

Abstract
The structure of the NH2-terminal region of troponin T (TnT) is hypervariable among the muscle type-specific isoforms and is also regulated by alternative RNA splicing. This region does not contain binding sites for other thin filament proteins but alteration of its structure affects the Ca2+-regulation of muscle contraction. Here we report a truncated cardiac TnT produced during myocardial ischemia-reperfusion. Amino acid sequencing and protein fragment reconstruction determined that it is generated by a posttranslational modification selectively removing the NH2-terminal variable region and preserving the conserved core structure of TnT. Triton X-100 extraction of cardiac muscle fibers promoted production of the NH2-terminal truncated cardiac TnT (cTnT-ND), indicating a myofibril-associated proteolytic activity. μ-Calpain is a myofibril-associated protease and is known to degrade TnT. Supporting a role of μ-calpain in producing cTnT-ND in myocardial ischemia-reperfusion, calpain inhibitors decreased the level of cTnT-ND in Triton-extracted myofibrils. μ-Calpain treatment of cardiac myofibril and troponin complex specifically reproduced cTnT-ND. In contrast, μ-calpain treatment of isolated cardiac TnT resulted in non-specific degradation, suggesting that this structural modification is relevant to physiological structures of the myofilament. Triton X-100 treatment of transgenic mouse cardiac myofibrils over-expressing fast skeletal muscle TnT produced similar NH2-terminal truncations of the endogenous and exogenous TnT, despite different amino acid sequences at the cleavage site. With the functional consequences of removing the NH2-terminal variable region of TnT, the μ-calpain-mediated proteolytic modification of TnT may act as an acute mechanism to adjust muscle contractility under stress conditions.
Cardiac and skeletal muscle contraction is activated by Ca2+ via troponin-tropomyosin in the actin thin filament regulatory system (1–3). Troponin T (TnT) 1 is the anchoring subunit of the troponin complex (4). Three muscle type-specific TnT isoform genes have evolved in higher vertebrates (5–7) and alternative RNA splicing further produces multiple protein isoforms (8–10). The various TnT isoforms mainly differ in their NH2-terminal structures. The amino acid sequence of the NH2-terminal region of TnT is hypervariable among the cardiac, slow and fast skeletal muscle TnT isoforms and regulated by alternative splicing during perinatal heart and muscle development (8, 9). The clearly regulated developmental switches from embryonic to adult TnT isoforms suggest a functional significance of the NH2-terminal structural variation of TnT.
Nonetheless, the NH2-terminal region of TnT does not contain any known binding sites for other thin filament proteins (11–13). Deleting the NH2-terminal variable region does not diminish the regulatory activity of troponin (14–16), suggesting that the NH2-terminal variable region of TnT may function as a modulatory structure. Supporting the hypothesis that alterations in TnT NH2-terminal structure affect the Ca2+-regulation of muscle contraction, a previous study demonstrated that NH2-terminal alternatively spliced TnT isoforms convey significant changes in the activation of actomyosin ATPase (17). Aberrant splicing of cardiac TnT (cTnT) in the NH2-terminal region is found in both hypertrophic and failing human hearts (18) and animal models with dilated cardiomyopathy (19, 20). Consistent with the functional effects, studies showed that NH2-terminal alterations in TnT affect the overall protein conformation (21, 22), and binding to tropomyosin, TnI and TnC (21, 23).
Altered TnT isoform gene expression (24) and alternative RNA splicing (18) have been found during muscle adaptation to stress conditions. In contrast to the relatively slow response at the gene regulation and RNA splicing levels, posttranslational regulation provides a mechanism for rapid adaptation to acute stress. The mostly prominently studied posttranslational mechanism of myofilament protein regulation is phosphorylation (25-27). Proteolysis is usually considered a deteriorating mechanism in muscle under physiological or pathological stress conditions (28, 29). However, restricted proteolysis of cardiac troponin I (TnI) with a deletion of the NH2-terminal phosphorylation sites has been found in rat cardiac muscle under simulated microgravity conditions (30). This specific structural modification of cardiac TnI has been demonstrated to enhance the relaxation of cardiac muscle as compensation to the decrease in cardiac preload in the microgravity model (31). It has been reported that NH2-terminal truncated fast skeletal muscle TnT is produced during postmortal proteolysis in rabbit (32) and porcine muscle (33). It is not known whether this modification occurs in vivo.
Calpain is a calcium activated cysteine protease that has been found to play regulatory modification of proteins (34). Two major calpain isoforms have been identified in muscle cells. The μ-calpain (calpain 1) requires micromolar concentrations of calcium to activate and the m-calpain (calpain 2) requires millimolar concentrations of calcium (34, 35). μ-Calpain is a myofibril-associated enzyme (34). It has been observed that μ-calpain can degrade TnT and TnI in vitro (36).
In the present study, we found a truncated cTnT produced during myocardial ischemia-reperfusion, a stress condition that results in cardiac muscle injuries (37). Amino acid sequencing and protein fragment reconstruction determined that it is generated by a posttranslational modification to selectively remove the NH2-terminal variable region and preserve the conserved core structure of TnT. Triton X-100 extraction of cardiac muscle fibers promoted the production of the NH2-terminal truncated cTnT (cTnT-ND), suggesting a myofibril-associated proteolytic activity. Supporting a role for μ-calpain in producing cTnT-ND, calpain inhibitors reduced cTnT-ND production in Triton-extracted myofibrils. μ-Calpain treatment of cardiac myofibril and troponin complex reproduced cTnT-ND. μ-Calpain treatment of isolated cTnT resulted in non-specific degradation, suggesting that this structural modification is relevant to physiological structures of the myofilament. Triton X-100 treatment of transgenic mouse cardiac myofibrils over-expressing fast skeletal muscle TnT produced similar NH2-terminal truncations of the endogenous and exogenous TnTs, despite the different amino acid sequences at the cleavage site, indicating that it is the myofilament structure that determines the specific cleavage. With the functional consequences of removing the NH2-terminal variable region of TnT, the μ-calpain-mediated proteolytic modification of TnT may act as an acute mechanism to adjust muscle contractility under stress conditions. In contrast to the commonly observed proteolytic destruction during ischemia-reperfusion injury (28), our finding demonstrates a novel specific modification of troponin structure as a potentially functional adaptation.
MATERIALS AND METHODS
Cardiac muscle tissues
Fresh bovine cardiac muscle was obtained from local slaughterhouse and kept on ice for ~1.5 hours before being frozen at -80 °C prior to use. Fresh rodent cardiac muscles were obtained from Sprague-Dawley rats, Balb/c mice and C57BL/6 transgenic mice that over-express the embryonic cTnT and/or exon 7-deleted cTnT (20) or chicken fast skeletal muscle TnT (38) in the heart.
Double transgenic mice expressing both embryonic and exon 7 deleted cTnT in the adult heart were produced by crossing between homozygous single transgenic parents bearing each of the transgene alleles. The F1 offspring was verified for the double transgenic genotype by PCR analysis of genomic DNA extracted from tail tissue as described previously (20).
All animal procedures were approved by the Institutional Animal Care and Use Committee and were conducted in accordance with the Guiding Principles in the Care and Use of Animals, as approved by the Council of the American Physiological Society.
