

Abstract
To cope with oxidative stress, the metabolic network of plant cells must be reconfigured either to bypass damaged enzymes or to support adaptive responses. To characterize the dynamics of metabolic change during oxidative stress, heterotrophic Arabidopsis (Arabidopsis thaliana) cells were treated with menadione and changes in metabolite abundance and 13C-labeling kinetics were quantified in a time series of samples taken over a 6 h period. Oxidative stress had a profound effect on the central metabolic pathways with extensive metabolic inhibition radiating from the tricarboxylic acid cycle and including large sectors of amino acid metabolism. Sequential accumulation of metabolites in specific pathways indicated a subsequent backing up of glycolysis and a diversion of carbon into the oxidative pentose phosphate pathway. Microarray analysis revealed a coordinated transcriptomic response that represents an emergency coping strategy allowing the cell to survive the metabolic hiatus. Rather than attempt to replace inhibited enzymes, transcripts encoding these enzymes are in fact down-regulated while an antioxidant defense response is mounted. In addition, a major switch from anabolic to catabolic metabolism is signaled. Metabolism is also reconfigured to bypass damaged steps (e.g. induction of an external NADH dehydrogenase of the mitochondrial respiratory chain). The overall metabolic response of Arabidopsis cells to oxidative stress is remarkably similar to the superoxide and hydrogen peroxide stimulons of bacteria and yeast (Saccharomyces cerevisiae), suggesting that the stress regulatory and signaling pathways of plants and microbes may share common elements.
Oxidative stress is a key underlying component of most abiotic stresses (Mittler, 2002; Apel and Hirt, 2004) and a major limiting factor of plant growth in the field (Mittler, 2006). Production of reactive oxygen species (ROS) and other radicals increases dramatically during abiotic stress conditions and oxidative stress occurs when the rate of ROS production outstrips the capacity of antioxidant systems to detoxify them. The result is oxidative damage to key biomolecules such as proteins, lipids, and DNA leading to cellular dysfunction and ultimately cell death (Halliwell, 2006). In specific cases, ROS may also directly induce genetic programs that lead to cell death (Wagner et al., 2004). To avoid these cell death scenarios, plants invoke a molecular response that allows them to cope with, and adapt to, the oxidative stress situation. The precise nature of this molecular response remains poorly characterized. Many stress-related genes have been identified and the list of antioxidant enzymes and metabolites present in plants continues to be extended (Mittler et al., 2004). It is also apparent that there are specific transcriptomic responses to oxidative stress (Gadjev et al., 2006) and specific regulatory elements of this transcriptomic response are beginning to be uncovered (Davletova et al., 2005). However, an intellectual reconstitution of the mechanism by which these myriad molecular changes lead to adaptation has yet to be realized.
In microbial systems it is well established that metabolic change is a key part of the response to oxidative stress. In yeast (Saccharomyces cerevisiae), for example, a distinct hydrogen peroxide (H2O2) stimulon is observed in which a key event is the rerouting of central carbohydrate metabolism with a diversion of carbon away from respiratory pathways and into the oxidative pentose phosphate pathway (OPPP) to provide reductant for antioxidant metabolism (Godon et al., 1998). Moreover, the gene expression events that underpin these changes in microbes appear to be regulated by specific redox-sensitive transcription factors (Liu et al., 2005). Despite the fact that metabolic change is central to the microbial response to oxidative stress, the role of metabolism during oxidative stress in plants has largely been overlooked. What little is known generally relates to inactivation of enzymes by oxidative damage. For example, enzymes of the tricarboxylic acid (TCA) cycle such as aconitase, pyruvate-, and 2-oxoglutarate-dehydrogenase are known to be sensitive to oxidative inhibition (Verniquet et al., 1991; Sweetlove et al., 2002). While proteomic approaches are beginning to systematically catalogue oxidative modification of proteins (Moller and Kristensen, 2006), only in a few instances has the connection between this oxidation and loss of protein function been demonstrated (Hancock et al., 2005).
Consequently, there are a number of important open questions regarding the role and nature of metabolic change during oxidative stress. How widespread is oxidative inhibition of metabolism? What are the consequences of this inhibition for continued growth and survival of the plant? To what extent is metabolism reprogrammed to bypass inhibited steps and to support antioxidant activity? What role does gene expression play in the regulation of such metabolic reprogramming? Without knowing the answers to such questions, it is difficult to gauge the physiological consequences of oxidative stress and impossible to define the nature of the coping strategies that might be implemented. In an attempt to address these issues, we have undertaken a systematic investigation of metabolic change in Arabidopsis (Arabidopsis thaliana) cells exposed to oxidative stress using established metabolomic approaches. However, to go beyond the usual metabolic snapshot taken at a single time point, we analyzed a time series of samples to capture the dynamics of the response. We also quantified changes in mass isotopomers in this time series as a result of the introduction of [U-13C]Glc at the beginning of the treatment. We demonstrate that the resultant dynamic metabolite labeling profiles provide an additional and sensitive measure of the metabolic phenotype (Harada et al., 2006) and reveals changes that are not apparent from the metabolite abundance changes alone. We relate these metabolic changes to transcriptomic changes measured in the same samples. The integrated datasets provide important new information about the extent of metabolic change during oxidative stress and reveal the nature of the molecular response strategy that allows the cell to overcome the oxidative insult.
RESULTS
Experimental System
The experimental strategy we wished to pursue was to induce oxidative stress in Arabidopsis cells and follow metabolic change over the initial period of the response by profiling changes in metabolite abundance and labeling (from exogenously supplied [U-13C]Glc). Parallel measurements of transcriptomic change would provide insight into the underlying regulatory mechanisms of this metabolic change. A well-established heterotrophic Arabidopsis cell suspension culture (May and Leaver, 1993) that has been the subject of previous investigations into the response to oxidative stress (Desikan et al., 2001; Sweetlove et al., 2002; Tiwari et al., 2002) was used. To cause oxidative stress, we added the redox-active quinone, menadione, to the growth medium (Sweetlove et al., 2002). The main site of ROS production due to addition of menadione to heterotrophic cells is the mitochondrial electron transport chain where menadione competes with the ubiquinone pool for electrons from complexes I and II (Thor et al., 1982). However, menadione may also be active in other membrane systems. The menadione treatment was further optimized for the heterotrophic Arabidopsis cell suspension culture used here to ensure that the treatments caused reproducible metabolic changes without having a detrimental effect on cell viability. It was found that 60 μm menadione led to an inhibition of the TCA cycle enzyme aconitase (Supplemental Fig. S1), a sensitive indicator of oxidative stress (Verniquet et al., 1991). This treatment did not significantly affect cell viability (Supplemental Fig. S1) and cells continued to grow and divide over a subsequent 4 d period (data not shown).
Preliminary experiments indicated that most metabolite abundance changes following the introduction of menadione stabilized after 6 h (data not shown). This suggests that the initial metabolic perturbation and subsequent response had occurred during this period and the system had established a new steady state. We therefore decided to investigate metabolic change during this period in more detail.
Metabolite Abundance Change following Oxidative Stress
A total of 50 polar metabolites were analyzed representing a range of pathways mainly of primary metabolism. Although additional metabolites could be identified in the gas chromatography-mass spectrometry (GC-MS) spectra (and the complete data set is given in Supplemental Table S1), this set of 50 represented those for which we could also reliably detect 13C-labeled mass isotopomers (see subsequent section: “Changes in Metabolite Labeling Kinetics following Oxidative Stress”). The amount of each metabolite at each time point following imposition of oxidative stress was compared to the equivalent control time point. Figure 1 shows the time series of those metabolites that were significantly different from control (t test, P < 0.05) in at least two consecutive time points. The complete dataset is presented in Supplemental Figure S2. A total of 23 metabolites (46% of those analyzed) were significantly affected by the oxidative stress treatment in at least two consecutive time points, demonstrating that oxidative stress had a considerable impact on primary metabolism. Of the 23 metabolites that were changed, 16 were decreased and seven were increased relative to the control. When these changes are mapped onto the metabolic network it can be seen that they are not randomly distributed, but instead occur coordinately in distinct localized regions of the network (Fig. 3).
