

Summary
Deletion of the mouse selenoprotein P gene (Sepp1) lowers selenium concentrations in many tissues. We examined selenium homeostasis in Sepp1−/− and Sepp1+/+ mice to assess the mechanism of this. The liver produces and exports selenoprotein P, which transports selenium to peripheral tissues, and urinary selenium metabolites, which regulate whole-body selenium. At intakes of selenium near the nutritional requirement, Sepp1−/− mice had whole-body selenium concentrations 72 to 75% of Sepp1+/+ mice. Genotype did not affect dietary intake of selenium. Sepp1−/− mice excreted in their urine approximately 1.5 times more selenium in relation to their whole-body selenium than did Sepp1+/+ mice. In addition, Sepp1−/− mice gavaged with 75SeO32− excreted 1.7 to 2.4 times as much of the 75Se in the urine as did Sepp1+/+ mice. These findings demonstrate that deletion of selenoprotein P raises urinary excretion of selenium. When urinary small-molecule 75Se was injected intravenously into mice, over 90% of the 75Se appeared in the urine within 24 h, regardless of selenium status. This shows that urinary selenium is dedicated to excretion and not to utilization by tissues. Our results indicate that deletion of selenoprotein P leads to increased urinary selenium excretion. We propose that the absence of selenoprotein P synthesis in the liver makes more selenium available for urinary metabolite synthesis, increasing loss of selenium from the organism and causing the decrease in whole-body selenium and some of the decreases observed in tissues of Sepp1−/− mice.
Keywords: selenium homeostasis, selenoprotein P, urinary selenium excretion
Introduction
Selenium is an essential micronutrient that exerts its biological functions through selenoproteins [1, 2]. Homeostasis of selenium is achieved by its distribution among tissues and its excretion in the urine [3, 4]; absorption of selenium is not regulated [5].
Newly absorbed selenium is presented to the liver in the portal vein blood and the major dietary form of the element, selenomethionine, is transsulfurated (or transselenated) in the liver to free selenocysteine. Free selenocysteine is rapidly catabolized [6] to yield ‘metabolically active’ selenium, the putative pool of selenium that supplies specific selenium pathways, including selenoprotein synthesis [7]. Thus, the liver is the portal of entry for selenium.
The liver is known to release two specific forms of selenium. One is selenoprotein P, a selenium-rich plasma protein that supplies selenium to brain, testis, and other tissues [8]. The other is 1β-methylseleno-N-acetyl-D-galactosamine, which has been identified in rat liver [9] and in rat and human urine [10]. The liver produces other methylated excretory forms under certain conditions [3]. It also incorporates selenomethionine into proteins in a nonspecific manner.
Mice with deletion of selenoprotein P (Sepp1−/− mice) have been produced [11, 12]. Sepp1−/− mice have lower selenium concentrations than do Sepp1+/+ mice in all tissues investigated except liver [11]. Because deletion of selenoprotein P affects distribution of selenium from the liver to peripheral tissues, we compared homeostasis of selenium in Sepp1−/− and Sepp1+/+ mice. Some of the work reported here has been published in abstract form [13].
Materials and Methods
Animals
Sepp1+/− mice that had been backcrossed 10 times with C57BL/6 mice were mated as reported previously [11]. Male Sepp1+/+ and Sepp1−/− offspring were selected for study by genotyping. In experiments that used selenium deficient mice, weanling male C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME).
In all experiments, mice were fed from the time of weaning Torula yeast-based diets that differed only in their selenium content [11, 14]. The basal diet was selenium deficient. Forms of the diet supplemented with 0.1 or 0.25 mg selenium/kg as sodium selenite were also fed. Using selenite, and not selenomethionine, as the supplementary form of selenium ensured that the selenium that accumulated in the animal would be in the form of selenoproteins. The diets were prepared to our specifications and pelleted by Harlan-Teklad (Madison, WI).
Mice were housed in plastic cages on aspen shavings in a room with a 10 h:14 h light:dark cycle. For food intake and urine collection experiments mice were housed individually in metabolic cages (Nalgene, Rochester, NY). Food cups and food scattered on the runway to the cup were weighed daily in experiments performed to determine food intake. Mice hadz free access to food and tap water except before administration of 75Se by gavage. Those mice were starved overnight prior to gavage but were allowed to drink.
