

Abstract
Lymphopenia and restricted T cell repertoires in humans are often associated with severe eosinophilic disease and a T cell Th2 bias. To examine the pathogenesis of this phenomenon, C57BL/6 Rag2−/− mice received limited (3 × 104) or large (2 × 106) numbers of CD4 T cells. Three to 5 months after transfer, mice that had received 3 × 104 T cells, but not those that received 2 × 106, developed fulminant macrophage pneumonia with eosinophilia, Ym1 deposition, and methacholine-induced airway hyperresponsiveness, as well as eosinophilic gastritis; esophagitis and other organ damage occurred in some cases. Donor cells were enriched for IL-4, IL-5, and IL-13 producers. When 3 × 104 cells were transferred into CD3ε−/− hosts, the mice developed strikingly elevated serum IgE. Prior transfer of 3 × 105 CD25+ CD4 T cells into Rag2−/− recipients prevented disease upon subsequent transfer of CD25− CD4 T cells, whereas 3 × 104 regulatory T cells (Tregs) did not, despite the fact that there were equal total numbers of Tregs in the host at the time of transfer of CD25− CD4 T cells. Limited repertoire complexity of Tregs may lead to a failure to control induction of immunopathologic responses, and limitation in repertoire complexity of conventional cells may be responsible for the Th2 phenotype.
Keywords: eosinophils, IgE, IL-4, macrophages, pneumonia
In primary human immunodeficiencies in which limited numbers of T cells are delivered to the periphery, a common phenotype is eosinophilia, occasionally markedly elevated levels of serum IgE, and lymphocytic infiltration of parenchymal tissues. This phenotype is seen in Omenn's syndrome, maternal engraftment in SCID, and atypical complete DiGeorge syndrome (1). Omenn's syndrome is perhaps the most well studied of these diseases. It is a severe combined immunodeficiency commonly caused by mutations in Rag 1 or Rag 2 that severely reduce, but do not eliminate, the recombinase's function (2). In patients with Omenn's syndrome the periphery is populated by oligoclonal T cell populations heavily weighted toward expression of the Th2 phenotype, despite the fact that there is no intrinsic defect in the peripheral T cells themselves. The limited T cell repertoire seen in Omenn's syndrome is also a feature of the other immunodeficiencies associated with erythroderma, hypereosinophilia and elevated serum IgE. In advanced HIV infection, the eosinophilia that often develops is associated with, and may be caused by, the limited T cell repertoire (3). Cutaneous T cell lymphoma (mycosis fungoides) has also been reported to be associated with a limited peripheral T cell receptor (TCR) repertoire, a Th2 phenotype, and peripheral eosinophilia (4, 5).
Examples of lymphopenia associated with a Th2 phenotype, hypereosinophilia, and/or elevated IgE have also been observed in mice. This phenotype is seen in lat mutants (6, 7). It has recently been reported that mice with limited numbers of CD4 T cells, such as MHC class II−/− mice and nu/nu mice, have elevated serum IgE (8). There are no mouse models, however, of lymphopenia in the context of normal thymic and peripheral development.
Upon transfer into lymphopenic hosts, T cells undergo a process termed homeostatic or lymphopenia-induced proliferation. This proliferation is thought to be driven by cytokines as well as TCR engagement. It is unclear whether the peptides recognized by the proliferating T cells are derived from self-proteins, from gut flora, or from foreign antigens (9).
We have reported that a reduced TCR repertoire with normal numbers of memory phenotype CD4 cells can be achieved by transferring small numbers of CD4+ T cells into lymphopenic recipients (10). The question arises as to whether this state of reduced repertoire could have deleterious effects for the recipient organism, much like the profound phenotype seen in the immunodeficient conditions mentioned above. Here we show that these mice develop a severe, multiorgan eosinophilic disease, strikingly elevated levels of serum IgE (when B cells are present), and a memory population of Th2-phenotype CD4 T cells. An important element in the development of this disease appears to be the limited repertoire of regulatory T cells (Tregs).
Results
Rag2−/− Mice Receiving a Small Number of CD4 T Cells Develop Severe Multiorgan Inflammatory Disease.
Transfer of CD4 T cells from C57BL/6 donors into syngeneic Rag2−/− recipients leads to rapid proliferation of a portion of the transferred cells. At 2 months after transfer, the number of CD44hi CD4 cells in the lymph nodes of the recipients is ≈1 × 106, independent of the number of cells transferred, over a range from 104 to 107 (10).