Figure 1.

Time series of metabolite abundance showing significantly altered metabolites following oxidative stress. White symbols are menadione-treated cells and black symbols are control cells. Values are means of four replicates ± sem. The asterisk (*) indicates significantly different to equivalent control point (t test, P < 0.05). All data are expressed relative to the 0 h value.
Figure 3.

Schematic showing qualitative changes of metabolite abundance and labeling mapped onto the metabolic network. Solid arrows in the network diagram indicate a single step connecting two metabolites, dashed arrows represent multiple steps. Gray shading indicates no significant change, blue shading a significant increase in value, and red shading a significant decrease, based on Figures 2 and 3. A change is deemed significant if there are significant changes following menadione treatment (t test, P < 0.05) in at least two consecutive time points. Green shading indicates unmeasured metabolites.
Most of the increases in metabolite levels occur in the linked pathways of glycolysis and the OPPP. In glycolysis, there were significant increases in the hexose phosphates, Glc-6-P, and Fru-6-P, as well as 3-phosphoglycerate (3-PGA). There were also major increases in gluconate and Rib, which are most likely derivitization-degradation products of the OPPP intermediates, 6-phosphogluconate and Rib-5-P, respectively. The accumulation of these metabolites suggests that there is a relative decrease in the flux through pathways downstream of glycolysis and the oxidative branch of the OPPP. Consistent with this suggestion are the observed decreases in the levels of amino acids linked to downstream glycolytic intermediates or the TCA cycle. There were decreases in Ser and Gly (linked to 3-PGA), Ala (linked to pyruvate), in the Asp branch of amino acids linked to oxaloacetate (Asp, β-Ala, homoserine, Met, and Thr), and in the Glu branch linked to 2-oxoglutarate (Glu, Gln, and Pro). There was also a pronounced decrease in malate that may indicate a perturbation of the TCA cycle.
The increases in gluconate and Rib suggest an increase in flux through the oxidative branch of the OPPP relative to the nonoxidative branch. The increases in both of these metabolites are substantial yet transient. Gluconate reaches a peak of nearly 2-fold control level at 1 h and thereafter declines back to control level by 4 h. Ribose reaches a peak of 3-fold control level slightly later, at 3 h, and similarly declines back to control level by 5 h. The time shift in the peak point of these two metabolite profiles suggests a sequential accumulation of metabolites within the OPPP. There is also a sequential accumulation of glycolytic intermediates, with 3-PGA showing a peak increase at 1 h before gradually declining, while Glc-6-P and Fru-6-P are not significantly increased until 3 h. This is consistent with a gradual backing up of glycolysis due to inhibition of downstream fluxes. The steady decline of metabolites back to control level suggests that adaptive regulatory changes in enzyme activity are occurring.
There is evidence of ascorbate turnover in both control and oxidative stress-treated cells, with a rapid decrease in ascorbate levels within the first hour. This indicates that there is a degree of oxidative stress even in the control cells at the beginning of the experiment, probably due to handling of the cells (the experiment demands that the cell culture medium is replaced with new medium containing the treatment and the 13C-Glc). However, in the control cells the ascorbate levels rapidly recover, indicating the oxidative stress is transient. In contrast, in the menadione-treated cells ascorbate levels fail to recover, indicating a more prolonged and severe oxidative stress. Moreover, there is a pronounced accumulation of threonate, a breakdown product of ascorbate. Ascorbate is one of the principal antioxidant molecules in the cell and the production of ascorbate breakdown products indicates a failure to recycle all of the oxidized ascorbate via the ascorbate-glutathione cycle. There was also a modest increase in glycerol-3-P after 3 and 4 h, although the levels of this metabolite were not changed at other points in the time course.
Change in 13C-Labeling Kinetics following Oxidative Stress
Metabolite abundance changes give a clear indication of perturbation in the metabolic network in the form of an altered relationship between influx into and efflux from a metabolite pool (Raamsdonk et al., 2001). However, it is possible for metabolic flux to change without affecting metabolite levels (Fell, 2005; Fernie et al., 2005). This occurs when the flux change is coordinated through a pathway such that change of influx into metabolite pools is exactly balanced by change in efflux. To identify additional areas of metabolic perturbation that are not apparent from metabolite abundance alone, we also followed the kinetics of distribution of 13C label from [U-13C]Glc into the various metabolite pools analyzed. Of the 50 metabolites analyzed, there were significant changes in the amount of label in at least two consecutive time points in 27 (54%) of the metabolites (Fig. 2). The complete dataset is presented in Supplemental Figure S3. When these changes are mapped onto the metabolic network (Fig. 3), it can immediately be seen that a qualitatively similar pattern of change to that of metabolite abundances is observed, confirming the areas of metabolic perturbation identified from metabolite abundance change.
Figure 2.

Time series of 13C-label accumulation showing metabolites with significant differences in labeling following oxidative stress. White symbols are menadione-treated cells and black symbols are control cells. Values are means of four replicates ± sem. The asterisk (*) indicates significantly different to equivalent control point (t test, P < 0.05).
However, there were additional changes in the labeling data that were not apparent in the metabolite abundance time series. In particular, there were significant decreases in the labeling of all measured TCA cycle acids which, with the exception of malate, were unaltered in abundance in response to the oxidative stress treatment (Figs. 1, 2, and 3). The fact that there were decreases in the abundance of the Glu and Asp families of amino acids led us to suggest that there was an inhibition of flux through the TCA cycle. The decreased labeling of TCA cycle acids while their abundance is unaltered is consistent with this idea and suggests coordinated decreases in flux through all steps of the TCA cycle. However, this interpretation of the labeling data must be made with caution. It is equally possible that there is no decrease in flux through the TCA cycle and a decrease in the labeling of pyruvate (the immediate precursor) is responsible for the decreased labeling of TCA cycle acids. To distinguish between these two possibilities, it is necessary to track backwards through the metabolic network and look at the abundance and labeling patterns of earlier metabolites. Although we do not have any information for the labeling of pyruvate, we do have data for 3-PGA, three steps upstream of pyruvate in the glycolytic sequence. 3-PGA is increased in abundance (Fig. 1) but its labeling pattern is essentially unchanged (Supplemental Fig. S3). This strongly suggests a decreased efflux from the 3-PGA pool that would lead to the observed increased abundance while having a limited impact on the accumulation of label. In other words, it is likely that there is a decreased demand for 3-PGA from downstream processes such as the TCA cycle. Although we cannot completely rule out changes in the labeling of pyruvate contributing to the labeling pattern of the TCA cycle acids, the available evidence (decrease of efflux from 3-PGA, decreased labeling of TCA cycle acids, and decreased abundance of Glu and Asp family amino acids) corroborates an inhibition of the TCA cycle.