Blood was removed from the inferior vena cava with a syringe and needle while the animal was anesthetized with isoflurane. EDTA (1 mg/ml) was added to the blood to prevent coagulation and plasma was separated by centrifugation at 16,000 g for 2 min. Tissues were removed, frozen in liquid nitrogen, and stored at −20°C until they were assayed for selenium. The remaining carcass was divided into 4 parts, which were frozen at −20°C until they were assayed for selenium. The Vanderbilt University Institutional Animal Care and Use Committee approved all experimental protocols.
Materials
75Se-selenite (specific activity: ∼2,250 Ci/g selenium) was purchased from the University of Missouri Research Reactor Facility (Columbia, MO). 75Se was quantitated using a Perkin-Elmer Wizard 3″ gamma counter (Perkin-Elmer, Shelton, CT). Superdex™ 30 was purchased from Amersham Biosciences Corp. (Piscataway, NJ). All other reagents were reagent grade quality or better.
Assays
Tissue and urine selenium was measured using a modification of the fluorometric assay of Koh and Benson [15, 16]. The limit of detection of this assay is 1 ng selenium. Glutathione peroxidase activity was determined as previously described [11]. Activities are reported as U/mg protein with 1 U = 1 μmol NADPH oxidized per min.
Metabolic Fate of 75Se-Labeled Compounds
Tracer doses of 75Seselenite (10 μCi) were administered by gavage to six Sepp1+/+ mice fed the diet supplemented with 0.25 mg selenium/kg and urine was collected for 24 h. One ml of that urine was chromatographed on a Superdex™ 30 column in phosphate-buffered saline (PBS; 0.14 M NaCl, 0.003 M KCl, 0.020 M phosphate, pH 7.4) and a single 75Se peak was observed just ahead of the inner volume of the column. The fractions that made up the 75Se peak were pooled. The selenium concentration of the pool was determined by fluorometric analysis to be 44 ng/ml.
Mice fed selenium deficient diet and diet supplemented with 0.25 mg selenium/kg for 10 weeks were administered 8.8 ng selenium each by tail vein as 75Se-selenite or 75Se-labeled urinary metabolite in PBS. Urine was collected for 24 h. Then blood was taken and tissues were harvested and weighed. 75Se was determined in urine, blood, tissues, and carcass.
Results
The whole-body selenium concentrations of Sepp1−/− mice fed 0.10 and 0.25 mg selenium/kg diet were 75% and 72%, respectively, of comparable Sepp1+/+ mice (Figure 1). This demonstrates that deletion of selenoprotein P causes a decrease in the whole-body selenium of mice fed amounts of the element near its nutritional requirement of 0.10 mg selenium/kg diet.
Figure 1.

Whole-body selenium concentration in Sepp1−/− (open bars) and Sepp1+/+ (shaded bars) mice. Mice were fed the respective diets as follows: The 0.1 mg selenium/kg group (Sepp1−/− mice weight 15.5 ± 0.7 g, n=6; Sepp1+/+ mice weight 20.8 ± 0.9 g, n=7) was fed the 0.25 mg selenium/kg diet for 2 weeks from weaning and then switched to the 0.1 mg selenium/kg diet for 2 weeks before being studied. This was done because Sepp1−/− mice develop nervous system injury if fed the 0.1 mg selenium/kg diet immediately after weaning [14]. The group fed 0.25 mg selenium/kg diet from weaning (Sepp1−/− mice weight 20.2 ± 1.7 g, n=9; Sepp1+/+ mice weight 22.6 ± 1.0 g, n=10) was fed the diet for 4 weeks. At the time of study, selenium was determined in selected tissues and in the rest of the carcass. Values shown are sums of all body parts. They are means ± S.D. Asterisks indicate pairs that were significantly different (P<0.05) by the Student t-test.
In addition to the modest loss of selenium in the form of selenoprotein P [17], potential causes of the genotype effect on whole-body selenium include decreased selenium intake and increased selenium excretion. Table 1 shows that food intake was not significantly different between Sepp1−/− and Sepp1+/+ mice. Therefore, a difference in selenium intake was not the cause of the genotype effect on whole-body selenium.