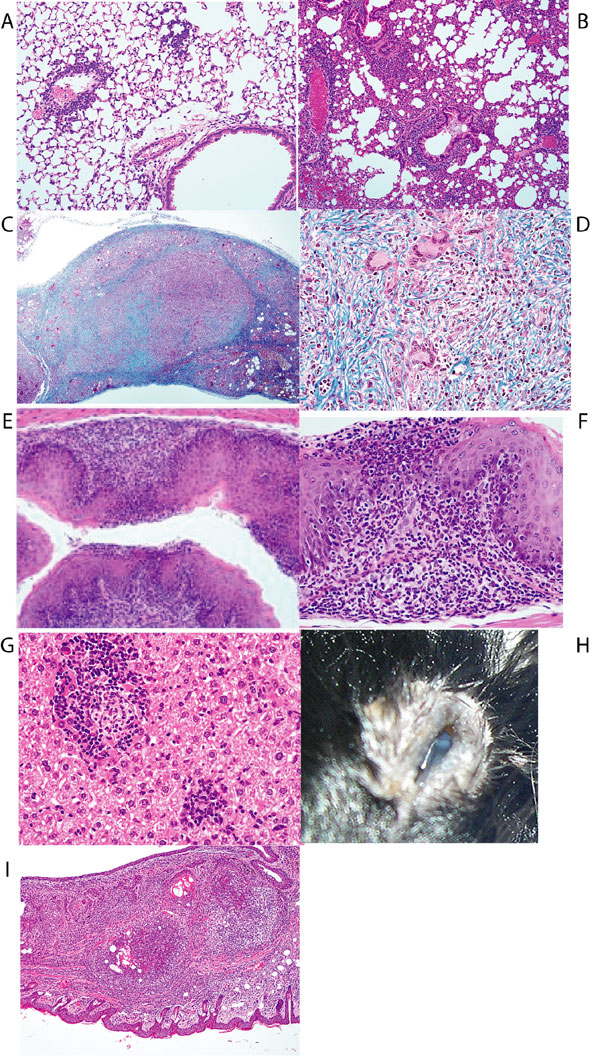
Although the number of CD44hi CD4 T cells present 6 weeks after transfer was independent of the number of transferred cells, the recipients of large and small numbers of cells showed a striking difference in their subsequent development of an eosinophilic inflammatory disease. C57BL/6 Rag2−/− mice received either 3 × 104 or 2 × 106 CD4 lymph node T cells from C57BL/6 donors. Three to 6 months after transfer, mice that had received 3 × 104, but not mice that had received 2 × 106, CD4 T cells had severe macrophage pneumonia with eosinophilic and lymphocytic infiltrates, mucus metaplasia of airway epithelium, and eosinophilic crystal formation, both within pulmonary macrophages and in the extracellular space (Figs. 1 A–C and A). The crystals were found to be Ym1-positive (Fig. 1 D and E). Ym1 is an eosinophilic and potentially eosinophilotactic crystal produced by “alternatively activated” macrophages (11). These mice also displayed methacholine airway hypersensitivity (Fig. 3A).
Fig. 1.

Pathology at 4 months after transfer of 3 × 104 CD4 T cells into Rag2−/−mice. (A) Gross pathology of lungs from normal Rag2−/−mice and mice that had received 3 × 104 CD4 T cells. (B and C) H&E stain (B) and Luna stain (C) for eosinophils (red arrow) and eosinophilic crystal-laden macrophages (black arrow). (D and E) Immunohistochemical anti-Ym1 stain of lungs from mice that had received 3 × 104 CD4 T cells (D) and of lungs from normal mice (E). (F) H&E stain of junction of forestomach and glandular stomach from mice that had received 3 × 104 CD4 T cells showing eosinophilic and lymphocytic infiltrate with parietal cell loss.
Fig. 2.

Representative H&E sections of whole lung (A) and junction of glandular stomach and forestomach (B) from Rag2−/−mice 4 months after transfer of 30,000 CD25− CD44lo CD4 T cells, 30,000 CD4 T cells, 2 million CD25− CD44lo CD4 T cells, or 2 million CD4 T cells and from an age-matched Rag2−/− control that had not received cells.
Fig. 3.