Anti-TnT antibodies
A mouse monoclonal antibody (mAb) CT3 was previously developed by immunization with purified bovine cTnT (23). mAb CT3 cross-reacts with slow skeletal muscle TnT but not fast skeletal muscle TnT. The distinct mobility of cTnT and slow TnT in SDS-polyacrylamide gel electrophoresis (SDS-PAGE) allows an easy identification of cTnT in Western blots. The CT3 epitope has been mapped in the central region of TnT (23).
A polyclonal rabbit anti-TnT antiserum (RATnT) was previously generated by immunization using purified chicken breast muscle TnT as the antigen (21). The RATnT antiserum recognizing multiple epitopes on TnT strongly reacts with chicken fast skeletal muscle TnT and cross-reacts with avian and mammalian cardiac and slow skeletal muscle TnTs.
A mouse mAb (2C8) was previously developed by immunization with human cTnT (39). mAb 2C8 recognizes cardiac, slow, and fast TnTs almost equally in Western blots (39). The 2C8 mAb epitope is located in the central region of TnT.
SDS-PAGE and Western blotting
Ventricular muscle tissues or myocytes were homogenized in Laemmli SDS-PAGE sample buffer containing 2% SDS, heated at 80°C for 5 min, and clarified by centrifugation. Total protein extracts were resolved by 14% Laemmli gel with an acrylamide:bisacrylamide ratio of 180:1 (low cross-linker) or by 15% Laemmli gel with an acrylamide:bisacrylamide ratio of 29:1 (high cross-linker). The gels were stained with Coomassie Brilliant Blue R250 to reveal the resolved protein bands. Duplicate gels were electrically blotted to nitrocellulose membranes, as described previously (21). After blocking in Tris-buffered saline (TBS) containing 1% bovine serum albumin (BSA), the membrane was incubated with anti-TnT mAbs CT3, 2C8, or polyclonal antibody RATnT. The membranes were then washed with high stringency using TBS containing 0.5% Triton X-100 and 0.05% SDS, incubated with alkaline phosphatase-conjugated anti-mouse IgG or anti-rabbit IgG second antibodies (Sigma), washed again, and developed in 5-bromo-4-chloro-3-indolylphosphate/nitro blue tetrazolium substrate solution, as described previously (21).
Ex vivo ischemia-reperfusion of working rat heart preparations
The Langendorff-Neely working heart preparation was used to perfuse isolated rat hearts and apply ex vivo ischemia-reperfusion. As described previously (31), rats were anesthetized with sodium pentobarbital (50 mg/kg body weight, intraperitoneally). The heart was removed and placed in chilled Krebs-Henseleit Buffer (118 mM NaCl, 4.7 mM KCl, 2.25 mM CaCl2, 2.25 mM MgSO4, 1.2 mM KH2PO4, 0.32 mM EGTA, 11 mM D-glucose and 25 mM NaHCO3) aerated with 95% O2, 5% CO2 (pH 7.4 at 37°C). The aorta was cannulated with a 16-gauge needle and the apex of the heart placed in 37°C Krebs-Henseleit Buffer to maintain physiological temperature. The heart was then perfused with aerated and warmed Krebs-Henselite Buffer in the retrograde at a pressure of 70 mmHg for 15 min to stabilize the heart. During this period, the left atrium was cannulated with a 16-gauge atrial cannula set at a filling pressure of 15 mmHg. The heart was then converted to working mode by switching the tube delivering retrograde perfusion from 70 mmHg to an open column and initiating flow through the atrial cannula. The heart was stabilized in the working mode for 30 min at an afterload pressure of 55 mmHg. Following stabilization, low-flow ischemia was initiated by decreasing the afterload, and hence the coronary filling pressure, to 20 mmHg for 20 min. At a filling pressure of 20 mmHg coronary flow is insufficient to maintain adequate O2 delivery to the myocardium and the heart becomes ischemic. During ischemia the heart continued to beat weakly. Following the ischemic period coronary perfusion was resumed by returning the afterload to 55 mmHg for 40 min. At the end of the perfusion protocol the heart was removed from the cannula, flash frozen in liquid nitrogen and stored at −80°C for SDS-gel and Western blot analysis. Samples from 105 min working hearts without ischemia-reperfusion were used as controls.
As a control of simple myocardial ischemia, eight-week-old C57/BL6 mice were euthanized by cervical dislocation and the bodies placed at room temperature (22 °C) in a sealed plastic bag to prevent dehydration. The hearts were removed at 0, 2, 4, and 8 hours postmortem and homogenized in SDS-PAGE sample buffer for SDS-gel and Western blot analysis to determine degradation of cTnT resulting from postmortem ischemia.
Ischemia-reperfusion treatment of mouse cardiomyocytes
To induce ischemia-reperfusion damage in cardiac myocytes, mouse ventricular myocytes were isolated in low oxygen buffer followed by incubation in oxygenated buffer. Similar to that previously described (20), cardiomyocytes were isolated from transgenic mouse hearts over-expressing embryonic cTnT or exon 7-deleted cTnT by retrograde perfusion with Ca2+ free Joklik solution containing 1% BSA and collagenase without oxygenation. Following isolation, the cardiac myocytes were incubated at room temperature without oxygenation for 30 min before initiating re-oxygenation and returning Ca2+ to 1.25 mM stepwise over a 35 min period. Once Ca2+ had been restored oxygenation was continued and the myocytes incubated at room temperature for an additional 30 min before the myocytes were collected by centrifugation, washed in TBS and lysed in SDS-Sample Buffer for Western blot analysis.
Isolation of cTnT fragment for NH2-terminal sequencing
To investigate the primary structure of the ischemia-reperfusion-produced cTnT fragment, mouse cardiac myocytes treated with ischemia-reperfusion were homogenized in TBS and fractionated by ammonium sulfate precipitation. The 30–50% saturation fraction was dialyzed against three changes of 100 volumes of 0.1 mM EDTA at 4°C. After dialysis, the precipitated material was collected by centrifugation at 25,000 × g, 4°C, for 30 min. The cTnT fragment in the low salt precipitate was further purified by a two step preparative SDS-PAGE procedure. The sample was first resolved by electrophoresis on high cross-linker gel (12% Laemmli gel with an acrylamide-to-bisacrylamide ratio of 29:1). The resulting gel was stained with Coomassie Brilliant Blue R-250 and the band containing the cTnT fragment as determined by parallel Western blot using the CT3 mAb was cut out. The protein contents were recovered from the gel slices by electrophoresis elution in SDS-gel running buffer. After dialysis against 0.1% formic acid and concentrated by lyophilization, the protein sample was re-dissolved in SDS-gel sample buffer and further resolved by electrophoresis on low cross-linker SDS-PAGE (14% Laemmli gel with an acrylamide-to-bisacrylamide ratio of 180:1). The resulting gel was electronically transferred to PVDF membrane and stained with Amido Black to visualize the protein bands. A parallel strip of the membrane was subjected to Western blot using the anti-cTnT mAb CT3 as described above. The CT3 positive band of the Western blot was aligned to the Amido Black stained membrane and the corresponding band was excised for NH2-terminal sequencing at the Biotechnology Resource Laboratory Protein Sequencing and Peptide Synthesis Facility, Medical University of South Carolina, Charleston, SC.
Expression of TnT and reconstructed TnT fragment in E. coli
Intact mouse cTnT (TnT4) was expressed in E. coli culture. The construction of pAED4 expression plasmid from a cloned cDNA (40), large scale expression, and purification were done as described previously for the turkey cTnT (19).
