the cone photoreceptors of the GC1 KO mouse are likely to be significantly reduced compared with WT animals. If they are, abnormally low levels of cGMP in the cone cells of the retinas of these animals would slow the dark current and lead to cellular hyperpolarization independent of light state, thereby inducing the biochemical equivalent of chronic light exposure. The cone arrestin staining pattern observed in untreated GC1 KO cones is reminiscent of the pattern observed in cone cells after exposure to light.
In the absence of GC1, cone cells fail to translocate cone arrestin and are unable to transduce light for vision, suggesting a level of dependency of these processes on the synthesis of cGMP. Recent studies aimed at uncovering the signaling cascade that drives protein translocation in vertebrate rod photoreceptors suggest that the signaling cascades driving protein translocation and vision are both triggered by the activation of rhodopsin. In these studies, mice carrying null mutations in RPE65, which is the isomerohydrolase that synthesizes the chromophore required for the regeneration of rod visual pigment,24 display altered rod transducin and rod arrestin translocation.25 Attempts to identify downstream components of the cascade driving protein translocation have revealed that the extent of rod arrestin translocation into ROS is dependent on the “amount” of signaling through the phototransduction cascade and not on the number of photoactivated rhodopsin molecules.26 Analyses of the R9AP knockout mouse, which lacks the ability to turn off transducin signaling,27 revealed that the threshold light intensity required to induce rod arrestin movement was drastically reduced in these mice compared with WT mice. In other words, fewer photoactivated rhodopsin molecules were required to trigger the movement of rod arrestin to ROS when transducin was constitutively activated.26 Although some reports suggest that rod arrestin translocation is unaffected by the absence of transducin,25,28 another study reports that its absence results in less efficient arrestin movement.26 Proteins associated with the recovery steps of phototransduction, such as rhodopsin phosphorylation, do not appear to be required for transducin or arrestin translocation in rod photoreceptors.25,28 Significantly less is known about the signaling cascade that drives protein translocation in cone cells. As in rods, cone arrestin translocation is unaffected by the disruption of cone opsin phosphorylation.28
The current debate in the literature is whether the mechanism of protein translocation in rod and cone photoreceptors is driven by passive diffusion or by the action of molecular motors. In support of the former, it has been demonstrated that the redistribution of arrestin between the rod inner and outer segment can proceed in adenosine triphosphate (ATP)–depleted photoreceptors.29 The theory that the redistribution of rod arrestin in light occurs passively through the binding of arrestin to photoactivated rhodopsin in the ROS presupposes that the amounts of photoactivated rhodopsin and rod arrestin molecules in ROS are equal.29,30 A recent report revealed, however, that the amount of rod arrestin that translocates into ROS after light exposure exceeds the amount of photoactivated rhodopsin by approximately 10-fold.26 This finding suggests that the process of arrestin translocation may not be entirely dependent on passive diffusion. Additional support for this notion comes from the observation that arrestin translocation is slower than the rate of diffusion of soluble proteins between rod compartments.26 It has also been shown that arrestin is confined to unknown binding sites in dark-adapted rod inner segments31 and that in dark-adapted conditions, arrestin is primarily associated with the microtubule cytoskeleton.29,32,33 These distribution patterns, which are not characteristic of soluble protein, GFP,26 suggest that the passive diffusion model may be too simple and that additional light-responsive mechanisms are likely to contribute to arrestin translocation.
Although we have established that rAAV-mediated GC1 expression in cone cells of the GC1 KO mouse is sufficient to restore light-driven cone arrestin translocation in these cells, the link between GC1 and translocation remains to be experimentally determined. The absence of GC1 is predicted, based on studies of the GUCY1*B retina,23 to lead to abnormally low levels of cGMP in the cone cells. Low cGMP levels would be expected to lead to closure of cGMP-gated channels and a chronic reduction in cytoplasmic levels of free calcium. We suggest that disruption of the ability of cone cells to effectively regulate intracellular levels of calcium could be the link between the absence of GC1 and the disruption of arrestin translocation in these cells. Further research into the light transduction cascade that triggers translocation and the translocation mechanism itself will undoubtedly provide insight into the link between GC1 and light-driven arrestin translocation in cones.
Clinical Implications
The results of this study show that GC1 is required for arrestin translocation in cone cells, a biochemical measure of the functionality of these cells. A corollary of these findings is that GC1 expression is not essential for the proper structural development of cone cells. Our inability to detect photopic ERGs or to prevent cone cell degeneration using our rAAV-bGC1 treatment in this study could be the result of detrimental effects of GC1 overexpression on the transduced cells, a possibility suggested by the observations that cone-rod dystrophy can result from mutations in GC1 or GCAP1 that lead to constitutive activation of GC1.34,35 In developing potential therapies, it will be imperative to identify promoters that drive GC1 expression at levels that can be tolerated by photoreceptors. Because the carrying capacity of rAAV precludes using large, native promoters in conjunction with GC1, it would be interesting to investigate the usefulness of other viral vectors in conjunction with native, cone-specific promoters to drive GC1 expression. Although additional studies should be conducted, we believe that the results of this study and of our recent study showing that GC1 expression in photoreceptors of the GUCY1*B chicken model of LCA1 restores vision to these animals, as assessed by behavioral and electrophysiological measures,19 support further efforts to develop gene therapies for the treatment of LCA1 in humans.3,4
Acknowledgments
The authors thank Vince Chiodo for the packaging and purification of recombinant AAV vectors. They also thank Sergei Zolotukhin for the generous gift of the smCBA promoter and Xuemei Zhu (DEI and Keck School of Medicine of the University of Southern California) for her contribution to the generation of theLUMIj/mouse cone arrestin antibody. They thank Adrian Timmers, John Alexander, Kristofer Eccles, and Amy Robinson for their excellent technical assistance, and Mary D. Allen for her continued, generous support.