Th2 phenotype predominates in diseased mice. (A) Methacholine hypersensitivity. Mice were placed individually in a whole-body plethysmograph (Buxco Electronics), and Penh values were calculated. There were three to five mice per group. Doses of methacholine >12 mg/ml resulted in death for a number of the mice in the groups receiving 30,000 T cells. (B) Peripheral lymph node cells from Rag2−/− mice that had received 3 × 104 or 2 × 106 CD4 T cells 4 months earlier were stimulated with PMA and ionomycin for 6 h; monensin was added for the last 2 h. Cells were stained for CD45.1 to identify transferred cells. Cells were then fixed, permeabilized, and stained for intracellular cytokines. Each group consisted of four to five mice; shown is a representative experiment of three similar experiments. (C) Elevated serum IgE. B10.A CD3ε−/−mice intravenously received the cells indicated and were bled at the times indicated, and serum IgE was measured by ELISA. There were four mice in each group; this experiment was repeated once with similar results. Standard error bars are shown.
A marked eosinophilic gastritis with inflammation of the glandular stomach and forestomach and with complete parietal cell loss with some vacuolization was also present in mice that received 3 × 104, but not 2 × 106, CD4 T cells (Figs. 1F and 2B). Some recipients of 3 × 104 CD4 T cells also developed eosinophilic esophagitis and variable small and large bowel lesions, not as consistently characterized by eosinophil infiltration. Other lesions commonly found in these mice were myeloid hyperplasia and chronic inflammation, with occasional eosinophilic infiltration of the liver, and mesenteric lymph node granulomas with fibrosis and giant cells [supporting information (SI) Fig. 6]. Sporadic lymphomas, sarcomatous change of mesenteric granulomata, conjunctivitis with eosinophils (SI Fig. 6), and spinal neuritis were also noted.
Even at 1 month after transfer of 3 × 104 CD4 T cells, sparse areas of perivascular lymphocytic infiltration in the lungs and eosinophilic and lymphocytic inflammation in the stomach were noted (SI Fig. 6). At 2 months after transfer of 3 × 104 CD4 T cells, more pronounced lesions with macrophage infiltration in the lungs and mucus metaplasia of bronchiolar endothelium were found, in some instances encompassing entire lung lobes. In the stomachs of these “2-month” mice there was more pronounced eosinophilic and lymphocytic inflammation, particularly of the glandular stomach. The inflammatory response was similar, if not more severe, in mice that had received 3 × 104 CD25− “naïve” (CD44lo) CD4 T cells. More variably, transfer of 2 × 106 CD25− naïve CD4 T cells also induced this inflammatory disease (Table 1 and Fig. 2). CD4 T cells derived from AND TCR transgenic mice on a Rag2−/− background, whose T cells are specific for a pigeon cytochrome c peptide, did not induce disease when either 3 × 104 or 2 × 106 cells were transferred, although these AND cells underwent vigorous proliferation in the lymphopenic host. This finding indicates that not all proliferating T cells with limited receptor diversity (here, monoclonal) can induce disease (Table 1).
Table 1.
Summary of disease induced by transfer of CD4 or CD25− CD44lo CD4 T cells into Rag2−/− mice
| Cells | Macrophage/eosinophil pneumonia | Eosinophilic gastritis | Eosinophilic esophagitis | Liver inflammation | Colitis | Mesenteric lymph node granuloma |
|---|---|---|---|---|---|---|
| 30,000 CD25− CD44lo cells | 13/13 | 13/13 | 5/8 | 8/8 | 4/13 | 7/13 |
| 30,000 CD4 cells | 7/7 | 4/7 | 1/7 | 5/7 | 5/7 | 3/7 |
| 2 million CD25− CD44lo cells | 3/6 | 4/6 | 3/6 | 2/6 | 0/6 | 0/6 |
| 2 million CD4 cells | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 |
| AND 30,000 CD4 cells | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 |
| AND 2 million CD4 cells | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 |
Shown are results from three or more separate experiments for each group, with pathology determined at least 4 months after transfer.
Disease Induction in Rag2−/− Recipients Appears to Be Related to Th2 Phenotype and Is Associated with Elevated Serum IgE in CD3ε−/− Recipients.
A larger proportion of the CD44hi CD4 T cells isolated from peripheral lymph nodes of mice that had received 3 × 104 cells produced IL-4, IL-5, and IL-13 upon ex vivo stimulation than those that had received 2 × 106 cells. The frequency of IFNγ-producing cells was similar in mice that had received small or large numbers of cells (Fig. 3B).