A cDNA template encoding an NH2-terminal deleted mouse cTnT was engineered by polymerase chain reaction (PCR) mutagenesis to create a translational initiation codon prior to the cleavage site (Leu72) as determined by NH2-terminal sequencing. As shown in Figure 4, cloned adult mouse cTnT cDNA in pBluescript SK(−) plasmid (40) was used as template and PCR was carried out using a custom designed 5’ oligonucleotide primer that contained an NdeI restriction site (underlined), a translational initiation codon ATG, and the region complimentary to the coding sequence for amino acid 72–77 (McTnT-NDF: 5′– AGCCCCATATGCTCTTCATGCCCAACTT–3′). The PCR product was modified at the 5’ and 3’ ends by NdeI and XhoI cuts and cloned into the pAED4 expression plasmid (41). The cDNA insert was sequenced by dideoxy chain termination method to verify the construction and sequence authenticity.
FIGURE 4.
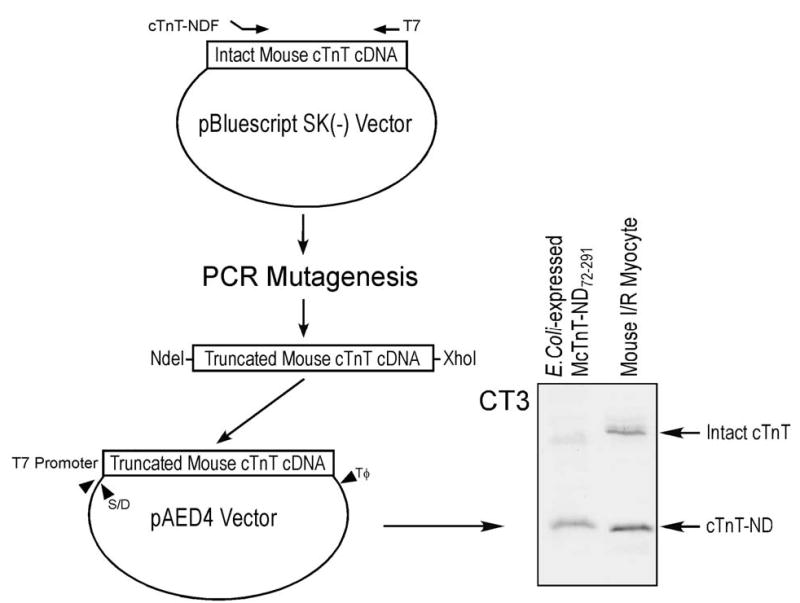
Bacterial expression of the reconstructed cTnT fragment. 5’-truncated mouse cTnT cDNA was constructed according to the NH2-terminal truncation site for protein expression in E. coli (see Materials and Methods). S/D and Tφ in the pAED4 expression vector indicate the Shine-Dalgarno ribosomal binding site and the transcription termination sequence, respectively. The cTnT fragment expressed from the truncated cDNA shows a size identical to that of the cTnT fragment produced in ischemia-reperfused cardiac muscle (the slightly slower gel mobility seen in the blot may be due to the addition of an NH2-terminal Met in the expression construct), indicating that the NH2-terminal truncation is the only primary structure modification.
The truncated mouse cTnT cDNA was expressed by transformation of BL21(DE3)pLyseS E. coli cells with the expression plasmid. Freshly transformed bacterial cells were cultured in 2x TY rich liquid media (16 g/L Tryptone, 10 g/L yeast extract, 5 g/L NaCl, 1.32 g/L Na2HPO4, pH 7.3) containing 100 mg/L ampicillin and 25 mg/L chloramphenicol at 37 °C with vigorous shaking and induced with 0.4 mM isopropyl-1-thiol-β-D-galactoside at mid-log phase. After three additional hours of culture, the bacterial cells were harvested by centrifugation at 4 °C. The bacterial pellet was suspended in 2.5 mM EDTA, 50 mM tris-HCl, pH 8.0 and lysed by three passes through a French Press cell. The bacterial lysate was clarified by centrifugation and precipitated with ammonium sulfate to obtain the 0–35% saturation fraction. Following dialysis against 0.1 mM EDTA containing 6 mM β-mercaptoethanol, the 0–35% fraction was brought to 6 M urea, 0.1 mM EDTA, 6 mM β-mercaptoethanol, 20 mM sodium acetate, pH 6.0 and fractionated by chromatography on a CM52 cation-exchange column equilibrated in the same buffer. The column was eluted by a 0–500 mM linear KCl gradient and the protein peaks analyzed by SDS-PAGE. Fractions containing the NH2-terminal truncated TnT were further purified by G75 gel filtration chromatography in 6 M urea, 500 mM KCl, 0.1 mM EDTA, 6 mM β-mercaptoethanol, 10 mM imidazole-HCl, pH 7.0. Protein peaks were analyzed by SDS-PAGE and the fractions containing pure NH2-terminal truncated TnT were dialyzed against 0.1% formic acid and lyophilized. All purification steps were carried out at 4 °C.
According to the NH2-terminal truncation site (between Thr45 and Ala46) reported in rabbit fast TnT (32), an expression vector encoding NH2-terminal truncated mouse fast skeletal muscle TnT was reconstructed by similar procedures and expressed in E. coli as described above.
Triton X-100 extraction of ventricular muscle strips
Operated on ice, ventricular muscle was cut with a sharp razor blade into fine pieces of approximately the size of isolated trabeculae. The muscle strips were washed in relaxing solution containing 0.1 KCl, 2 mM MgCl2, 2 mM EGTA, 10 mM Tris, 0.5 mM DTT, 0.1 mM PMSF and 2 mM Na4P2O7. After centrifugation at 2,800 × g at 4 °C for 15 min, the pellet was skinned in relaxing solution plus 0.5% (w/w) Triton X-100 at 4 °C with rotation for 10 min. After centrifugation at 14,000 × g at 4 °C for 20 min, the pellet was suspended in relaxing solution without Triton X-100 and incubated at 37 °C with rotation. Samples were collected at a series of time points for SDS-PAGE and Western blotting to examine the modifications of cTnT and other myofibrillar proteins.
Calpain inhibition
To test whether endogenous calpain in the cardiac muscle contributes to TnT NH2-terminal modification, several different calpain inhibitors were applied to the Triton X-100 extraction procedure. It has been reported that μ-calpain proteolytic activity is strongly inhibited by the application of an oxidant, e.g. 100 μM of hydrogen peroxide (42). Triton treatment of mouse ventricular muscle strips was carried out as above in the presence or absence of 100 μM hydrogen peroxide and the effect on cTnT modification was examined by Western blotting.
We also tested the effects of two cell membrane permissible non-peptide calpain inhibitors, PD150606 and PD151746 (Calbiochem, San Diego, CA). PD150606 exhibits similar apparent inhibition constants against μ-calapin (Ki 0.21 ± 0.01 μM) and m-calpain (Ki 0.37 ± 0.03 μM), whereas PD151746 has a 20-fold selectivity for μ-calpain (Ki 0.26 ± 0.03 μM) over m-calpain (Ki 5.33 ± 0.77 μM) (43). PD150606 and PD151745 were separately added to the relaxing solution before Triton treatment and incubated with the minced ventricular muscle at 4 °C for 10 min to allow the inhibitor to penetrate cell membrane and bind to calpain. After Triton X-100 extraction, samples were collected for SDS-PAGE and Western blotting to examine the effect on cTnT modification.