Table 1.
Food intake of Sepp1−/− and Sepp1+/+ mice*
| dietary selenium supplement |
Sepp1−/− | Sepp1+/+ | ||
|---|---|---|---|---|
| n | 24-h food intake (g/g body weight) |
n | 24-h food intake (g/g body weight) |
|
| 0.1 mg/kg | 4 | 0.23 ± 0.07 | 5 | 0.19 ± 0.01 |
| 0.25 mg/kg | 6 | 0.17 ± 0.04 | 6 | 0.21 ± 0.02 |
Values are means ± S.D. All values were obtained 4 weeks after weaning. Mice fed 0.1 mg selenium/kg had been fed 0.25 mg selenium/kg for the first 2 weeks and then 0.1 mg selenium/kg for the 2 weeks before the measurement was made. The other mice were fed the 0.25 mg selenium/kg diet for the full 4 weeks. The values for the genotypes fed the same diet were not significantly different by the Student t-test.
Because the selenium intakes of the Sepp1 genotypes were the same, urinary selenium excretion was not significantly different between them (Table 2). However, the Sepp1−/− mice, with a whole-body selenium concentration that was 72-75% that of the Sepp1+/+ mice, achieved this steady state of selenium metabolism only because they had a lower whole-body selenium concentration (Figure 1). This indicates that they excreted a greater fraction of their whole-body selenium, and, by inference, of their ‘metabolically active’ selenium, than did the Sepp1+/+mice.
Table 2.
Urinary selenium excretion by Sepp1−/− and Sepp1+/+ mice*
| dietary selenium supplement |
Sepp1−/− | Sepp1+/+ | ||
|---|---|---|---|---|
| n | selenium/24-h (ng/g body weight) |
n | selenium/24-h (ng/g body weight) |
|
| 0.1 mg/kg | 6 | 9.2 ± 4.1 | 6 | 7.9 ± 1.5 |
| 0.25 mg/kg | 5 | 12 ± 4.4 | 6 | 11 ± 3.0 |
Values are means ± S.D. All values were obtained 4 weeks after weaning. Mice fed 0.1 mg selenium/kg were fed 0.25 mg selenium/kg for the first 2 weeks and then 0.1 mg selenium/kg for the second 2 weeks. The other mice were fed the 0.25 mg selenium/kg diet for the full 4 weeks. Then urine was collected for 24 hours in plastic metabolic cages. The values for the genotypes fed the same diet were not significantly different by the Student t-test.
A pool of ‘metabolically active” selenium is presumably available in the liver for selenoprotein and urinary metabolite synthesis (Scheme 1). This pool receives selenium from dietary intake and from breakdown of selenocysteine derived from selenoproteins that are turning over. The fate of the metabolically active selenium pool was evaluated in Sepp1−/− and Sepp1+/+ mice after it had been labeled by gavage of a tracer dose of 75SeO32−. Absorption of the 75Se was 90-94% of the dose administered as judged by the 75Se recovered in the carcass and urine (data not shown). Sepp1−/− mice fed 0.1 mg selenium/kg diet excreted 2.4 times as much 75Se in the urine as did Sepp1+/+ mice and the corresponding figure for mice fed 0.25 mg selenium/kg diet was 1.7 times as much (Figure 2). This strengthens the conclusion that deletion of selenoprotein P increases urinary selenium excretion.
Scheme 1.
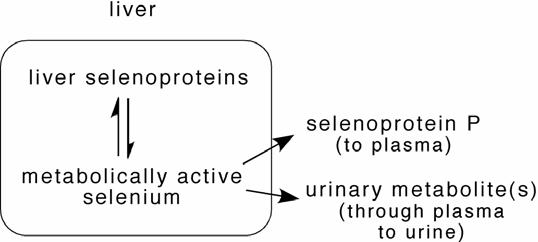
Relationships of “metabolically active” selenium pool in the liver with synthesis of selenoproteins and urinary selenium metabolites.
Figure 2.