When limited numbers of CD4 T cells were transferred into CD3ε−/− recipients, which lack T cells but have B cells, striking elevation in serum IgE levels was noted. Thirty thousand total CD4 T cells or naïve CD25− CD4 T cells or 2 × 106 total CD4 T cells or naïve CD25− CD4 T cells were transferred into CD3ε−/− recipients. By 1 week after transfer, mice that had received the larger numbers of T cells displayed detectable serum IgE. At 4 weeks after transfer, all mice had significant amounts of serum IgE (1–10 μg/ml). However, at 8 weeks and 12 weeks after transfer, mice that had received 3 × 104 CD4 T cells displayed massive increases in serum IgE, with a mean concentration of ≈350 μg/ml in mice that had received 3 × 104 naïve CD25− CD4 T cells 12 weeks earlier (Fig. 3C). The induction of IgE depended on IL-4 produced by the transferred cells, because Il4−/− donor CD4+ T cells were unable to elicit detectable IgE in the serum of CD3ε−/−hosts up to 8 weeks after transfer (data not shown). As noted above, recipients of 3 × 104 CD4 T cells displayed airway hypersensitivity as did recipients of 3 × 104 and 2 × 106 naïve CD25− CD4 T cells. Mice that received 2 × 106 CD4 T cells did not display airway hypersensitivity (Fig. 3A).
Failure of Lymphocytes from Affected Mice to Transfer Accelerated Disease Onset to Naïve Recipients.
To determine whether lymph node T cells from mice that had severe macrophage pneumonia would cause an accelerated onset of disease when transferred to Rag2−/−recipients, 2 × 105 lymph node T cells from such mice were transferred to Rag2−/− hosts. In two experiments involving 10 recipients, no disease was noted up to 1.5 months after the transfer, implying that these cells did not cause an accelerated onset of disease.
Disease Is Associated with Autoantibody Formation.
To determine whether CD3ε−/− recipients of small numbers of CD4 T cells developed antiparietal cell antibodies, we incubated normal stomach sections with serum from such recipients and used FITC anti-IgG to detect autoantibodies. Mice that had received 2 × 106 total or CD25− naïve CD4 T cells had no detectable antiparietal cell antibodies (0/4 and 0/3, respectively) whereas almost all of those receiving either 3 × 104 total or CD25− naïve CD4 T cells were strongly positive for such antibodies (3/4 and 4/4, respectively) (Fig. 4).
Fig. 4.

Antiparietal cell antibodies in mice receiving 30,000 CD4 T cells. Fluorescence microscopy to detect antiparietal cell antibodies in serum from CD3ε−/−recipient mice 12 weeks after transfer of 30,000 (Upper Left) or 2 million (Upper Right) sorted CD44lo CD25− CD4 T cells. Mouse serum was incubated on normal mouse stomach sections, and a FITC-labeled F(ab′)2 goat anti-mouse IgG antibody was used for detection. Results from the staining experiment are summarized in Lower.
CD25+ Tregs Play a Role in Controlling Lymphopenia-Associated Disease.
When limited numbers of CD4 T cells are transferred into lymphopenic recipients, the memory cells present 1 month after transfer have very limited TCR repertoire diversity (10). Because the relative expansion of CD25+ and CD25− cells in these mice is similar (10), it is likely that the TCR repertoires of both the conventional and regulatory T cells in mice that received small numbers of CD4 T cells are of limited complexity. We asked whether pretransfer of CD25+ cells could protect mice against the induction of the eosinophilic inflammatory disease induced by CD25− naïve CD4 T cells and whether the number of initially transferred CD25+ cells would determine whether there was protection.
A total of 3 × 104 or 3 × 105 CD25+ CD4 T cells were injected into Rag2−/− mice. At 2 months, mice were killed and the total number of CD25+ T cells in the lymph nodes and spleen were similar (Fig. 5A), consistent with a previous report (12). We then introduced 2 × 106 CFSE-labeled CD25− naïve CD4 T cells into other mice that had initially received similar transfers of CD25+ T cells. Although the number of CD25+ cells at the time of the secondary transfer was similar in mice that had initially received 3 × 104 or 3 × 105 CD25+ cells, only in mice that had received the larger number of CD25+ cells was the rapid division of the newly introduced, CD25− naïve CD4 T cells prevented (Fig. 5B).
Fig. 5.