Muscle protein purifications
Bovine cTnT was purified from left ventricular muscle as previously described (8).
Bovine cardiac TnI was purified from ventricular muscle as described previously (19).
Rabbit α-tropomyosin was purified from ventricular muscle as described previously (44).
Preparation of cardiac myofibrils
Cardiac myofibrils were prepared from ventricular muscle according to the method described previously (45) with modifications. All steps were conducted at 4 °C. The ventricular muscle was pulverized in a food blender in 10 volumes (w/v) of the above relaxing buffer. The homogenization was passed through two layers of cheesecloth, and centrifuged at 2,000 × g for 15 min. After three washes using the relaxing buffer without Na4P2O7, the pellet was suspended in the washing buffer containing 0.5% (w/w) Triton X-100 for 10 min with occasional stirring. Four more washes were performed to remove Triton X-100. The myofibrils were stored −20 °C in the washing buffer containing 50% glycerol until use.
Isolation of troponin complex from cardiac muscle
Cardiac troponin complex was isolated by immunoaffinity chromatography using a mouse mAb (TnI-1) against the COOH-terminus of TnI (46). The TnI-1 epitope is exposed in the troponin complex and can be used as a handle to isolate the troponin complex from muscle homogenates (30). The TnI-1 mAb (IgG1) was purified from hybridoma ascites fluid using a Protein G-Sepharose (Amersham Pharmacia Biotech) affinity column and coupled to CNBr-activated Sepharose 4B (Amersham Pharmacia Biotech) according to the manufacturer’s protocols. Bovine left ventricular muscle was minced into small pieces and extracted by 20 volumes (w/v) of Guba-Straub solution containing 300 mM KCl, 100 mM K2HPO4, 50 mM KH2PO4, 2.5 mM MgCl2, 1 mM EGTA, and 0.1 mM phenylmethylsulfonyl fluoride (PMSF), pH 6.5, on ice for 15 min. After centrifugation at 16,000 × g at 4°C for 20 min, the supernatant containing mainly myosin was removed. The pellet was extracted in 20 volumes (w/v) of 1 M KCl, 10 mM Tris-HCl, pH 8.0, 0.1 mM PMSF by stirring on ice for 30 min. After centrifugation as above, the extract was diluted 5-fold in TBS and loaded on a TnI-1 mAb affinity column of 0.5 ml bed volume equilibrated in TBS. The column was washed with TBS, and the proteins bound to the TnI-1 affinity beads were eluted with 50 mM glycine-HCl, pH 2.7. 0.3 ml fractions were collected into tubes containing 0.05 ml of neutralizing buffer containing 1 M Tris-HCl, 1.5 M NaCl, 1 mM EDTA, pH 8.0. The fractions were analyzed by SDS-PAGE and Western blotting as described above to identify the troponin peak. The fractions containing the three troponin subunits were examined by Sepharose G75 column (Amersham Pharmacia Biotech) under non-denaturing conditions to verify their formation of troponin complex.
μ-Calpain treatment of cTnT
Purified bovine cTnT and troponin complex were incubated at 37°C in 50 mM sodium borate buffer, pH 7.5, containing 3 mM MgCl2, 1.25 mM CaCl2 and various concentrations (0.25 U/ml to 4 U/ml) of μ-calpain (Calbiochem, San Diego, CA). After 30 min incubation, the reaction was stopped by adding 3x SDS-PAGE sample buffer and heating at 80 °C for 5 min. The samples were analyzed by SDS-PAGE and Western blotting as described above.
The isolated bovine cardiac myofibrils were centrifuged at 3,000 × g for 15 min to remove glycerol. The pellet was suspended in 50 mM sodium borate buffer, pH 7.5, containing 3 mM MgCl2, and 1.25 mM CaCl2. The calpain treatment conditions were the same as that for purified proteins except for the use of higher concentrations of μ-calpain (5 U/ml to 20 U/ml). The effects on myofilament proteins were examined by SDS-PAGE and mAb CT3 Western blotting as above.
Protein binding assays
Enzyme-linked immunosorbant assay (ELISA) solid phase protein binding experiments (21) were performed to investigate the interactions of the NH2-terminal truncated cTnT with TnI and tropomyosin. Purified intact and NH2-terminal truncated mouse cTnT or BSA control were dissolved at 5 μg/mL in Buffer A (0.1 M KCl, 3 mM MgCl2, 20 mM PIPES, pH 7.5) and coated onto 96-well microtitering plates by incubation at 4 °C overnight. After washes with Buffer T (Buffer A containing 0.05% Tween-20) at pH 7.5 to remove the unbound protein, the plates were blocked with Buffer T at pH 7.0 or 6.2 containing 1% BSA. The immobilized cTnT was incubated with serial dilutions of bovine cardiac TnI or rabbit α-tropomyosin in Buffer T (at the blocking pH) containing 0.1% BSA at room temperature for 2 h. After one wash with Buffer T of the blocking pH and two washes with Buffer T at pH 7.0, the bound TnI or tropomyosin was quantified via incubation at pH 7.0 and room temperature for 1 h with the anti-TnI mAb TnI-1 (46) or an anti-tropomyosin mAb CH1 (47), respectively. The plates were then processed by a standard ELISA procedure, including pH 7.0 Buffer T washes, horse radish peroxidase-conjugated anti-mouse immunoglobulin second antibody (Sigma) incubation, and H2O2/2,2'-azinobis-(3-ethylbenzthiazolinesulfonic acid) substrate reaction (21). A405nm of each assay well was recorded at a series of time points by an automated microtiter plate reader (BioRad Benchmark). The A405nm values in the linear course of the color development were used to plot the protein binding affinity curves. All experiments were done in triplicate.
Data analysis
The DNA and protein sequence analyses were done using computer programs from DNAStar. Statistical analysis of the SDS-gel and Western blot densitometry and the protein binding data was done by Student t test. All values are presented as mean ± SD.
RESULTS
A cTnT fragment produced in myocardial ischemia-reperfusion
Western blots using mAb CT3 recognizing cardiac and slow TnT (23) detected a significant amount of an additional protein band in ischemia-reperfused rat heart (Figure 1A). This band has a significantly lower apparent molecular weight than that of the slow skeletal muscle TnT or any known alternatively spliced cTnT isoforms. This band is also recognized by several other anti-cTnT mAbs as well as the anti-TnT rabbit polyclonal antibody RATnT (data not shown), indicating that it is a modified TnT protein. Considering the facts that slow and fast skeletal muscle TnT are not expressed in postnatal cardiac muscles (48), this low Mr TnT is likely a proteolytic fragment of cTnT.
FIGURE 1.