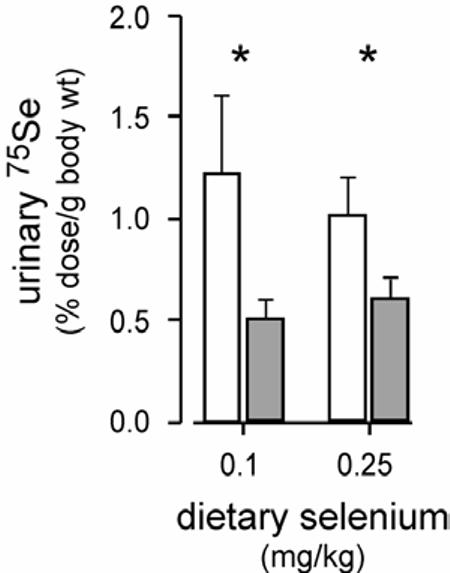
Urinary 75Se excretion by Sepp1−/− (open bars) and Sepp1+/+ (shaded bars) mice administered 75SeO32− by gavage. Mice were fed the respective diets as described in the legend to figure 1. They were then gavaged and urine was collected for 24 hours in plastic metabolic cages. Values are means ± S.D., n=4-8. Asterisks indicate pairs that were significantly different (P<0.05) by the Student t-test.
Urinary selenium metabolite(s) might serve, alternatively, to provide selenium to tissues. We evaluated this possibility. 75Se-labeled urinary metabolites were injected into selenium deficient and control mice by tail vein. Figure 3 shows that both groups of mice excreted over 90% of the 75Se in the urine within 24 hours. The same amount of selenium as 75Se-labeled selenite, a form of selenium that is available for tissue uptake, was injected as a control. Urinary excretion of the 75Se from selenite was much lower than excretion of the 75Se injected as urinary metabolite(s), reflecting the retention in tissues of 75Se injected as selenite (not shown). Moreover, urinary excretion of 75Se injected as selenite by selenium deficient mice was less than excretion by control mice (Figure 3). These results demonstrate that once selenium is converted to the urinary metabolites very little of it is available for uptake by tissues.
Figure 3.
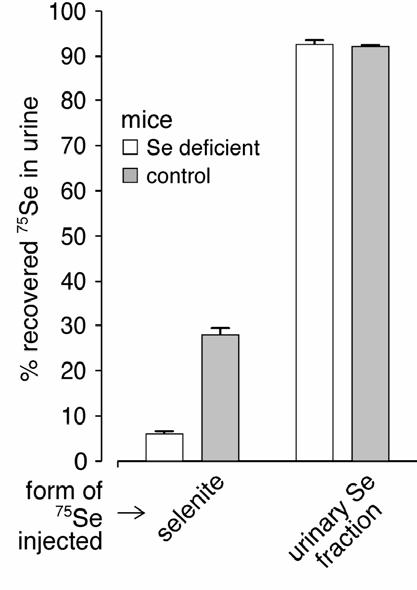
Urinary 75Se excretion by selenium deficient and control mice administered 75Se-labeled selenite or 75Se-labeled urinary metabolite by tail vein. The amount of selenium administered to each mouse was 8.8 ng. After injection, mice were put into metabolic cages and urine was collected for 24 hours. The results shown are percentages of the total recovered 75Se that was in the urine. Values are means ± S.D., n=4-8. Excretions by the groups given 75Se-labeled selenite were significantly different (P<0.05) from one another by the Student t-test.
Discussion
The initial studies of Sepp1−/− mice indicated that the absence of selenoprotein P affected selenium concentration in tissues [11, 12]. Testis and brain selenium concentrations were sharply decreased in the Sepp1−/− genotype and those in other tissues, such as kidney and heart, were more mildly decreased. These variable effects from tissue to tissue suggested that deletion of selenoprotein P affects tissue selenium by more than one mechanism.
One of the mechanisms is likely to be direct supply of selenium by selenoprotein P to selected tissues. Testis selenium in Sepp1−/− mice is depressed to below 20% of that in Sepp1+/+ mice and responds little to increasing the selenium intake of the animal [11]. Brain selenium is depressed to below 50% in Sepp1−/− mice, and raising selenium intake has no effect on brain selenium levels in them until very high intakes are reached [11]. In addition, the efficiency of brain selenium uptake from selenoprotein P is increased by selenium deficiency [18]. These findings suggest specific uptake of selenium from selenoprotein P by testis and brain, possibly mediated by receptors.