There are several other metabolites unaltered in abundance but that show altered labeling patterns (Supplemental Fig. S2; Fig. 2). Most notable among these is Suc, which despite unaltered abundance following oxidative stress is labeled at a massively slower rate than in control cells. Suc is synthesized from the hexose phosphate Fru-6-P and UDPGlc that is derived indirectly from Glc-6-P. Since the labeling of both Fru-6-P and Glc-6-P are unchanged (Supplemental Fig. S3) a decreased labeling of precursor molecules does not seem to be responsible for the decreased labeling of Suc. Thus, it is likely that the rate of Suc synthesis is significantly decreased in response to oxidative stress. The fact that the amount of Suc does not change may indicate a balancing decrease in the rate of breakdown. There is also a major decrease in labeling of Gal and a smaller decrease of the related sugar raffinose, suggesting a decrease in cell wall biosynthesis. Trehalose, another sugar derived from Glc-6-P, is also labeled more slowly following oxidative stress, similarly suggesting a decreased rate of synthesis. Finally, Phe falls into this category of metabolites unaltered in abundance but with a reduced rate of labeling. The labeling pattern of shikimate, a precursor metabolite of Phe (albeit some distance upstream) is unchanged, indicating that there is a decreased rate of synthesis (and utilization/turnover) of Phe.
The labeling data also allows a more precise interpretation of the metabolic changes in several specific instances. For example, there are many metabolites that decrease in abundance following oxidative stress and the most straightforward interpretation is a decrease in flux through the relevant pathways. However, it is possible for there to be an increase in flux through a pathway that would still lead to a decrease in metabolite levels. For example, if the influx and efflux into a metabolite both increase, but the efflux is increased more relative to influx. To assess whether a qualitative interpretation of labeling patterns can help resolve such issues, we constructed a simple model of metabolite labeling patterns based on mass balance equations (Morgan and Rhodes, 2002; Ratcliffe and Shachar-Hill, 2006). We examined the possible changes in influx and efflux that could lead to decreasing metabolite abundance (Fig. 4). Generally speaking, a decreasing metabolite pool resulted in a more rapid labeling of the pool. Distinguishing between the different influx/efflux scenarios that can lead to these increased labeling patterns would require numerical solution of the mass balance equations for flux using the labeling data, which is beyond the scope of this study. However, one of the scenarios can be qualitatively discriminated. That is when both influx and efflux into the metabolite pool are decreased, but influx is decreased relatively more. This is the only scenario that results in a decrease in rate of labeling (Fig. 4). This pattern (decrease in metabolite pool size and decrease in rate of labeling) is seen for several metabolites following oxidative stress. Where it is reasonable to assume that labeling of the precursor metabolite is unchanged, this provides evidence of a decrease of both influx into and efflux from that metabolite. Thus the labeling data provides evidence for a decrease of flux both into and from the citrate pool, lending further support to the notion that TCA cycle flux is decreased. In addition the data suggest that the flux of synthesis and utilization of Ala is decreased.
Figure 4.

Model examining effect of decreasing pool size on labeling kinetics. A simple linear system is modeled in which a labeled substrate (S) is introduced and is converted into a product (P) via two intermediates, A and B. The fractional enrichment of label in B is estimated by numerical iteration of mass balance differential equations. At steady state the influx into and efflux from pool B are given arbitrary values of 10 (JSA = 10, JAB = 10, JBP = 10). Several scenarios are examined that all lead to the same rate of decrease of pool B while the upstream JSA flux is constant: case 1 (JSA = 10, JAB = 8, JBP = 10), case 2 (JSA = 10, JAB = 10, JBP = 12), case 3 (JSA = 10, JAB = 12, JBP = 14), and case 4 (JSA = 10, JAB = 2, JBP = 4).
Transcriptomic Changes in Response to the Stress Treatments
The metabolite analysis clearly indicates that oxidative stress leads to a major perturbation in the metabolic network. We wanted to understand whether these changes are driven by gene expression or if the transcriptome is instead responding to the metabolic perturbation. If it is the latter, we wanted to investigate the nature of the adaptive change in the metabolic transcriptome. To this end, we analyzed changes in the transcriptome following oxidative stress using a 29 K oligonucleotide microarray (http://www.ag.arizona.edu/microarray/). Two time points were analyzed: 2 and 6 h postimposition of the stress and compared to a control sample at the equivalent time point (for more details of array experimental design and analysis see “Materials and Methods”). The 2 h time point was chosen because the labeling data indicated that widespread perturbation of the metabolic network was apparent by this point (Fig. 3). The 6 h time point was chosen because the majority of the metabolite changes had stabilized at this stage (Fig. 2). The complete transcriptomic dataset is available in Supplemental Table S2. A total of 10,710 transcripts passed quality control criteria. Significantly altered transcripts (P < 0.05, false discovery rate [FDR] < 0.1%) were identified using a moderated one-sample t test (Smyth, 2004). Oxidative stress led to significant changes in 32 transcripts after 2 h and 1,063 after 6 h. The complete list of significantly altered transcripts is presented in Supplemental Table S2.
Changes in Metabolic Transcripts after 2 h of Oxidative Stress
Of the 32 transcripts significantly changed after 2 h of oxidative stress, none encode enzymes of metabolic proteins that are linked to the pathways that are the sites of the major metabolic perturbation (Table I). This strongly suggests that the metabolic change occurring following oxidative stress is not driven by changes in the metabolic transcriptome. Rather, the initial metabolic change is more likely a direct result of the oxidative insult leading to enzyme inactivation. Of the 32 altered transcripts at 2 h (30 of which were increased in abundance) most have regulatory or signaling functions. For example, several transcription factors were identified including MYB transcription factors, a bZIP transcription factor, and a putative ethylene-responsive transcriptional coactivator. Other regulatory transcripts include a PPR family transcript (PPR proteins are thought to play a role in regulation of organellar RNA processing [Lurin et al., 2004]), a calcium-dependent protein kinase, and a GTP-binding protein. Collectively these transcripts are likely to underlie the regulation of the more widespread gene expression changes that are evident after 6 h. Interestingly, the list does not contain previously identified regulatory genes that are commonly induced in response to a variety of abiotic stresses. For example, the ZAT12 transcription factor (Davletova et al., 2005) is not induced at either 2 or 6 h (Table I; Supplemental Table S2). Similarly two genes of unknown function (At2g21640 and At1g1920) identified as markers of a wide range of abiotic stresses (Gadjev et al., 2006) were not altered at either 2 or 6 h (Table I; Supplemental Table S2). However, other stress-responsive regulatory proteins such as a defensin (At2g43510) and WRKY transcription factors (Gadjev et al., 2006) were significantly induced at 6 h (Supplemental Table S2). The list of regulatory genes here represents potential targets for engineering improved tolerance of oxidative stress.
Table I.
Significantly altered transcripts after 2 h of oxidative stress
Transcript data was analyzed using a moderated one-sample t test to generate P values that were FDR corrected such that FDR was <0.1%. The transcript abundance ratios (oxidative stress/control) are shown for transcripts with a P value < 0.05. For comparison, the significance of the same transcripts after 6 h of oxidative stress is also shown. NS, Not significant; NA, not analyzed.