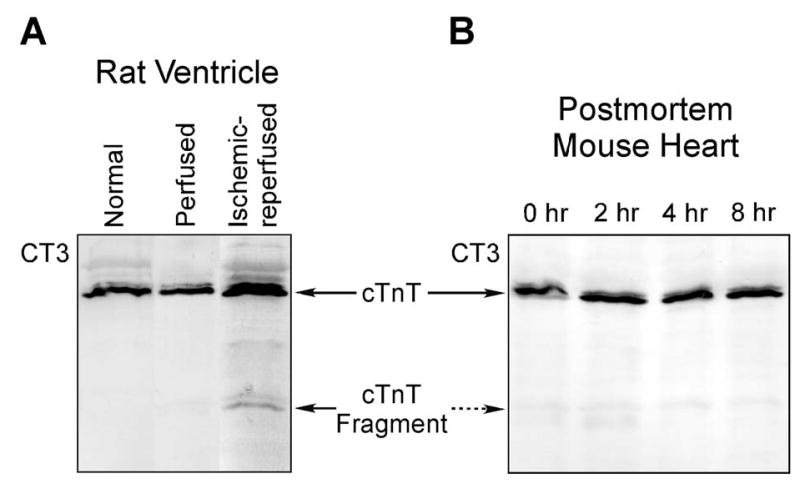
A cTnT fragment found in ischemia-reperfused cardiac muscle. A. While there was no detectable degradation of cTnT in rat heart after 105 min ex vivo perfusion, CT3 mAb Western blot showed that a specific cTnT fragment was produced in working rat heart preparations after ischemia-reperfusion treatment. B. Cardiac muscle samples from adult C57BL/6 mice were prepared at 0, 2, 4, and 8 hours postmortem and analyzed by SDS-PAGE and CT3 mAb Western blotting. The results showed that there was no detectable degradation of cardiac TnT due to the postmortem ischemia.
The cTnT fragment is present in normal cardiac muscle although at very low levels (Figure 6B), suggesting a physiological relevance. Simple postmortal ischemia for up to 8 hours did not increase the cTnT fragmentation (Figure 1B). There was no detectable change in the cTnT fragment in the rat heart after 105 min ex vivo perfusion, demonstrating a correlation to the ischemia-reperfusion stress conditions.
FIGURE 6.
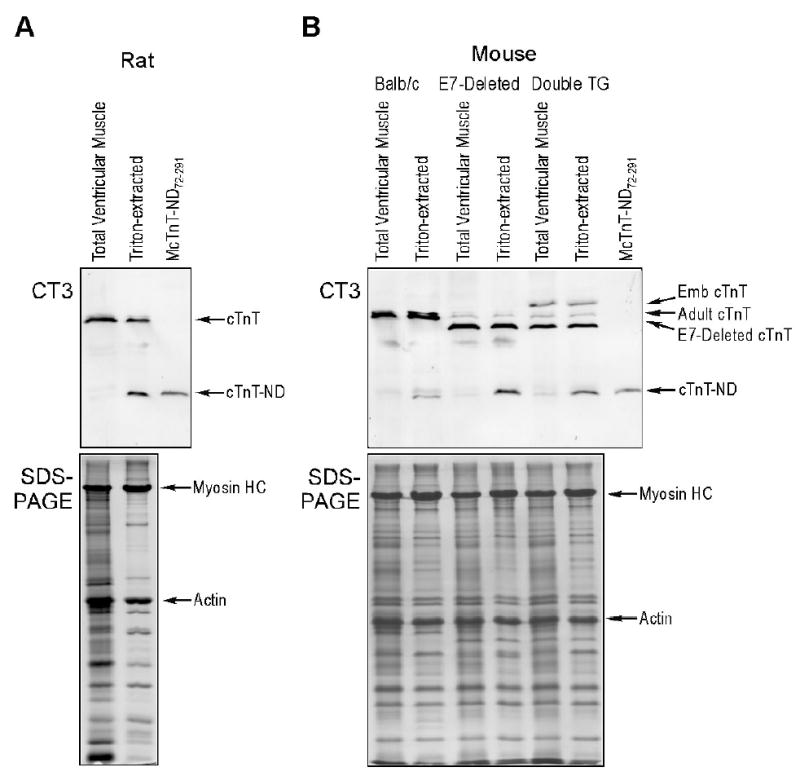
Triton X-100 extraction promotes the production of the NH2-terminal truncated cTnT. Rat (A) and mouse (B) ventricular muscle strips before and after Triton X-100 extraction were examined by SDS-PAGE and Western blot using anti-TnT mAb CT3. The results revealed that Triton X-100 extraction induced the production of a specific cTnT fragment in both rat and mouse cardiac muscle samples. In the mouse model, adult, exon 7 (E7)-deleted and embryonic (Emb) cTnTs with different NH2-terminal structures produced a single fragment that has the same size as the reconstructed McTnT-ND72–291 protein, consistent with a selective NH2-terminal truncation. The production of NH2-terminal truncated cTnT (cTnT-ND) by Triton X-100 extraction suggests the role of activation of a myofibril-associated protease. The accompanying SDS-PAGE showed comparable amounts of protein loading normalized to that of actin and that other major myofibril proteins were not affected by the Triton treatment.
The cTnT fragment is produced by a restricted NH2-terminal truncation
Suggesting an NH2-terminal deletion that produces the cTnT fragment, ischemia-reperfusion treatment of transgenic mouse cardiomyocytes expressing embryonic or exon 7-deleted cTnT that differ from the wild type cTnT only in the length of the NH2-terminal variable region produced cTnT fragments with identical size (Figure 2A).
FIGURE 2.
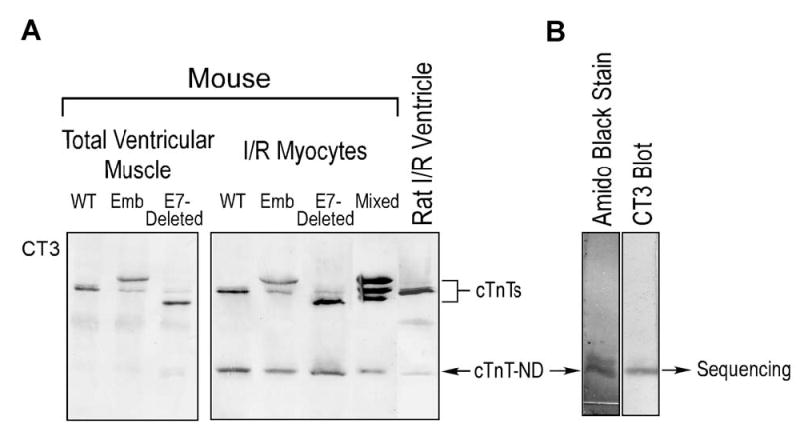
Isolation of the cTnT fragment from ischemia-reperfused mouse cardiomyocytes to determine the NH2-terminal sequence. A. The cTnT fragment was produced in isolated mouse cardiomyocytes that had undergone ischemia-reperfusion conditions. Samples from transgenic mouse hearts expressing embryonic cTnT and/or exon 7-deleted cTnT that differ from the endogenous adult cTnT in the NH2-terminal region (Figure 5) showed a cTnT fragment identical in size to that produced in the wild type adult mouse cardiomyocytes, indicating that a removal of the NH2-terminal variable region produces the cTnT fragment. B. The cTnT fragment band isolated from low cross-linker preparative SDS-PAGE gel (see Materials and Methods) was resolved on a high cross-linker SDS-gel and transferred to PVDF membrane for NH2-terminal sequencing. Amido Black staining revealed the yield of the cTnT fragment that is confirmed by mAb CT3 Western blot.