Transfer of large but not small numbers of Tregs controls disease. CD45.2 Rag2−/−mice received either 3 × 104 or 3 × 105 sorted CD25+ CD45.1 CD4 T cells intravenously. (A) Lymph nodes and spleens were harvested, and transferred CD45.1 cells were counted 12 weeks later. (B) Ten weeks after primary CD45.2, CD25+ CD4 T cell transfer, 1 million CFSE-labeled CD45.1 CD25− CD4 T cells were transferred, and lymph nodes were harvested 1 week later. Representative CFSE profiles of transferred CD45.1 T cells are shown. (C) Ten weeks after primary CD45.2+ Treg transfer, mice received a second transfer of 1 × 105 CD25− CD45.1+ CD4 T cells. Cells were harvested 10 weeks later. CD45.1+ and CD45.2+ CD4 T cells were enumerated, and pathology of mice initially receiving 3 × 104 Tregs (D and F) or 3 × 105 Tregs (E and G) was examined. Cell yields and representative pathology are shown. There were two to three mice in each group. This experiment was repeated once with similar results. Standard error bars are shown.
To test whether the pretransfer of Tregs would regulate disease induction, 3 × 104 or 3 × 105 sorted, CD25+ CD4 T cells were injected into Rag2−/− mice. Two months later 2 × 105 CD25− CD4 T cells were transferred into these mice, and the animals were killed a further 2 months later. The number of CD25+ T cells was similar at the time of death, but there were five times as many CD25− CD4 T cells in the mice that had initially received 3 × 104 CD25+ CD4 T cells as in those that received an initial transfer of 3 × 105 CD25+ CD4 T cells (Fig. 5C). Mice that had initially received 3 × 104 CD25+ CD4 T cells developed severe eosinophilic lung and stomach disease, whereas the mice that had initially received 3 × 105 CD25+ CD4 T cells developed far fewer lesions (Fig. 5 D–G).
Discussion
We describe here a severe multiorgan eosinophilic disease that develops in the context of lymphopenia with reduced T cell repertoire. This disease appears to be, in part, due to the lack of a diverse TCR repertoire among the Foxp3+ Tregs. It can be argued that limitation in TCR diversity in the Treg population would make these cells less efficient in inhibiting the action of conventional (effector) T cells that mediate the immunopathology observed in this transfer system. The implication is not necessarily that the conventional and regulatory T cells populations must have matching repertoires for normal control of autoimmunity/immunopathology but rather that some level of intersection of repertoires is needed, if only to ensure that potentially pathogenic T cells and regulatory T cells are activated in the same place, at the same time, so that the regulatory cells could have the opportunity to control the potential effectors.
Although the importance of regulatory T cells in the control of the immunopathology observed when small numbers of CD4 T cells are transferred into lymphopenic recipients seems clear, the reason for the Th2 diathesis in the resulting immunopathology is not obvious. It is possible that the absence of effective regulatory cells by itself is sufficient to induce a Th2-like immunopathology, independent of the number of “effector” cells transferred. Foxp3−/− mice demonstrate severe Th2 disease with markedly elevated serum Th2 (and Th1) cytokines as well as IgE (13). The fact that disease is more sporadic and serum IgE levels are not as high in mice that received 2 × 106 sorted naïve CD25− CD4 effector T cells than in mice that received 3 × 104 sorted cells could possibly be accounted for not by an inherent Th2 predisposition in mice receiving lower cell numbers but rather by the transfer of more contaminating Tregs with the larger number of sorted cells. There is a population of Foxp3+, CD25− CD4 T cells (≈10% of Foxp3+ cells) (14). Thus, substantially more regulatory cells (≈1 × 104, assuming that Foxp3+ cells are ≈5% of CD44dull CD4 T cells) (B. Min, personal communication) would be transferred with the larger number of sorted cells than with the smaller number, where the number of Foxp3+ cells may be anticipated to have been ≈150. The latter may be too small a frequency to inhibit a Th2 response even after homeostatic expansion whereas the former may have been partially active. Furthermore, lymphopenia-driven expansion has been associated with conversion of CD25− Foxp3− to the Foxp3+ phenotype (15), suggesting that the diversity of TCRs in the regulatory population may be increased by the presence of a complex TCR repertoire among conventional cells undergoing rapid proliferation. In a mouse immunization model, elevated serum IgE and allergy have been noted when only monoclonal T and B cell populations specific for the antigen are present, but the phenotype is markedly inhibited when regulatory T cells are present (16). Although the absence or reduced repertoire of natural or peripherally converted regulatory T cells alone may explain the Th2 disease, it is still possible that an effector population of limited TCR diversity may intrinsically differentiate to the Th2 phenotype much more efficiently than a similar population of greater TCR diversity.