| Locus | Transcript Abundance Ratio
|
Gene Annotation | |
|---|---|---|---|
| 2 h | 6 h | ||
| At5g48070 | 8.5 | NS | Xyloglucan endotransglycosylase, putative |
| At3g30843 | 6.0 | NS | Hypothetical protein |
| At5g14730 | 5.9 | 4.6 | Putative protein |
| At5g46460 | 5.6 | NS | PPR repeat-containing protein |
| At2g36220 | 5.3 | NS | Expressed protein |
| At2g04135 | 5.3 | NS | Hypothetical protein |
| At3g29470 | 4.7 | NS | Hypothetical protein |
| At5g13650 | 4.5 | NS | GTP-binding protein typA (Tyr phosphorylated protein A) |
| At2g22590 | 4.5 | NS | Putative anthocyanidin-3-glucoside rhamnosyltransferase |
| At1g11905 | 4.3 | NS | Expressed protein |
| At1g10060 | 4.2 | NS | Branched chain amino acid transaminase |
| At5g14270 | 4.1 | NS | Global transcription factor group E |
| At1g25270 | 4.0 | NA | MtN21 nodulin protein, putative |
| At3g62420 | 4.0 | NS | bZIP family transcription factor |
| At3g24500 | 3.9 | NS | Ethylene-responsive transcriptional coactivator, putative |
| At1g68440 | 3.8 | NS | Expressed protein |
| At4g01360 | 3.5 | NS | Hypothetical protein |
| At5g10695 | 3.5 | 6.5 | Putative protein |
| At1g48330 | 3.4 | NS | Hypothetical protein |
| At5g02240 | 3.2 | NS | Putative protein |
| At5g15410 | 2.8 | NS | Cyclic nucleotide-regulated ion channel (CNGC2) |
| At2g38090 | 2.7 | NS | Putative MYB family transcription factor |
| At2g22710 | 2.6 | NS | Myb family protein |
| At1g25350 | 2.6 | NS | Gln-tRNA synthetase |
| At5g54940 | 2.6 | NS | Translation initiation factor-like protein |
| At1g56400 | 2.5 | NS | Hypothetical protein |
| At5g42570 | 2.4 | NS | Expressed protein |
| At2g41730 | 2.3 | NS | Hypothetical protein |
| At5g12480 | 2.1 | 0.6 | Calcium-dependent protein kinase-like protein |
| At4g38360 | 0.5 | NS | Putative protein |
| At1g69530 | 0.4 | NS | Expansin (At-EXP1) |
| At4g31290 | 0.2 | NA | Unknown protein |
Changes in Metabolic Transcripts after 6 h of Oxidative Stress
In contrast to the picture at 2 h, after 6 h of oxidative stress, there is widespread alteration in the abundance of metabolic transcripts (Supplemental Table S2). To gain an overview of these changes, the significantly altered transcripts at 6 h were displayed using MapMan software (Thimm et al., 2004; Usadel et al., 2005; Fig. 5). It can be seen that, in general, there was a decrease in transcripts encoding enzymes of inhibited pathways. For example, there were decreases in two transcripts encoding TCA cycle enzymes: At1g34430 and At2g13560 encoding subunits of pyruvate dehydrogenase and NAD-malic enzyme, respectively (Fig. 5; Supplemental Table S2). There were also wholesale decreases in transcripts encoding enzymes of amino acid biosynthesis (Fig. 5). The metabolic transcript changes provide striking evidence of a switch from anabolic to catabolic metabolism. Transcripts encoding the enzymes of a variety of anabolic pathways including lipid and amino acid biosynthesis and nucleotide metabolism are decreased in abundance at 6 h (Fig. 5). At the same time, there are coordinated increases in transcripts associated with lipid and amino acid breakdown (Fig. 5). Thus, alternative internal stores of carbon are being mobilized to provide additional respiratory substrates to supplement those normally produced by the TCA cycle. The transcriptome data also suggests that the mitochondrial electron transport chain is being reconfigured with a general decrease in abundance of transcripts encoding cytochrome pathway components and an increase in external NADH dehydrogenases and ATP synthase subunits (Fig. 5). This probably occurs to bypass oxidatively damaged electron transport chain components (Sweetlove et al., 2002) and to overcome TCA cycle inhibition by allowing cytosolic NADH to be used as a source of electrons for respiration.
Figure 5.

Metabolic transcripts that are significantly altered after 6 h of oxidative stress. For details of statistical tests see legend to Table I. The abundance ratio (log [2] abundance treatment/control) of significantly altered transcripts (P < 0.05, FDR < 0.1%) are shown using the color scale indicated on the diagram. Key genes are labeled. DHAR, Dehydroascorbate reductase; GPX, glutathione peroxidase; GS, glutathione synthetase; MDHAR, monodehydroascorbate reductase.
There was also evidence of a transcriptomic response in the antioxidant gene network after 6 h of oxidative stress. For example, several genes of the ascorbate-glutathione cycle, including a dehydroascorbate reductase (At1g75270), a monodehydroascorbate reductase (At5g03630), and a glutathione synthase (At5g27380) are strongly induced (Fig. 5; Supplemental Table S2). There were a number of other antioxidant-related changes, including a significant increase in several glutathione-S transferases (Supplemental Table S2) that are known to be involved in stress responses and detoxification (Dixon et al., 2002), a glutathione peroxidase (At4g11600), and several oxidoreductases (Supplemental Table S2).
Many of the metabolic transcriptomic changes we observed here were not apparent in a previous microarray study of oxidative stress in Arabidopsis cell cultures (Desikan et al., 2001). For example, in the Desikan et al. (2001) study, no significant differences were found in the expression of TCA cycle enzymes or enzymes of amino acid or lipid metabolism. This is probably a reflection of the different means of inducing oxidative stress; the Desikan study used exogenous H2O2 whereas we used menadione to generate superoxide. Application of exogenous H2O2 may generate a very mild intracellular oxidative stress in comparison to menadione due to the activity of extracellular peroxidases (Amor et al., 2000) and will predominantly affect extramitochondrial subcellular compartments. There may also be specific differences in the response to superoxide and H2O2 (Gadjev et al., 2006).
Analysis of Transcriptomic Change by Functional Class Sorting
Although the t statistic used here gives a robust measure of significant differences, it may miss more subtle changes in the transcriptome. Two alternative statistical tests were therefore used to identify such changes. First, functional class scoring (Pavlidis et al., 2002) was used to identify groups of functionally related transcripts in which there was an overall coordinated change (Table II). In this analysis, relatively few significantly altered functional classes of genes were regulated in the same way at the 2 and 6 h time points, indicating a distinct temporal response. Of the classes that showed similar regulation at both time points, there was evidence for sustained down-regulation of nucleotide and DNA metabolism and a sustained induction of transcripts related to development and those of a NAC domain transcription factor family. The NAC domain transcription factor is plant specific and is known to play a role in the response of plants to stress (Hu et al., 2006). The bZIP class of stress-related transcription factors was also up-regulated, but only after 2 h, confirming the results of the gene-by-gene t test analysis (Table I). Of the metabolic classes, there were several changes that only occurred after 6 h that also confirmed the t test analysis (Table II; Fig. 5; Supplemental Table S2), including a decrease in transcripts associated with purine metabolism and protein synthesis and an increase in those associated with sulfur assimilation. However, there were other changes in metabolic classes that occurred after 2 h and were not apparent in the t test analysis. These included the TCA cycle-associated transcripts and those involved in amino acid biosynthesis, both of which showed a significant decrease in transcript abundance across the functional class after 2 h. Thus, for TCA cycle and amino acid biosynthesis there is a coordinated decrease of transcripts at 2 h but no significant change in individual transcripts (as judged by t test), whereas at 6 h there is no coordinated change in transcripts but a significant decrease of specific individual transcripts. To a certain extent this merely reflects the different nature of the two statistical tests but may also suggest that there is a subtle and more general early response that occurs across the metabolic pathways and specific changes directed at specific parts of the pathways that occurs later.
Table II.