We successfully isolated the low molecular weight cTnT fragment from ischemia-reperfused mouse cardiomyocytes for NH2-terminal sequencing. Figure 2B shows the enrichment of the cTnT fragment by preparative SDS-PAGE. The NH2-terminal sequencing result revealed that this low molecular weight TnT protein is indeed a cTnT fragment with a deletion of the NH2-terminal amino acids 1–71 (Figure 3).
FIGURE 3.

NH2-terminal truncations of cTnT. NH2-terminal amino acid sequencing of the cTnT fragment revealed a single truncation site between residues Arg71 and Leu72. The Met in bracket indicates the addition of an initiation codon for expressing cTnT-ND72-291 in E. coli (Figure 4). Amino acid sequence alignment demonstrated that the cTnT NH2-terminal truncation removes the entire variable region (Figure 5), similar to the NH2-terminal truncated fast TnT previously isolated from rabbit skeletal muscle (rabbit 26-kDa fsTnT, 32) and a model protein previously studied (Rat cTnT77-298, 16). In contrast, a caspase cleavage-produced cTnT fragment (Rat 25-kDa cTnT, 29) involves the deletion of a part of the conserved region. The number of amino acids (A.A.) and calculated molecular weights of these proteins are indicated after the sequences.
To investigate the integrity of the COOH-terminus in the cTnT fragment, we re-constructed the NH2-terminal truncation in mouse cTnT by generating a 5’-truncated mouse cTnT cDNA (Figure 4). Expression of the truncated cDNA in E. coli produced a cTnT protein with a size identical to that of the cTnT fragment produced in ischemia-reperfused cardiac muscle. The results demonstrate that there was no COOH-terminal deletion in this cTnT fragment.
The NH2-terminal truncation site (Arg71-Leu72) is not at an exon boundary and, therefore, the cTnT-ND72-291 fragment is not generated by alternative RNA splicing but by proteolytic cleavage. Sequence alignment (Figure 3) demonstrates that the cleavage site is different from the previously reported caspase cleavage site in cTnT (the rat 25-kDa cTnT) under ischemia-reperfusion conditions (29).
The restricted NH2-terminal truncation of cTnT preserves the core functional structure of TnT
Figure 5 compares the NH2-terminal truncated cTnT with several intact cTnT variants and demonstrates that the NH2-terminal cleavage of the cTnT polypeptide chain specifically removes entirely the hypervariable region encoded by exons 2 to 7. The central and COOH-terminal conserved regions that contain binding sites for other thin filament regulatory proteins, TnI, TnC and tropomyosin are preserved in this proteolytic modification. The integrity of the conserved core structure of TnT in the NH2-terminal truncated cTnT (cTnT-ND72–291) implies a functional effect in myocardial ischemia-reperfusion. This notion is consistent with the fact that cTnT-ND72–291 was retained in the isolated cardiac myofibrils with a proportion identical to that in the total muscle extract, indicating its full ability to incorporate into the myofilament (Figure 6A).
FIGURE 5.
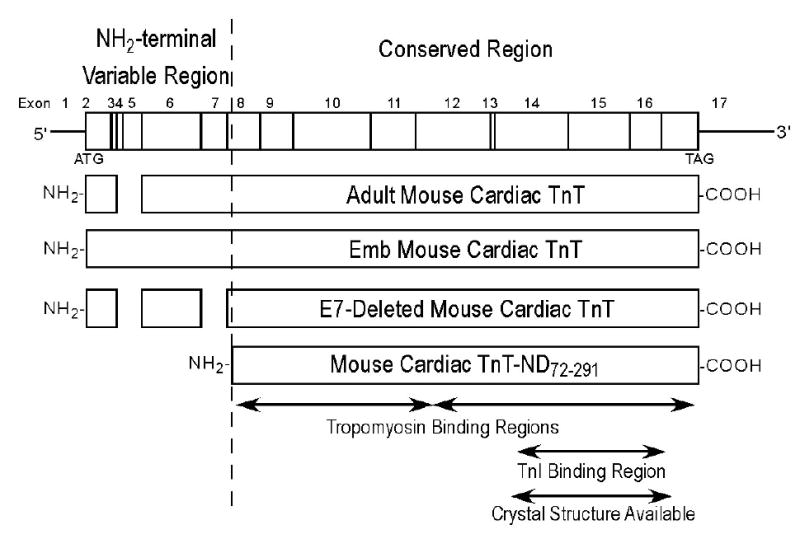
Structural comparison of the NH2-terminal truncated cTnT with cTnT splicing variants. A cTnT mRNA map noted with exon boundaries is shown on the top. Protein primary structural maps of the cTnT variants studied are aligned with the coding region of the mRNA map. The NH2-terminal and adjacent regions of intact wild type adult mouse cTnT and two alternatively spliced variants are compared with the NH2-terminal truncated cTnT. The NH2-terminal truncation specifically removes of the entire variable region that is alternatively spliced to produce the adult mouse cardiac TnT (Adult cTnT-4, exclusion of exons 4 and 5), embryonic isoform (Emb cTnT-1, contains all the exons), and E7-deleted mouse cardiac TnT (E7-deleted TnT, exclusion of exons 4, 5 and exon 7). The central and COOH-terminal conserved regions of TnT contain the core functional structure that binds other thin filament regulatory proteins, TnI, TnC and tropomyosin (Tm). These protein binding sites and the portion of TnT with X-ray crystallography structure available are outlined. The NH2-terminal truncated cardiac (McTnT-ND72-291) retains the integrity of the conserved core structure, implying a functional role in myocardial ischemia-reperfusion.
Triton X-100 extraction of cardiac muscle activates an endogenous proteolytic activity that produces the NH2-terminal truncated cardiac TnT
Western blotting using mAb CT3 demonstrated that Triton X-100 treatment of rat and mouse cardiac muscle strips reproduced the specific cTnT-ND fragment (Figure 6). In the mouse model, Triton X-100 extraction of transgenic cardiac muscle containing wild type adult, embryonic and exon 7-deleted cTnTs that are different in the NH2-terminal region produced a single fragment with the same size as that of McTnT-ND72–291. This was most clearly shown in the double transgenic mouse heart that simultaneously expresses all of the three cTnT variants (Figure 6B). This result indicates that the cTnT fragment produced by Triton extraction is the same as the NH2-terminal truncated cTnT-ND72–291 identified in ischemia reperfusion. Triton X-100 treatment is known to remove lipid contents from the muscle fiber without disruption of the myofibril structure. SDS-PAGE in Figure 6 showed that other major myofibril proteins were not affected by Triton X-100 treatment. The Triton treatment may have activated a myofibril-associated protease that is responsible for the specific production of cTnT-ND72–291 under stress conditions.
The production of NH2-terminal truncated cTnT by myofibril associated proteolytic activity is suppressed by calpain inhibitors
Calpain has been reported to cleave cTnT (36). Therefore, we tested the effects of calpain inhibitors on the myofilament-associated endogenous proteolytic activity. Western blot examination showed that the presence of 100 μM hydrogen peroxide resulted in a ~35% decrease in the Triton X-100-induced production of cTnT-ND72–291 in mouse ventricular muscle strips (Figure 7A). Hydrogen peroxide is known to inhibit the proteolytic activity of μ-calpain (42). Therefore, this result suggests that cardiac myofilament associated μ-calapin may be responsible for the production of cTnT-ND72-291 fragment during ischemia-reperfusion.
FIGURE 7.