Footnotes
Disclosure: S.E. Haire, None; J. Pang, None; S.L. Boye, None; I. Sokal, None; C.M. Craft, None; K. Palczewski, None; W.W. Hauswirth, P; S.L. Semple-Rowland, P
Supported by NIH Grants EY11388 (SLS-R), EY15851 (CMC), and EY11123 (WWH), Core Grant EY08571, Mary D. Allen Endowment (CMC), Core Grant EY03040 (Doheny Eye Institute), Core Grant EY08061 (KP), and research funds from the University of Florida McKnight Brain Institute. CMC is the Mary D. Allen Chair in Vision Research (DEI).
References
- 1.Yang RB, Robinson SW, Xiong WH, Yau KW, Birch DG, Garbers DL. Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J Neurosci. 1999;19:5889–5897. doi: 10.1523/JNEUROSCI.19-14-05889.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Milam AH, Barakat MR, Gupta N, et al. Clinicopathologic effects of mutant GUCY2D in Leber congenital amaurosis. Ophthalmology. 2003;110:549–558. doi: 10.1016/S0161-6420(02)01757-8. [DOI] [PubMed] [Google Scholar]
- 3.Perrault I, Rozet JM, Calvas P, et al. Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat Genet. 1996;14:461–464. doi: 10.1038/ng1296-461. [DOI] [PubMed] [Google Scholar]
- 4.Perrault I, Rozet JM, Gerber S, et al. Spectrum of retGC1 mutations in Leber’s congenital amaurosis. Eur J Hum Genet. 2000;8:578–582. doi: 10.1038/sj.ejhg.5200503. [DOI] [PubMed] [Google Scholar]
- 5.Coleman JE, Semple Rowland SL. GC1 deletion prevents light-dependent arrestin translocation in mouse cone photoreceptor cells. Invest Ophthalmol Vis Sci. 2005;46:12–16. doi: 10.1167/iovs.04-0691. [DOI] [PubMed] [Google Scholar]
- 6.Shyjan AW, de Sauvage FJ, Gillett NA, Goeddel DV, Lowe DG. Molecular cloning of a retina-specific membrane guanylyl cyclase. Neuron. 1992:727–737. doi: 10.1016/0896-6273(92)90035-c. [DOI] [PubMed] [Google Scholar]
- 7.Lowe DG, Dizhoor AM, Liu K, et al. Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc Natl Acad Sci USA. 1995;92:5535–5539. doi: 10.1073/pnas.92.12.5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang RB, Foster DC, Garbers DL, Fulle HJ. Two membrane forms of guanylyl cyclase found in the eye. Proc Natl Acad Sci USA. 1995;92:602–606. doi: 10.1073/pnas.92.2.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang RB, Garbers DL. Two eye guanylyl cyclases are expressed in the same photoreceptor cells and form homomers in preference to heteromers. J Biol Chem. 1997;272:13738–13742. doi: 10.1074/jbc.272.21.13738. [DOI] [PubMed] [Google Scholar]
- 10.Coleman JE, Zhang Y, Brown GTAJ, Semple-Rowland SL. Cone cell survival and downregulation of GCAP1 protein in the retinas of GC1 knockout mice. Invest Ophthalmol Vis Sci. 2004;45:3397–3403. doi: 10.1167/iovs.04-0392. [DOI] [PubMed] [Google Scholar]
- 11.Yang GS, Schmidt M, Yan Z, et al. Virus-mediated transduction of murine retina with adeno-associated virus: effects of viral capsid and genome size. J Virol. 2002;76:7651–7660. doi: 10.1128/JVI.76.15.7651-7660.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flannery JG, Zolotukhin S, Vaquero MI, LaVail MM, Muzyczka N, Hauswirth WW. Efficient photoreceptor-targeted gene expression in vivo by recombinant adeno-associated virus. Proc Natl Acad Sci USA. 1997;94:6916–6921. doi: 10.1073/pnas.94.13.6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawicki JA, Morris RJ, Monks B, Sakai K, Miyazaki J. A composite CMV-IE enhancer/beta-actin promoter is ubiquitously expressed in mouse cutaneous epithelium. Exp Cell Res. 1998;244:367–369. doi: 10.1006/excr.1998.4175. [DOI] [PubMed] [Google Scholar]
- 14.Goraczniak RM, Duda T, Sitaramayya A, Sharma RK. Structural and functional characterization of the rod outer segment membrane guanylate cyclase. Biochem J. 1994;302:455–461. doi: 10.1042/bj3020455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zolotukhin S, Potter M, Zolotukhin I, et al. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 16.Timmers AM, Zhang H, Squitieri A, Gonzalez-Pola C. Subretinal injections in rodent eyes: effects on electrophysiology and histology of rat retina. Mol Vis. 2001;7:131–137. [PubMed] [Google Scholar]