Significantly altered functional classes of transcripts
Genes were sorted into bins based on the ontology developed for the MapMan software and significantly altered bins were identified by functional class sorting. In each case, it is indicated whether the overall change of transcript abundance in that bin relative to control is an increase (up) or decrease (down). For each significant bin a P value is shown. NS, Not significant (P > 0.05).
| Bin | Name | Bin Size | 2 h
|
6 h
|
||
|---|---|---|---|---|---|---|
| Change | P | Change | P | |||
| 8 | TCA/org. transformation | 76 | Down | 0.005 | NS | 0.217 |
| 8.1 | TCA/org. transformation.TCA | 42 | Down | 0.004 | NS | 0.074 |
| 13 | Amino acid metabolism | 297 | Down | 0.001 | NS | 0.765 |
| 14 | S-assimilation | 13 | NS | 0.441 | Up | 0.001 |
| 16.2 | Secondary metabolism. phenylpropanoids | 100 | Down | 0.001 | NS | 0.992 |
| 23 | Nucleotide metabolism | 164 | Down | 0.041 | Down | 0.001 |
| 23.1 | Nucleotide metabolism. synthesis | 37 | Down | 0.031 | Down | 0.001 |
| 26.03 | Misc.gluco- galacto- and mannosidases | 102 | Down | 0.001 | NS | 0.290 |
| 26.1 | Misc.cytochrome P450 | 249 | Up | 0.013 | Up | 0.001 |
| 26.21 | Misc.protease inhibitor/seed storage/lipid transfer protein (LTP) family protein | 88 | Down | 0.008 | Down | 0.001 |
| 28 | DNA | 3,065 | Down | 0.010 | Down | 0.001 |
| 29.7 | Protein.glycosylation | 22 | Down | 0.006 | NS | 0.162 |
| 33 | Development | 583 | Up | 0.001 | Up | 0.006 |
| 33.99 | Development. unspecified | 554 | Up | 0.001 | Up | 0.008 |
| 34.1 | Transport.p- and v-ATPases | 45 | Down | 0.001 | NS | 0.653 |
| 13.1.6.1 | Amino acid metabolism.synthesis.aromatic aa.chorismate | 13 | Down | 0.001 | NS | 0.915 |
| 16.2.1 | Secondary metabolism. phenylpropanoids.lignin biosynthesis | 34 | Down | 0.001 | NS | 0.993 |
| 23.1.2 | Nucleotide metabolism.synthesis.purine | 17 | NS | 0.063 | Down | 0.001 |
| 27.3.27 | RNA.regulation of transcription.NAC domain transcription factor family | 108 | Up | 0.001 | Up | 0.001 |
| 27.3.35 | RNA.regulation of transcription.bZIP transcription factor family | 72 | Up | 0.001 | NS | 0.097 |
| 28.1.3 | DNA.synthesis/chromatin structure.histone | 44 | Up | 0.001 | NA | – |
| 29.2.3 | Protein.synthesis.initiation | 90 | NS | 0.051 | Down | 0.001 |
| 29.3.4 | Protein.targeting.secretory pathway | 104 | Down | 0.004 | NS | 0.845 |
| 29.3.4.99 | Protein.targeting.secretory pathway.unspecified | 56 | Down | 0.001 | NS | 0.793 |
| 29.5.11.2 | Protein.degradation.ubiquitin.proteasome | 62 | Down | 0.002 | NS | 0.650 |
Identification of Reporter Metabolites from 6 h Transcriptomic Changes
We also identified reporter metabolites: metabolites around which there is significant change in the abundance of transcripts connected to that metabolite (i.e. enzymes that produce and consume that metabolite; Patil and Nielsen, 2005; Table III). Several of the identified reporter metabolites are involved in amino acid biosynthesis, including aspartyl-4-P and Asp semialdehyde, which are both intermediates of Lys/homoserine biosynthesis. There were coordinated decreases of transcripts both upstream and downstream of both of these metabolites, indicating a strong and coordinated decrease in transcripts associated with this pathway. Arg was also identified as a reporter metabolite with a decrease in transcripts encoding enzymes that produce Arg and a decrease in transcripts encoding those that consume it. Other reporter metabolites of central metabolism were identified with evidence of a down-regulation of transcripts around the OPPP intermediate, Rib-5-P, and an increase in transcripts around Suc. The relationship between these transcriptional changes and the measured metabolic changes reveals whether the former are homeostatic in nature or whether they are designed to further alter metabolic flux (see “Discussion”). There were also changes in secondary metabolites with an increase in transcripts around folate and a decrease around spermidine and putrescine.
Table III.
Reporter metabolites (metabolite nodes around which significant changes in transcript abundance have occurred)
Expression change values are based on mean ratios of transcript abundance (treatment/control) for all enzymes that either produce or consume that metabolite. All data is 6 h after the imposition of the stress condition. ND, Not determined.
| Metabolite | P | Pathway | Mean Expression Change of Producing Enzymes | Mean Expression Change of Consuming Enzymes |
|---|---|---|---|---|
| Aspartyl-4-P | 0.0001 | Lys/homoserine synthesis | 0.6 | 0.6 |
| Asp semialdehyde | 0.008 | Lys/homoserine synthesis | 0.6 | 0.9 |
| Arg | 0.027 | Arg metabolism | 0.4 | 1.5 |
| Spermidine | 0.006 | Spermidine synthesis | 0.3 | ND |
| Putrescine | 0.044 | Spermidine synthesis | ND | 0.3 |
| Rib-5-P | 0.02 | Oxidative pentose phosphate | 0.6 | 0.5 |
| Dehydro-neo-pterin | 0.03 | Folate synthesis | 1.5 | 3.4 |
| Suc | 0.03 | Suc metabolism | 2.5 | 3.2 |
DISCUSSION
Oxidative Stress Imposes Profound Restrictions on Primary Metabolic Pathways
In this study we followed changes in metabolite abundance and labeling patterns to assess the metabolic phenotype caused by oxidative stress induced by addition of menadione. Even though only mild oxidative stress was induced (we used a concentration of menadione of 60 μm, considerably lower than the 400 μm required to cause oxidative degradation of mitochondrial proteins in the same cell culture line; Sweetlove et al., 2002), a pronounced metabolic phenotype was uncovered. This phenotype seemed to originate in the mitochondrion with an inhibition of the TCA cycle. This is not unexpected. One of the main sites of ROS production upon addition of menadione to nonphotosynthetic cells is the mitochondrion (Thor et al., 1982) and a number of TCA cycle enzymes including aconitase and the lipoic acid-containing subunits of pyruvate and 2-oxogutarate dehydrogenase are known to be sensitive to oxidative inactivation. What this study does emphasize, however, is the far-reaching consequences of TCA cycle inhibition. In particular, the ability of the cell to maintain pools of amino acids whose synthesis depends upon TCA cycle intermediates is severely compromised. Oxidative stress also led to decreases in amino acids such as Gly, Ser, and Ala that are not directly connected to the TCA cycle. Collectively, these metabolic inhibitions will have a major impact upon the continued functioning and survival of the cell: inhibition of the TCA cycle will restrict ATP generation and a failure to maintain the pools of proteinogenic amino acids will ultimately prevent synthesis of new proteins.
Cells Respond by Implementing an Emergency Survival Strategy
Although the metabolic consequences of the menadione treatment are severe, they do not kill the cells that ultimately continue to grow and divide. Clearly this is not possible in the state of metabolic inhibition we have described, implying that the cells are able to respond and adapt to the stress situation. Using transcriptomic analysis we have been able to probe this response. Focusing on the metabolic transcriptome, we have uncovered dramatic changes in gene expression that will lead to a coordinated reconfiguration of the metabolic network. Taken together, these changes can be seen as an organized and decisive emergency response that gives the cell the best chance of surviving the oxidative insult. First, transcripts encoding enzymes of inhibited pathways such as the TCA cycle and amino acid biosynthesis are down-regulated, the strategy seemingly being one of avoiding wastage of energy in producing proteins that will only be immediately oxidatively damaged or will have no function in anabolic pathways starved of precursors. Second, there is wholesale induction of transcripts of catabolic pathways (such as lipid and amino acid breakdown) that will lead to a mobilization of internal carbon reserves. This is coupled with a reorganization of the respiratory pathways to permit ATP to be derived from these catabolic processes in the absence of the normal respiratory route through the TCA cycle. Finally, an antioxidant stress response is mounted, with increased expression of key antioxidant enzymes and a rerouting of glycolytic carbon flow into the OPPP possibly to provide reductant for this antioxidant effort.