There are a number of possible explanations to account for Th2 pathology as due to an intrinsic property of effector cells transferred at lower frequencies. It may well be that the larger number of divisions that would be required in mice receiving fewer CD4 T cells may in some way predispose the cells to Th2 differentiation, perhaps in part because of a greater propensity of differentiating Th1 cells to undergo apoptosis (17). Alternatively, TCR affinity for antigen may determine that a Th2-like differentiation is dominant when there is a limited TCR diversity. Thus, on the average any newly formed pMHC complex is less likely to find a complementary high-affinity TCR when diversity is low, and thus most interactions in mice that receive small numbers of CD4 T cells are likely to be of low affinity. It has been shown in several systems that priming with peptides of low affinity for the receptor of the responding T cell (i.e., altered peptide ligands) (18) or with low concentrations of peptides (19, 20) favors Th2 differentiation. If such low-affinity responses are normally “outcompeted” when high-affinity interactions also occur, then one could anticipate that, in a population of limited diversity, on the average, Th2 differentiation of activated CD4 T cells would be favored. A related possibility is raised by a recent report that Tregs exert preferential control of CD5low effectors (21). Such cells presumably have lower affinity for self; thus, in the absence of Tregs their “self” response would be relatively favored, and, if they tended to develop into Th2 cells, then a preferential self-specific Th2 response in the absence of Tregs would be expected, which might be even more marked with a limited repertoire diversity.
Many of the lymphopenic C57BL/6 mice receiving CD25+ Treg-depleted cell populations did not develop colitis, which would have been expected with such transfers (22). Differences in gut microflora could potentially explain this. Interestingly, in a different National Institutes of Health animal facility, transfer of 5 × 105 CD45RBhi CD4 cells into C57BL/6 Rag2−/− recipients resulted in colitis only 20% of the time, but in C57BL/10 Rag2−/− recipients such transfer resulted in colitis 80% of the time. Furthermore, an eosinophilic, Ym1+ pneumonia was seen 20% of time in C57BL/10 recipients but not in C57BL/6, Rag2−/− recipients (B. Kelsall, personal communication).
Although we were able to detect specific antiparietal cell autoantibodies in CD3ε−/− recipient mice with eosinophilic immunopathology, we were not able to rapidly induce the disease by transferring cells from the draining nodes of affected mice to healthy Rag2−/− mice. This finding suggests that the immunopathologic response may be due to “bystander” effector functions as opposed to cognate TCR-based recognition of “autoantigens.” This is not surprising given the uniform induction of disease despite the very low numbers, and hence the limited TCR diversity, of cells transferred. Bystander, noncognate antigen-driven hyperIgE production in nu/nu or MHC class II−/− mice has recently been reported and has been attributed to IL-4 production from the few remaining CD4+ T cells in these immunodeficient mice (8). Furthermore, the simple introduction of IL-13 or IL-4 into airways can induce airway hypersensitivity and mucus metaplasia (23), implying that local cytokine production in a “sensitive” environment in and of itself can induce immunopathology.
While not mimicking every phenotype of disease associated with primary human immune deficiencies associated with markedly reduced T cell repertoires, we have created a model for lymphopenia and Th2 disease. This model may also be useful in studying other Th2-associated immunopathologic states such as allergy and “extrinsic” asthma. Antigen encounters during periods of limited TCR repertoire may predispose CD4 T cells toward Th2 differentiation, due to the lack of TCR specificity-matching between regulatory and effector T cells, an effector T cell intrinsic predisposition toward Th2 phenotype when TCR repertoires are reduced, or both. Future study of such antigen-specific encounters in the context of lymphopenia are needed to elucidate the mechanism by which the Th2 phenotype emerges.
Materials and Methods
Mice.
B10.A, Ly5.1 B10.A, C57BL/10, C57BL/10 AND TCR Tg, C57BL/10 Rag2−/−, B10.A Rag 2−/−, B10.A CD3ε−/−, Ly5.1 C57BL/6, C57BL/6 Rag2−/−, and C57BL/6 IL-4−/− mice were obtained from the National Institute of Allergy and Infectious Diseases contract facility at Taconic Farms (Germantown, NY). C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were maintained under pathogen-free conditions at the National Institute of Allergy and Infectious Diseases animal facility.
Adoptive Transfer.
CD4, CD25+ CD4, CD25− CD4, or CD25−/CD44dull CD4 lymph node cells were obtained by sorting on a FACSVantage SE or FACSAria (Becton Dickinson, Franklin Lakes, NJ). Purity was >99%. In some cases, cells were labeled with CFSE (Molecular Probes, Carlsbad, CA) at a final concentration of 1.25 μM. Cells suspended in PBS were transferred via tail vein injection into recipient mice.