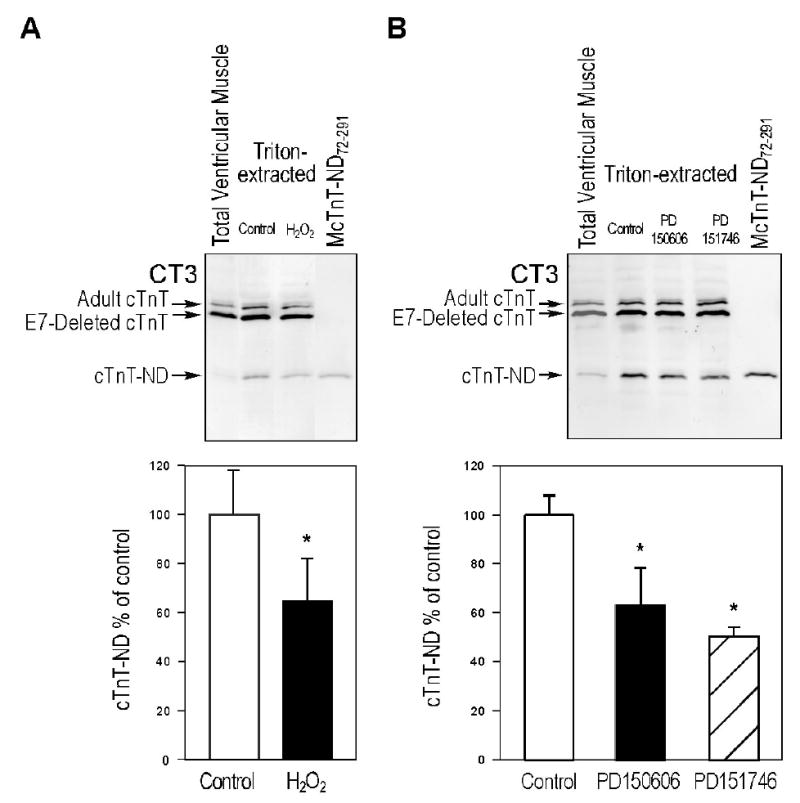
Calpain inhibitors reduce the production of NH2-terminal truncated cTnT by myofibril associated proteolytic activity. A. Western blots were used to examine Triton X-100 extracted exon 7 (E7)-deleted transgenic mouse ventricular muscle strips in the absence or presence of 100 μM hydrogen peroxide. Densitometric analysis showed that cTnT-ND production was reduced to 65% in the presence of hydrogen peroxide (*, P < 0.01), indicating an effect of inhibiting the activity of μ-calpain (42). B. Calpain-specific inhibitors, PD150606 and PD151746, also decreased the production of cTnT-ND in Triton X-100 extracted mouse cardiac muscle strips (to 63% and 50%, respectively, P < 0.01), further supporting the role of myofibril associated μ-calpain in the production of cTnT-ND. Data are shown as mean ± S.D. The results were summarized from the three individual experiments.
Consistently, calpain-specific inhibitors, PD150606 and PD151746, also decreased the productions of cTnT-ND72–291 in Triton X-100 extracted mouse cardiac muscle strips (Figure 7B). PD150606 and PD151746 resulted in ~37% and ~50% decreases in cTnT-ND72–291 production, respectively, further supporting the role of myofibril associated μ-calpain.
μ-Calpain treatment of myofibrils reproduced the NH2-terminal truncated cTnT fragment
The results in Figure 8A demonstrate that μ-calpain treatment of purified bovine cTnT effectively decreased the level of intact cTnT in a concentration-dependent manner, consistent with that observed in a previous study (36). However, no specific cTnT fragment was produced at a significant amount from μ-calpain treatment of isolated cTnT, demonstrating a non-specific degradation effect.
FIGURE 8.
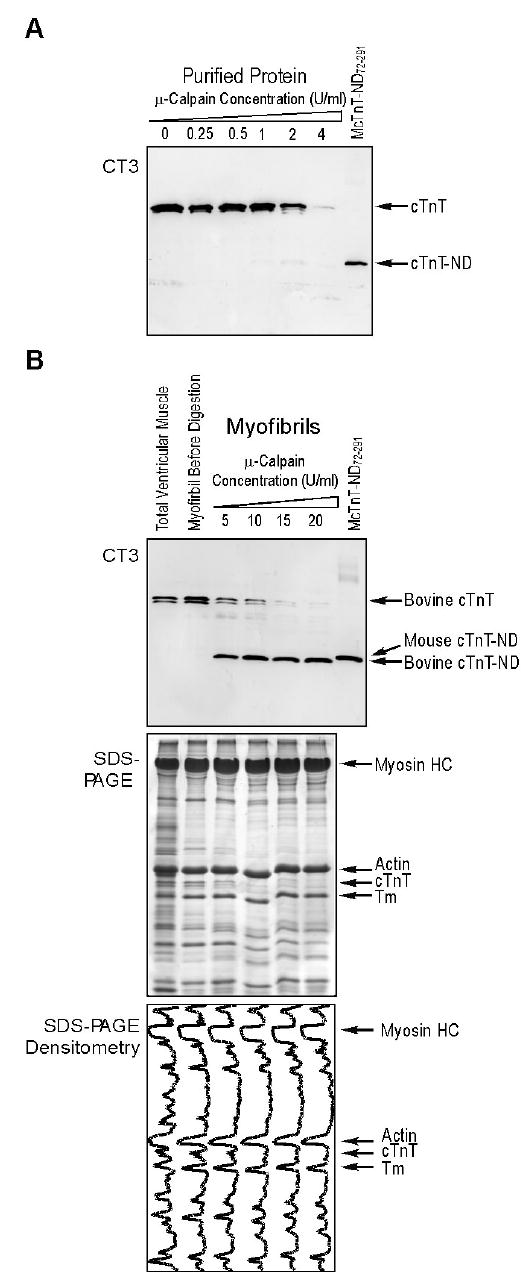
μ-Calpain treatment of cardiac myofibrils reproduced the NH2-terminal truncated cTnT fragment. A. Western blot using anti-cTnT mAb CT3 showed that μ-calpain treatment degraded purified bovine cTnT as that reported previously (36), but did not produce a specific fragment. B. In contrast, μ-calpain treatment of bovine cardiac myofibrils effectively reproduced the cTnT-ND fragment. The results demonstrate that the cTnT-ND modification by μ-calpain is dependent on the myofibril structure. Densitometry traces of the accompanying SDS-PAGE gel showed no apparent degradation of other major myofibrillar proteins, including myosin, actin and Tm, under the μ-calpain treatment conditions.
On the other hand, μ-calpain treatment of bovine cardiac myofibril effectively reproduced the cTnT-ND72–291 fragment (Figure 8B). SDS-PAGE and the gel densitometry plots (Figure 8B) further showed that other major myofibrillar proteins, including myosin, actin and tropomyosin, were not significantly affected by the μ-calpain treatment. This result supports the observation that the production of cTnT-ND72–291 during myocardial ischemia-reperfusion is by endogenous μ-calpain cleavage. In contrast to the non-specific degradation of purified cTnT by μ-calpain, the specific re-production of cTnT-ND72–291 by μ-calpain treatment of cardiac myofibrils demonstrates that this posttranslational modification of cTnT structure is dependent on the physiological structure of the myofilament, consistent with a physiological relevance.