Gene Expression for Metabolic Homeostasis
The identification of reporter metabolites is a powerful way of identifying transcriptomic change that is centered on a particular metabolite. However, the identification of such transcriptional changes alone does not necessarily reveal the purpose of the change. Are they homeostatic or are they designed to alter metabolic flux or metabolite levels? By comparison of identified reporter metabolites with the metabolite abundance and labeling changes, it is possible to identify homeostatic regulation of metabolism. For example, the OPPP intermediate Rib-5-P is a reporter metabolite with decreases in abundance of transcripts encoding enzymes both upstream and downstream of this metabolite at 6 h. The amount of Rib-5-P initially increases but then returns back to its initial levels. This suggests that the transcriptomic changes around Rib-5-P are designed to balance the oxidative and nonoxidative branches of the OPPP and bring metabolite levels back to their initial steady-state levels. Another example of homeostatic transcriptional regulation occurs around Arg. Arg is also a reporter metabolite, but in this case pool size is unaltered. The transcriptional change around this metabolite is reciprocal with a decrease in upstream transcripts and an increase in downstream transcripts. This suggests that the transcriptomic changes act to maintain a constant Arg pool size in the face of a reduced flux into this amino acid.
The Metabolic Response of Plant Cells to Oxidative Stress Is Remarkably Similar to That of Microbial Systems
In bacteria and yeast, exposure to mild oxidative stress leads to induction of specific genes that allow adaptation such that subsequent more severe oxidative stresses can be tolerated (Pomposiello and Demple, 2002). In Escherichia coli, the expression of these genes is regulated by two ROS-sensitive transcription factors, OxyR and SoxR/S (Liu et al., 2005). Although many oxidative stress-responsive promoters have been identified in plants, specific ROS regulatory DNA sequences and their cognate transcription factors have yet to be identified and this remains a major goal of plant stress research. A proteomic analysis of the response of yeast metabolism to low doses of H2O2 revealed a pronounced adaptive shift in metabolism (Godon et al., 1998) that is remarkably similar to the one we present in this article. In yeast, it was observed that enzymes associated with the TCA cycle and glycolysis were repressed while those catalyzing reactions of the OPPP were induced, indicating a redirection of carbon from respiration and into the OPPP for NADPH production. In addition, enzymes of the polyamine pathway and those involved in the synthesis of a range of amino acids were repressed. We also observed suppression of TCA cycle, a possible induction of the oxidative branch of the OPPP as well as suppression of amino acid synthesis and suppression of synthesis of the polyamine, spermidine (Table III). This observation of a common metabolic response to mild oxidative stress in Arabidopsis and yeast provides a new basis for comparative research aimed at identifying the regulatory components of a ROS signaling pathway in plants.
Summary
This article presents a detailed and comprehensive analysis of the short-term effects of oxidative stress on the metabolism of heterotrophic Arabidopsis cells and analyzes the transcriptomic response to those changes. In addition to metabolomic analysis, the value of analysis of 13C-labeling kinetics in the interpretation of the metabolic phenotype is demonstrated. Based on this analysis it is clear that oxidative stress has an immediate and severe inhibitory impact on several central metabolic pathways. We provide evidence that the extent of metabolic perturbation is much more widespread than previously appreciated. To cope with the resultant metabolic restrictions there is a dramatic change in the abundance of transcripts involved in metabolism, that serves both to mobilize alternative carbon reserves and to reconfigure metabolic fluxes to bypass some of the inhibited pathways. Finally, the discovery of a close similarity between the metabolic and transcriptomic response of Arabidopsis cells and microbial systems to oxidative stress will provide new impetus to comparative research aimed at identifying the key regulatory molecules involved.
MATERIALS AND METHODS
Cell Suspension Cultures
Arabidopsis (Arabidopsis thaliana), ecotype Landsberg erecta, suspension cultures (May and Leaver, 1993) were maintained in Murashige and Skoog basal medium (Duchefa) containing 0.5 mg L−1 naphthaleneacetic acid, 0.05 mg L−1 kinetin, and 3% (w/v) Glc, pH 5.6. The cell suspension cultures were maintained in 250 mL conical flasks in the dark at 22°C in an orbital shaker (150 rpm). At 4 d, each flask contained 8 to 10 g fresh weight cells and growth was approximately in the middle of the log phase. Subculture of 15 mL of culture to 100 mL of fresh media was conducted every 7 d.
Imposition of Stress Conditions, Introduction of 13C Label, and Sampling
Oxidative stress conditions were induced by the addition of menadione. The effectiveness of the stress treatment was monitored by measuring the activity of aconitase (Smith et al., 2004) throughout a 24 h time course. Growth of the cultures was assessed prior to and during all experiments by monitoring the optical density (OD600) of the cells. In addition, cell viability was determined using the method described by Swidzinski et al. (2002). Living cells, stained with the vital stain fluorescein diacetate and dead, nonfluorescent cells were counted using fluorescence microscopy. A minimum of 200 cells were counted for each measurement. To provide starting material for replicated metabolite and transcript experiments 1.2 L of cell suspension culture was produced for each individual replicate. This provided sufficient material for sampling for GC-MS and microarray analysis. Thus, data from the two analytical methods are directly comparable because they are derived from the exact same cell culture. To account for flask to flask variability, suspension cultures for different replicates came from independently subbed mother cultures. Stress conditions were imposed in 4-d-old cultures by allowing the cells to settle from the original culture medium and resuspending them in fresh medium containing 60 μm menadione and [U-13C-Glc] to a constant OD600 of 0.6. The Glc and nutrient content of the replacement suspension medium was adjusted to match that of 4-d-old cultures. Glc in the replacement suspension medium was 13C Glc (106 mm 13C6 Glc [99% atom 13C]; Sigma-Aldrich). Cells from each treatment, replicate, and time point (0, 1, 2, 3, 4, 5, and 6 h) were collected at staggered intervals by filtration and snap frozen in liquid nitrogen.
GC-MS Analysis of 13C Labeling and Metabolite Abundance
Metabolites for GC-MS analysis were extracted using a methanol/chloroform extraction procedure. A total of 200 mg of powdered frozen Arabidopsis cell suspension material was homogenized in 1,400 μL of 100% (v/v) methanol with 60 μL of ribitol (200 mg L−1) as an internal standard. Extracts were incubated for 15 min at 70°C on an orbital shaker (100 rpm) and then centrifuged at 4°C, 12,000g for 10 min. The supernatant was transferred to a new tube and mixed with 750 μL chloroform and 1,500 μL water. Extracts were centrifuged at 4°C, 12,000g for 15 min and aliquots of the resulting polar (upper) phase were taken for further analysis. A total of 100 μL and 200 μL aliquots of the polar phase were freeze dried and metabolites were subsequently derivatized and analyzed using an established protocol (Roessner et al., 2000). The chromatograms and mass spectra were evaluated using the Masslab program (ThermoQuest). Metabolite abundances and the 13C percentage enrichments of metabolites were determined using previously reported methods (Roessner et al., 2000; Roessner-Tunali et al., 2004). Briefly, 13C enrichment was calculated by measuring the changing abundance of isotopic spectral fragments of the molecular ion of each metabolite. Natural abundance of 13C was corrected for by reference to an unlabeled control sample. The percentage label present in each metabolite was calculated from the total abundance of 12C and 13C ions in a particular metabolite pool.