The rather modest decreases in selenium that occur in kidney, heart, and some other tissues of Sepp1−/− mice are readily repaired by increasing selenium intake [11] and thus appear to have a different nature from the decreases in testis and brain. Therefore, a second mechanism is needed to explain the effects of selenoprotein P deletion on selenium supply to these tissues.
The present work demonstrates an effect of the Sepp1−/− genotype on whole-body selenium homeostasis that might explain at least part of its effect on selenium concentration in tissues other than brain and testis. Scheme 1 outlines the mechanism we propose to explain this effect. Once selenium enters the metabolically active selenium pool in the liver, it has two potential fates. One is selenoprotein synthesis, which is genetically regulated. The other is the production of metabolites for urinary excretion. This second pathway appears to be largely confined to the liver [3], with a possible contribution by kidney, and to be regulated by selenium availability [4]. Urinary metabolite synthesis presumably competes with selenoprotein synthesis for selenium in the metabolically active hepatic selenium pool.
Deletion of selenoprotein P eliminates one of the pathways that remove selenium from the liver. This deletion would be expected to make more of the metabolically active selenium in the liver available for production of urinary metabolites and thereby lead to greater urinary excretion of selenium. This result is what we found in this study (Figure 2).
In addition to increasing urinary metabolite synthesis, deletion of selenoprotein P would be predicted to make more of the metabolically active selenium in the liver available for synthesis of liver selenoproteins. We observed an increase in liver selenium and glutathione peroxidase activity in Sepp1−/− mice fed a diet marginal in selenium [11], supporting this prediction.
We postulate, therefore, that the size of the metabolically active selenium pool in the liver dictates the rate of urinary metabolite synthesis and, as a consequence, regulates selenium excretion in the urine. Deletion of selenoprotein P would be predicted to enlarge this pool and thereby to increase the production of urinary metabolite(s), which would appear in the urine. This would account for the higher than expected urinary selenium excretion observed in Sepp1−/− mice (Figure 2). Deletion of liver selenoproteins such as glutathione peroxidase-1 would not be expected to cause increased urinary selenium excretion because such selenoproteins turn over within the liver and deletion of them would not be expected to affect the size of the metabolically active selenium pool. Our unpublished results using mice with deletion of glutathione peroxidase-1 are consistent with this expectation.
Mice with liver-specific inactivation of the gene for tRNA[ser]sec and, thus, with inability to synthesize selenoproteins in liver, have been shown to have sharply decreased selenium levels in several tissues [19, 20]. Our hypothesis predicts that those mice would produce excessive urinary metabolite in their livers, leading to increased selenium excretion in the urine and decreased whole-body selenium content. Thus, we speculate that excessive urinary excretion was at least part of the cause of low tissue selenium levels in those mice [20].
Our study also suggests that urinary selenium metabolites do not serve in transporting selenium to tissues (Figure 3). However, because we did not demonstrate complete excretion of injected metabolites (figure 3), it remains possible that some utilization of them occurs.
In conclusion, these experiments support the concept that the process of converting ‘metabolically active’ selenium to urinary metabolites is in competition with the use of that selenium for the synthesis of selenoproteins, especially selenoprotein P. This competition appears to take place principally in the liver. Work is needed to characterize the enzymes involved in production of the urinary metabolite(s) and the nature of the competition between that process and selenoprotein synthesis.
Acknowledgement
This work was supported by NIH grant ES02497.