Flow Cytometry.
Anti-CD25-allophycocyanin (APC) (PC61), CD4-FITC (3T4), CD44-phycoerythrin (PE), CD44 PE-cy5.5, CD45.1-PE, CD45.2-FITC, CD45.1 PE-cy5, IL-4-PE, IL-5-PE, and IFNγ-APC were purchased from BD Pharmingen (San Diego, CA). Anti-FoxP3 was purchased from eBiosciences (San Diego, CA). Anti-IL-13 (clone 38213) was purchased from R & D Systems (Minneapolis, MN) and conjugated to APC at the National Institute of Allergy and Infectious Diseases core custom antibody facility. All flow cytometry was performed on a Becton Dickinson FACSCalibur and analyzed by using FloJo software.
Pathology/Immunohistochemistry.
Immediately after killing mice in a CO2 chamber, organs were removed, fixed in neutral buffered formalin, and embedded in paraffin. Sections were stained with hematoxylin and eosin. Selected tissues were stained with Masson's trichrome (for collagen) or Luna stain (for eosinophils). Immunohistochemistry was performed on some tissues with antibodies to Ym-1 (24) by the ABC method (Vector Laboratories, Burlingame, CA). Antiparietal cell antibodies were detected by immunofluorescence on cryostat sections of normal BALB/c stomach as described (25). Briefly, sections were blocked with 2% FBS in 5% dry milk in PBS and incubated with a 1/50 dilution of serum for 1 h at room temperature. The presence of autoantibodies was visualized by adding FITC-goat F(ab′)2 anti-mouse Ig (BioSource, Camarillo, CA). Slides were examined under a fluorescence microscope and given a score of 0–4 depending on the extent of parietal cell staining by an observer who did not have knowledge of the treatment the mice had received.
Methacholine Hypersensitivity.
Airway hyperresponsiveness of control Rag2−/− mice and recipients of cell transfers was measured by challenging the mice with increasing doses of nebulized methylcholine (0, 6, and 12 mg/ml) in PBS and measuring “enhanced pause” by using whole body plethysmography (Buxco Electronics, Wilmington, NC) following the manufacturer's instructions. Doses above 12 mg/ml resulted in significant morbidity in compromised mice consistent with asphyxia.
IgE ELISA.
Immulon 4 96-well microtiter plates were coated with 100 μl of anti-mouse IgE (Pharmingen) per well at 2 μg/ml in PBS overnight at 4°C. Plates were washed with wash buffer (PBS plus 0.05% Tween 20) and blocked for 1 h at room temperature with diluent/blocking buffer (PBS plus 0.5% BSA plus 0.05% Tween 20). Samples [1/50 or 1/500 dilutions of mouse serum or mouse IgE standard (Pharmingen)] were added and incubated for 2 h at room temperature. Plates were washed, and biotin-anti mouse IgE (Pharmingen) was added in diluent/blocking buffer for 2 h at room temperature. Plates were washed again, and streptavidin-HRP (Pharmingen) (1/4,000 dilution in diluents/blocking buffer) was added for 1 h. Plates were washed again, TMB solution (Sigma, St. Louis, MO) was added for 2–3 min, and Stop Solution (2N H2SO4) was then added. Plates were read at 450 nm, and the optical densities of the samples were then plotted on the linear part of the standard curve and multiplied by the dilution factor to calculate IgE concentration in serum samples.
Supplementary Material
Acknowledgments
The excellent histotechnology assistance of Larry Faucette and Cindy Erexson is greatly appreciated. We thank Drs. Brian Kelsall, Warren Strober, and Hidehiro Yamane for critical readings of the manuscript. J.D.M. is a fellow of the Pediatric Scientist Development Program (K12 HD00850). This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases and, in part, by a National Institute of Allergy and Infectious Diseases contract to SoBran.
Abbreviations
- Treg
regulatory T cell
- TCR
T cell receptor
- PE
phycoerythrin.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0610289104/DC1.