Modeling of 13C Labeling
A linear system is envisaged in which a 13C-labeled substrate (S) is converted to a product (P) via two sequential intermediates, A and B, where JSA is the flux of S to A, JAB of A to B, and JBP of B to P.
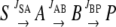 |
For intermediates A and B, the following mass balance equations can be written:
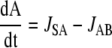 |
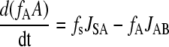 |
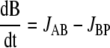 |
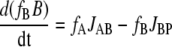 |
where fs, fA, and fB are the fractional enrichment of label in S, A, and B, respectively. fs is assumed to be fixed and was given an arbitrary value of 0.1. If the fluxes are constant (although not necessarily equal) during the time interval being considered, then a time-dependent fA and fB can be computed. In all calculations the initial value of both A and B was 60 and the initial value of both fA and fB was 0. The fluxes were set according to the case being examined. The computation was done using Euler's method.
Microarray Analysis
A common reference experimental design was used in which each sample was compared to the same 0 h control sample. Four replicates of samples from the control and menadione treatment at 2 and 6 h were analyzed, with two replicates in each case being dye swapped. Total RNA was isolated from homogenized, powdered Arabidopsis cells using a TRIzol (Invitrogen) method (Giegé et al., 2005). Glass slides containing elements from the Qiagen-Operon Arabidopsis genome array ready oligo set (AROS) version 3.0 were obtained directly from David Galbraith (University of Arizona). The Arabidopsis array contains 29,000 oligonucleotides. Technical details of the spotting of the slides and annotation of the array elements are provided on the University of Arizona Web site (http://www.ag.arizona.edu/microarray/). Total RNA was reverse transcribed and labeled with Cy-dCTP (Amersham) using standard protocols (Baxter et al., 2005). Oligonucleotide array elements were immobilized according to the manufacturer's protocol (http://ag.arizona.edu/microarray/methods.html) on the day of hybridization. For hybridization, pelleted, labeled cDNA was resuspended in 19 μL of hybridization solution (2× SSC, 0.08% [w/v] SDS) and the Cy3- and Cy5-labeled probes were combined. A total of 2.4 μL of liquid block solution (GE Healthcare) was added to the labeled probes and these were denatured at 95°C for 2 min. Probe solution was added to the spotted surface of the slide and hybridization carried out under a hybri-slip (Sigma-Aldrich Chemie GmbH) in a humidified hybridization cassette (Telechem International) for 16 h at 55°C. Following hybridization, slides were washed for 5 min at 55°C in 2× SSC, 0.1% (w/v) SDS, 5 min at room temperature in 0.1× SSC, 0.1% SDS, and 5 min at room temperature in 0.1× SSC. Microarrays were scanned using an Affymetrix 428 Array scanner (Affymetrix) and acquisition software according to the manufacturer's instructions. After, scanning images were analyzed in Genepix Pro v. 4.1 software (Axon Instruments). Print-tip loess normalization was utilized as implemented in Smyth (2004). Quality control of the microarrays was done using the ArrayMagic package (Buness et al., 2005). Background subtraction was deemed necessary for reduction of regional bias according to the guidelines given in Scharpf et al. (2005). Spots flagged as present by GenePix in fewer than 10 of the 16 arrays were discarded. Spots with more than two missing values for either time point were not tested for differential expression. Following data normalization and quality control all values were expressed relative to the 0 h reference value and ratios were log2 transformed prior to further analysis.
Statistical Analysis of Microarray Data
To gain a qualitative overview of transcript changes microarray data was visualized using MapMan (http://gabi.rzpd.de/projects/MapMan/; Thimm et al., 2004). Data from individual time points of oxidative stress treatments were directly compared to that of control cultures by creating a log ratio of treatment versus control data. These datasets were sorted into functional category bins and visualized using the MapMan program. To identify bins with a significantly high proportion of up- or down-regulated genes a mean log p approach was utilized (Pavlidis et al., 2002) that was recently shown to be preferrable to discrete overrepresentation tests (Allison et al., 2006; Raghavan et al., 2006). An arbitrary statistic for differential expression was formed as sd=−clog(p) where p is the P value from hypothesis test assessing the differential expression and c is −1 if the transcript is down-regulated and 1 if its up-regulated. To test a bin the average sd was calculated and its significance determined by taking 1e5 samples of the same size from the rest of the transcripts. Bootstrap P values were calculated as the amount of random samples larger than the observed average sd divided by the amount of random samples drawn. Reporter metabolites were identified from transcript data utilizing metabolic network topology and the algorithm described by Patil and Nielsen (2005) using a metabolite and enzyme interaction graph constructed from information in the Aracyc database (Zhang et al., 2005). Ratios of 6 h transcript data (6/0 h transcripts) were used to identify reporter metabolites between treatments.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Optimization and characterization of oxidative stress treatment.
Supplemental Figure S2. Time series of selected metabolite abundances following oxidative stress treatment.
Supplemental Figure S3. Time series of 13C labeling of selected metabolites in cells incubated with [U-13C]Glc.
Supplemental Table S1. Complete dataset of relative metabolite abundance over a 6 h time course in control cells and cells treated with menadione.
Supplemental Table S2. Transcript abundance changes after 2 h and 6 h of menadione treatment analyzed by microarray.
Supplementary Material
Acknowledgments
The authors would like to thank Bjoern Usadel, Max Planck Institute for Molecular Plant Physiology for assistance with the MapMan analysis, and Professor George Ratcliffe, Department of Plant Sciences, University of Oxford for useful discussions about interpretation of 13C-labeling kinetics.
This work was supported by the Biotechnology and Biological Sciences Research Council, United Kingdom (to L.J.S.), the Bundesministerium für Bildung und Forschung in the framework of a Deutsche Israeli Project award and by the Max Planck Gesellschaft (to N.S. and A.R.F.), the Center for Microbial Biotechnology, who receive funding from the Danish Agency for Science, Technology, and Innovation (to K.P.), and the Scottish Executive Environment and Rural Affairs Department (to J.L.).
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Lee J. Sweetlove (lee.sweetlove@plants.ox.ac.uk).
The online version of this article contains Web-only data.