References
- 1.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–43. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 2.Stadtman TC. Biosynthesis and function of selenocysteine-containing enzymes. J. Biol. Chem. 1991;266:16257–16260. [PubMed] [Google Scholar]
- 3.Bopp BA, Sonders RC, Kesterson JW. Metabolic fate of selected selenium compounds in laboratory animals and man. Drug Metab. Rev. 1982;13:271–318. doi: 10.3109/03602538209030000. [DOI] [PubMed] [Google Scholar]
- 4.Burk RF, Brown DG, Seely RJ, Scaief CC. Influence of dietary and injected selenium on whole-body retention, route of excretion, and tissue retention of 75Se032- in the rat. J. Nutr. 1972;102:1049–1055. doi: 10.1093/jn/102.8.1049. [DOI] [PubMed] [Google Scholar]
- 5.Brown DG, Burk RF, Seely RJ, Kiker KW. Effect of dietary selenium on the gastrointestinal absorption of 75Se032- in the rat. Int. J. Vit. Nutr. Res. 1972;42:588–591. [PubMed] [Google Scholar]
- 6.Esaki N, Nakamura T, Tanaka H, Soda K. Selenocysteine lyase, a novel enzyme that specifically acts on selenocysteine. Mammalian distribution and purification and properties of pig liver enzyme. J. Biol. Chem. 1982;257:4386–91. [PubMed] [Google Scholar]
- 7.Veres Z, Kim IY, Scholz TD, Stadtman TC. Selenophosphate synthetase. Enzyme properties and catalytic reaction. J. Biol. Chem. 1994;269:10597–10603. [PubMed] [Google Scholar]
- 8.Burk RF, Hill KE. Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 2005;25:215–235. doi: 10.1146/annurev.nutr.24.012003.132120. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi Y, Ogra Y, Ishiwata K, Takayama H, Aimi N, Suzuki KT. Selenosugars are key and urinary metabolites for selenium excretion within the required to low-toxic range. Proc Natl Acad Sci U S A. 2002;99:15932–6. doi: 10.1073/pnas.252610699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gammelgaard B, Bendahl L. Selenium speciation in human urine samples by LC- and CE-ICP-MS - separation and identification of selenosugars. J. Anal. At. Spectrom. 2004;19:135–142. [Google Scholar]
- 11.Hill KE, Zhou J, McMahan WJ, Motley AK, Atkins JF, Gesteland RF, Burk RF. Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 2003;278:13640–13646. doi: 10.1074/jbc.M300755200. [DOI] [PubMed] [Google Scholar]
- 12.Schomburg L, Schweizer U, Holtmann B, Flohé L, Sendtner M, Kohrle J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 2003;370:397–402. doi: 10.1042/BJ20021853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burk RF, Hill KE, Motley AK, Hu M, Austin LM. Deletion of selenoprotein P disrupts selenium homeostasis in the mouse. FASEB J. 2004;18:A849. [Google Scholar]
- 14.Hill KE, Zhou J, McMahan WJ, Motley AK, Burk RF. Neurological dysfunction occurs in mice with targeted deletion of selenoprotein P gene. J. Nutr. 2004;134:157–161. doi: 10.1093/jn/134.1.157. [DOI] [PubMed] [Google Scholar]
- 15.Koh TS, Benson TH. Critical re-appraisal of fluorometric method for determination of selenium in biological materials. J. Assoc. Off. Anal. Chem. 1983;66:918–926. [PubMed] [Google Scholar]
- 16.Sheehan TMT, Gao M. Simplified fluorometric assay of total selenium in plasma and urine. Clin. Chem. 1990;36:2124–2126. [PubMed] [Google Scholar]
- 17.Read R, Bellew T, Yang J-G, Hill KE, Palmer IS, Burk RF. Selenium and amino acid composition of selenoprotein P, the major selenoprotein in rat serum. J. Biol. Chem. 1990;265:17899–17905. [PubMed] [Google Scholar]
- 18.Burk RF, Hill KE, Read R, Bellew T. Response of rat selenoprotein P to selenium administration and fate of its selenium. Am. J. Physiol. 1991;261:E26–E30. doi: 10.1152/ajpendo.1991.261.1.E26. [DOI] [PubMed] [Google Scholar]
- 19.Carlson BA, Novoselov SV, Kumaraswamy E, Lee BJ, Anver MR, Gladyshev VN, Hatfield DL. Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J. Biol. Chem. 2004;279:8011–7. doi: 10.1074/jbc.M310470200. [DOI] [PubMed] [Google Scholar]
- 20.Schweizer U, Streckfuss F, Pelt P, Carlson BA, Hatfield DL, Kohrle J, Schomburg L. Hepatically derived selenoprotein P is a key factor for kidney but not for brain selenium supply. Biochem J. 2005;386:221–6. doi: 10.1042/BJ20041973. [DOI] [PMC free article] [PubMed] [Google Scholar]