References
- 1.Markert ML, Alexieff MJ, Li J, Sarzotti M, Ozaki DA, Devlin BH, Sempowski GD, Rhein ME, Szabolcs P, Hale LP, et al. J Allergy Clin Immunol. 2000;113:734–741. doi: 10.1016/j.jaci.2004.01.766. [DOI] [PubMed] [Google Scholar]
- 2.Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, Gatta LB, Ochs HD, Schwarz K, Notarangelo LD, et al. Cell. 1998;93:885–896. doi: 10.1016/s0092-8674(00)81448-8. [DOI] [PubMed] [Google Scholar]
- 3.Tietz A, Sponagel L, Erb P, Bucher H, Battegay M, Zimmerli W. Eur J Clin Microbiol Infect Dis. 1997;16:675–677. doi: 10.1007/BF01708558. [DOI] [PubMed] [Google Scholar]
- 4.Yamanaka K, Yawalkar N, Jones DA, Hurwitz D, Ferenczi K, Eapen S, Kupper TS. Clin Cancer Res. 2005;11:5748–5755. doi: 10.1158/1078-0432.CCR-04-2514. [DOI] [PubMed] [Google Scholar]
- 5.Yawalkar N, Ferenczi K, Jones DA, Yamanaka K, Suh KY, Sadat S, Kupper TS. Blood. 2003;102:4059–4066. doi: 10.1182/blood-2003-04-1044. [DOI] [PubMed] [Google Scholar]
- 6.Aguado E, Richelme S, Nunez-Cruz S, Miazek A, Mura AM, Richelme M, Guo XJ, Sainty D, He HT, Malissen B, Malissen M. Science. 2002;296:2036–2040. doi: 10.1126/science.1069057. [DOI] [PubMed] [Google Scholar]
- 7.Sommers CL, Park CS, Lee J, Feng C, Fuller CL, Grinberg A, Hildebrand JA, Lacana E, Menon RK, Shores EW, et al. Science. 2002;296:2040–2043. doi: 10.1126/science.1069066. [DOI] [PubMed] [Google Scholar]
- 8.McCoy KD, Harris NL, Diener P, Hatak S, Odermatt B, Hangartner L, Senn BM, Marsland BJ, Geuking MB, Hengartner H, et al. Immunity. 2006;24:329–339. doi: 10.1016/j.immuni.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 9.Surh CD, Boyman O, Purton JF, Sprent J. Immunol Rev. 2006;211:154–163. doi: 10.1111/j.0105-2896.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- 10.Min B, Foucras G, Meier-Schellersheim M, Paul WE. Proc Natl Acad Sci USA. 2004;101:3874–3879. doi: 10.1073/pnas.0400606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raes G, De Baetselier P, Noël W, Beschin A, Brombacher F, Hassanzadeh G. J Leukocyte Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- 12.Almeida AR, Legrand N, Papiernik M, Freitas AA. J Immunol. 2002;169:4850–4860. doi: 10.4049/jimmunol.169.9.4850. [DOI] [PubMed] [Google Scholar]
- 13.Lin W, Truong N, Grossman WJ, Haribhai D, Williams CB, Wang J, Martin MG, Chatila TA. J Allergy Clin Immunol. 2005;116:1106–1115. doi: 10.1016/j.jaci.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 14.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. J Immunol. 2004;173:7259–7268. doi: 10.4049/jimmunol.173.12.7259. [DOI] [PubMed] [Google Scholar]
- 16.Curotto de Lafaille MA, Muriglan S, Sunshine MJ, Lei Y, Kutchukhidze N, Furtado GC, Wensky AK, Olivares-Villagomez D, Lafaille JJ. J Exp Med. 2001;194:1349–1359. doi: 10.1084/jem.194.9.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu CY, Kirman JR, Rotte MJ, Davey DF, Perfetto SP, Rhee EG, Freidag BL, Hill BJ, Douek DC, Seder RA. Nat Immunol. 2002;3:852–858. doi: 10.1038/ni832. [DOI] [PubMed] [Google Scholar]
- 18.Pfeiffer C, Stein J, Southwood S, Ketelaar H, Sette A, Bottomly K. J Exp Med. 1995;181:1569–1574. doi: 10.1084/jem.181.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamane H, Zhu J, Paul WE. J Exp Med. 2005;202:793–804. doi: 10.1084/jem.20051304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. J Exp Med. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen S, Ding Y, Tadokoro CE, Olivares-Villagomez D, Camps-Ramirez M, Curotto de Lafaille MA, Lafaille JJ. J Clin Invest. 2005;115:3517–3526. doi: 10.1172/JCI25463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Int Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 23.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 24.Ward JM, Yoon M, Anver MR, Haines DC, Kudo G, Gonzalez FJ, Kimura S. Am J Pathol. 2001;158:323–332. doi: 10.1016/S0002-9440(10)63972-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suri-Payer E, Cantor H. J Autoimmun. 2001;16:115–123. doi: 10.1006/jaut.2000.0473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}