References
- Allison DB, Cui X, Page GP, Sabripour M (2006) Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet 7 55–65 [DOI] [PubMed] [Google Scholar]
- Amor Y, Chevionb M, Levinea A (2000) Anoxia pretreatment protects soybean cells against H(2)O(2)-induced cell death: possible involvement of peroxidases and of alternative oxidase. FEBS Lett 477 175–180 [DOI] [PubMed] [Google Scholar]
- Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55 373–399 [DOI] [PubMed] [Google Scholar]
- Baxter CJ, Sabar M, Quick WP, Sweetlove LJ (2005) Comparison of changes in fruit gene expression in tomato introgression lines provides evidence of genome-wide transcriptional changes and reveals links to mapped QTLs and described traits. J Exp Bot 56 1591–1604 [DOI] [PubMed] [Google Scholar]
- Buness A, Huber W, Steiner K, Sültmann H, Poustka A (2005) Arraymagic: two-colour cdna microarray quality control and preprocessing. Bioinformatics 21 554–556 [DOI] [PubMed] [Google Scholar]
- Davletova S, Schlauch K, Coutu J, Mittler R (2005) The zinc-finger protein Zat12 plays a central role in reactive oxygen and abiotic stress signaling in Arabidopsis. Plant Physiol 139 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan R, A-H-Mackerness S, Hancock JT, Neill SJ (2001) Regulation of the Arabidopsis transcriptome by oxidative stress. Plant Physiol 127 159–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon DP, Lapthorn A, Edwards R (2002) Plant glutathione transferases. Genome Biol 3 REVIEWS3004 [DOI] [PMC free article] [PubMed]
- Fell DA (2005) Enzymes, metabolites and fluxes. J Exp Bot 56 267–272 [DOI] [PubMed] [Google Scholar]
- Fernie AR, Geigenberger P, Stitt M (2005) Flux: an important, but neglected component of functional genomics. Curr Opin Plant Biol 8 174–182 [DOI] [PubMed] [Google Scholar]
- Gadjev I, Vanderauwera S, Gechev TS, Laloi C, Minkov IN, Shulaev V, Apel K, Inze D, Mittler R, Van Breusegem F (2006) Transcriptomic footprints disclose specificity of reactive oxygen species signaling in Arabidopsis. Plant Physiol 141 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giegé P, Sweetlove LJ, Cognat V, Leaver CJ (2005) Coordination of nuclear and mitochondrial genome expression during mitochondrial biogenesis in Arabidopsis. Plant Cell 17 1497–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godon C, Lagniel G, Lee J, Buhler JM, Kieffer S, Perrot M, Boucherie H, Toledano MB, Labarre J (1998) The H2O2 stimulon in Saccharomyces cerevisiae. J Biol Chem 273 22480–22489 [DOI] [PubMed] [Google Scholar]
- Halliwell B (2006) Reactive species and antioxidants: redox biology is a fundamental theme of aerobic life. Plant Physiol 141 312–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock JT, Henson D, Nyirenda M, Desikan R, Harrison J, Lewis M, Hughes J, Neill SJ (2005) Proteomic identification of glyceraldehyde 3-phosphate dehydrogenase as an inhibitory target of hydrogen peroxide in Arabidopsis. Plant Physiol Biochem 43 828–835 [DOI] [PubMed] [Google Scholar]
- Harada K, Fukusaki E, Bamba T, Sato F, Kobayashi A (2006) In vivo 15N-enrichment of metabolites in suspension cultured cells and its application to metabolomics. Biotechnol Prog 22 1003–1011 [DOI] [PubMed] [Google Scholar]
- Hu H, Dai M, Yao J, Xiao B, Li X, Zhang Q, Xiong L (2006) Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc Natl Acad Sci USA 103 12987–12992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Colavitti R, Rovira II, Finkel T (2005) Redox-dependent transcriptional regulation. Circ Res 97 967–974 [DOI] [PubMed] [Google Scholar]
- Lurin C, Andres C, Aubourg S, Bellaoui M, Bitton F, Bruyere C, Caboche M, Debast C, Gualberto J, Hoffmann B, et al (2004) Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16 2089–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May M, Leaver C (1993) Oxidative stimulation of glutathione synthesis in Arabidopsis thaliana suspension cultures. Plant Physiol 103 621–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittler R (2002) Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci 7 405–410 [DOI] [PubMed] [Google Scholar]
- Mittler R (2006) Abiotic stress, the field environment and stress combination. Trends Plant Sci 11 15–19 [DOI] [PubMed] [Google Scholar]
- Mittler R, Vanderauwera S, Gollery M, Van Breusegem F (2004) Reactive oxygen gene network of plants. Trends Plant Sci 9 490–498 [DOI] [PubMed] [Google Scholar]
- Moller IM, Kristensen BK (2006) Protein oxidation in plant mitochondria detected as oxidized tryptophan. Free Radic Biol Med 40 430–435 [DOI] [PubMed] [Google Scholar]
- Morgan JA, Rhodes D (2002) Mathematical modeling of plant metabolic pathways. Metab Eng 4 80–89 [DOI] [PubMed] [Google Scholar]
- Patil KR, Nielsen J (2005) Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc Natl Acad Sci USA 102 2685–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Lewis DP, Noble WS (2002) Exploring gene expression data with class scores. Pac Symp Biocomput 474–485 [PubMed]
- Pomposiello PJ, Demple B (2002) Global adjustment of microbial physiology during free radical stress. Adv Microb Physiol 46 319–341 [DOI] [PubMed] [Google Scholar]
- Raamsdonk LM, Teusink B, Broadhurst D, Zhang N, Hayes A, Walsh MC, Berden JA, Brindle KM, Kell DB, Rowland JJ, et al (2001) A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat Biotechnol 19 45–50 [DOI] [PubMed] [Google Scholar]
- Raghavan N, Amaratunga D, Cabrera J, Nie A, Qin J, McMillian M (2006) On methods for gene function scoring as a means of facilitating the interpretation of microarray results. J Comput Biol 13 798–809 [DOI] [PubMed] [Google Scholar]
- Ratcliffe RG, Shachar-Hill Y (2006) Measuring multiple fluxes through plant metabolic networks. Plant J 45 490–511 [DOI] [PubMed] [Google Scholar]
- Roessner-Tunali U, Liu J, Leisse A, Balbo I, Perez-Melis A, Willmitzer L, Fernie AR (2004) Kinetics of labelling of organic and amino acids in potato tubers by gas chromatography-mass spectrometry following incubation in C labelled isotopes. Plant J 39 668–679 [DOI] [PubMed] [Google Scholar]
- Roessner U, Wagner C, Kopka J, Trethewey RN, Willmitzer L (2000) Simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J 23 131–142 [DOI] [PubMed] [Google Scholar]
- Scharpf R, Iacobuzio-Donahue C, Sneddon J, Parmigiani G (2005) When should one subtract background florescence in two color microarrays. Johns Hopkins University, Department of Biostatistics Working Papers. Working Paper 50. http://www.bepress.com/jhubiostat/ [DOI] [PubMed]
- Smith AMO, Ratcliffe RG, Sweetlove LJ (2004) Activation and function of mitochondrial uncoupling protein in plants. J Biol Chem 279 51944–51952 [DOI] [PubMed] [Google Scholar]
- Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3 Article3 [DOI] [PubMed]
- Sweetlove LJ, Heazlewood JL, Herald V, Holtzapffel R, Day DA, Leaver CJ, Millar AH (2002) The impact of oxidative stress on Arabidopsis mitochondria. Plant J 32 891–904 [DOI] [PubMed] [Google Scholar]
- Swidzinski JA, Sweetlove LJ, Leaver CJ (2002) A custom microarray analysis of gene expression during programmed cell death in Arabidopsis thaliana. Plant J 30 431–446 [DOI] [PubMed] [Google Scholar]
- Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37 914–939 [DOI] [PubMed] [Google Scholar]
- Thor H, Smith MT, Hartzell P, Bellomo G, Jewell SA, Orrenius S (1982) The metabolism of menadione (2-methyl-1,4-naphthoquinone) by isolated hepatocytes: a study of the implications of oxidative stress in intact cells. J Biol Chem 257 12419–12425 [PubMed] [Google Scholar]
- Tiwari BS, Belenghi B, Levine A (2002) Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol 128 1271–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel B, Nagel A, Thimm O, Redestig H, Blaesing OE, Palacios-Rojas N, Selbig J, Hannemann J, Piques MC, Steinhauser D, et al (2005) Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol 138 1195–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verniquet F, Gaillard J, Neuburger M, Douce R (1991) Rapid inactivation of plant aconitase by hydrogen peroxide. Biochem J 276 643–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner D, Przybyla D, Op den Camp R, Kim C, Landgraf F, Lee KP, Wursch M, Laloi C, Nater M, Hideg E, et al (2004) The genetic basis of singlet oxygen-induced stress responses of Arabidopsis thaliana. Science 306 1183–1185 [DOI] [PubMed] [Google Scholar]
- Zhang P, Foerster H, Tissier CP, Mueller L, Paley S, Karp PD, Rhee SY (2005) MetaCyc and AraCyc: metabolic pathway databases for plant research. Plant Physiol 138 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.