- 17.Campbell AM. Laboratory Techniques in Biochemistry and Molecular Biology. New York: Elsevier; 1984. Monoclonal antibody technology. [Google Scholar]
- 18.Zhu X, Li A, Brown B, Weiss ER, Osawa S, Craft CM. Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Mol Vis. 2002;8:462–447. [PubMed] [Google Scholar]
- 19.Williams ML, Coleman JE, Haire SE, et al. Lentiviral expression of retinal guanylate cyclase-1 (RetGC1) restores vision in an avian model of childhood blindness. PLoS Med. 2006;3:e201. doi: 10.1371/journal.pmed.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carroll J, Neitz M, Hofer H, Neitz J, Williams DR. Functional photoreceptor loss revealed with adaptive optics: an alternate cause of color blindness. Proc Natl Acad Sci USA. 2004;101:8461–8466. doi: 10.1073/pnas.0401440101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geller AM, Sieving PA. Assessment of foveal cone photoreceptors in Stargardt’s macular dystrophy using a small dot detection task. Vision Res. 1993;33:1509–1524. doi: 10.1016/0042-6989(93)90144-l. [DOI] [PubMed] [Google Scholar]
- 22.Seiple W, Holopigian K, Szlyk JP, Greenstein VC. The effects of random element loss on letter identification—implications for visual-acuity loss in patients with retinitis-pigmentosa. Vision Res. 1995;35:2057–2066. doi: 10.1016/0042-6989(94)00289-x. [DOI] [PubMed] [Google Scholar]
- 23.Semple-Rowland SL, Lee NR, Van Hooser JP, Palczewski K, Baehr WA. A null mutation in the photoreceptor guanylate cyclase gene causes the retinal degeneration chicken phenotype. Proc Natl Acad Sci USA. 1998;95:1271–1276. doi: 10.1073/pnas.95.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci USA. 2005;102:12413–12418. doi: 10.1073/pnas.0503460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendez A, Lem J, Simon M, Chen J. Light-dependent translocation of arrestin in the absence of rhodopsin phosphorylation and transducin signaling. J Neurosci. 2003;23:3124–3129. doi: 10.1523/JNEUROSCI.23-08-03124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keresztes G, Martemyanov KA, Krispel CM, et al. Absence of the RGS9.Gβ5 GTPase-activating complex in photoreceptors of the R9AP knockout mouse. J Biol Chem. 2004;279:1581–1584. doi: 10.1074/jbc.C300456200. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Huang W, Zhang H, et al. Light-dependent redistribution of visual arrestins and transducin subunits in mice with defective phototransduction. Mol Vis. 2003;9:231–237. [PubMed] [Google Scholar]
- 29.Nair KS, Hanson SM, Mendez A, et al. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mangini NJ, Garner GL, Okajima TI, Donoso LA, Pepperberg DR. Effect of hydroxylamine on the subcellular distribution of arrestin (S-antigen) in rod photoreceptors. Vis Neurosci. 1994;11:561–568. doi: 10.1017/s0952523800002467. [DOI] [PubMed] [Google Scholar]
- 31.Peet JA, Bragin A, Calvert PD, et al. Quantification of the cytoplasmic spaces of living cells with EGFP reveals arrestin-EGFP to be in disequilibrium in dark adapted rod photoreceptors. J Cell Sci. 2004;117:3049–3059. doi: 10.1242/jcs.01167. [DOI] [PubMed] [Google Scholar]
- 32.Peterson JJ, Tam BM, Moritz OL, et al. Arrestin migrates in photo-receptors in response to light: a study of arrestin localization using an arrestin-GFP fusion protein in transgenic frogs. Exp Eye Res. 2003;76:553–563. doi: 10.1016/s0014-4835(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 33.Peterson JJ, Orisme W, Fellows J, et al. A role for cytoskeletal elements in the light-driven translocation of proteins in rod photoreceptors. Invest Ophthalmol Vis Sci. 2005;46:3988–3998. doi: 10.1167/iovs.05-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tucker CL, Woodcock SC, Kelsell RE, Ramamurthy V, Hunt DM, Hurley JB. Biochemical analysis of a dimerization domain mutation in RetGC-1 associated with dominant cone-rod dystrophy. Proc Natl Acad Sci USA. 1999;96:9039–9044. doi: 10.1073/pnas.96.16.9039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Payne AM, Downes SM, Bessant DA, et al. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet. 1998;7:273–277. doi: 10.1093/hmg/7.2.273. [DOI] [PubMed] [Google Scholar]