Quantitative densitometry analysis of the Western blots shows that the μ-calpain modification of cTnT in bovine cardiac myofibril had a non-linear (reverse exponential) concentration relationship (Figure 9). The sensitive responses to the initial increasing concentrations of μ-calpain imply a preferred selective cleavage of cTnT in the myofibrils. On the other hand, the reaching of a plateau at the higher concentration of μ-calpain suggests a restricted proteolytic modification.
FIGURE 9.
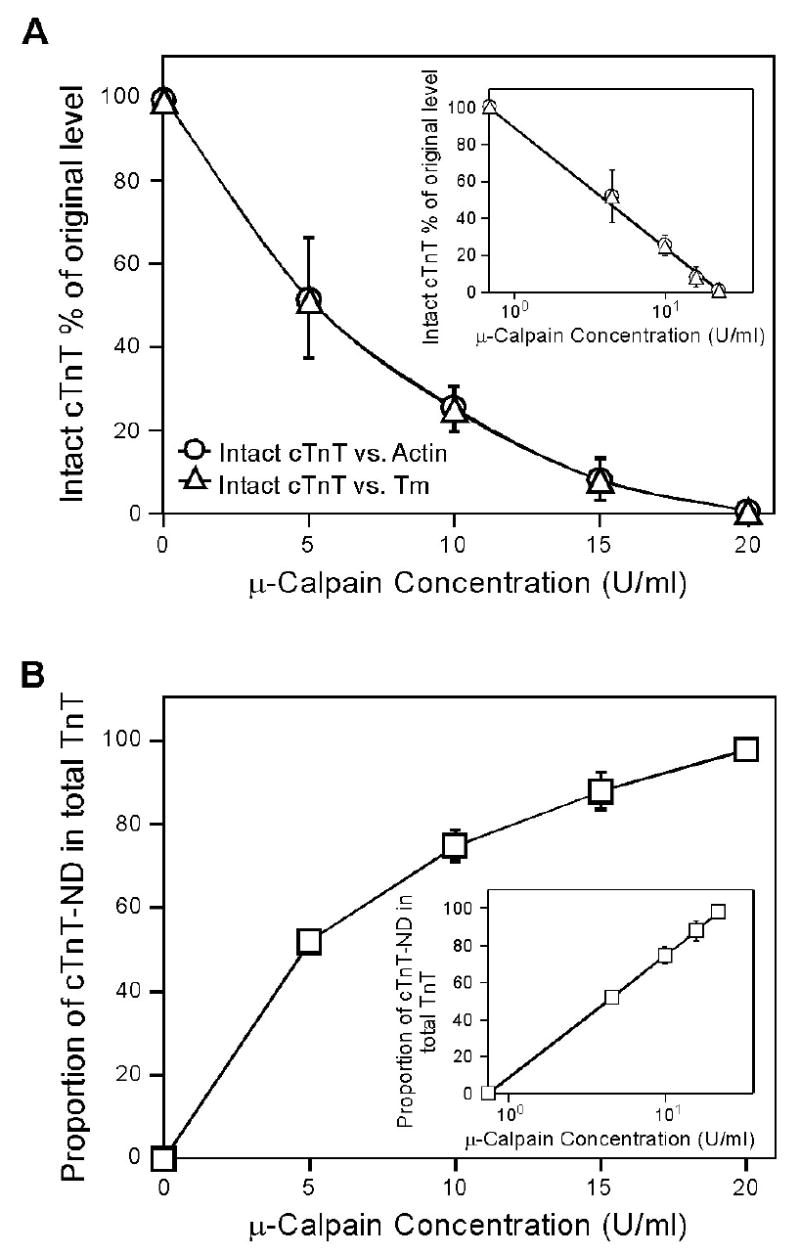
μ-Calpain production of NH2-terminal truncated cTnT suggests a restricted proteolytic modification. Densitometry analysis of multiple copies of mAb CT3 Western blots (Figure 8) determined the relative amounts of intact and NH2-terminal truncated cTnT in μ-calpain treated bovine cardiac myofibrils. The results show that intact cTnT decreased from 100% to 1.5% when the concentration of μ-calpain increased from 0 U/ml to 20 U/ml (A), while the amount of the NH2-terminal truncated cardiac TnT, cTnT ND, increased from 0% to 98% of the total cTnT (truncated plus intact) (B). The reverse exponential concentration responses of the cleavage of intact cTnT as shown by the semi-log plot inserts and the specific production of the NH2-terminal truncated cTnT by μ-calpain treatment suggest a selective and restricted proteolytic modification.
μ-Calpain treatment of isolated bovine cardiac troponin complex also showed a selective cleavage of the NH2-terminal domain of TnT (Figure 10). The difference between free TnT and troponin complex in μ-calpain proteolysis is consistent with the determining role of the substrate structural conformation. Nonetheless, the higher sensitivity and less effective preservation of the TnT core structure seen in the μ-calpain treatment of isolated troponin complex than that of intact myofibrils (Figure 8B) suggest that this selective structural modification of TnT is most effective under physiological conditions.
FIGURE 10.
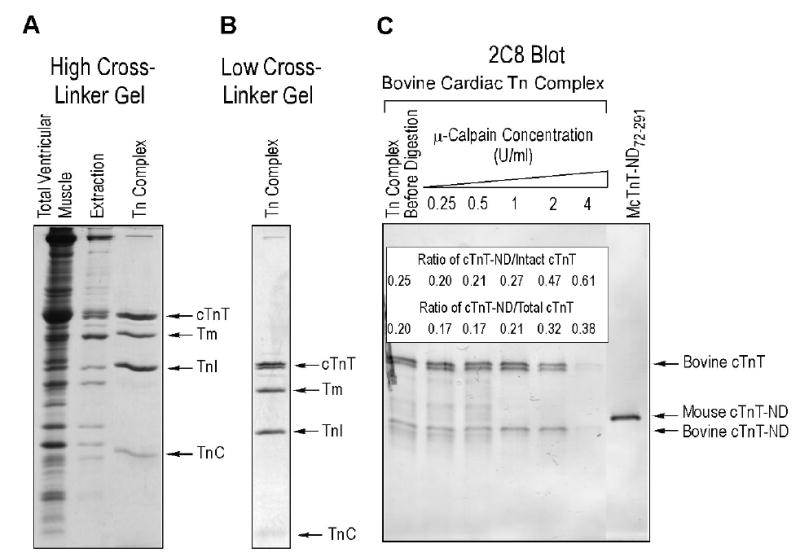
Isolation and μ-calpain treatment of bovine cardiac troponin complex. A. SDS-PAGE (15% high cross-linker gel) showed the effective isolation of troponin (Tn) complex from bovine ventricular muscle homogenate by immunoaffinity chromatography. The TnI-1 mAb affinity column fraction contained 1:1:1 ratio of TnT, TnI and TnC. Some tropomyosin (Tm) was co-isolated with the troponin complex, reflecting the native binding between troponin and tropomyosin in the thin filament. B. 14% Low cross-linker SDS-PAGE gel shows the isolated bovine troponin complex contains both of the two adult cTnT isoforms (8), indicating a native state. C. Western blot using anti-cTnT mAb 2C8 showed that μ-calpain treatment of isolated bovine cardiac troponin reproduced a single cTnT-ND fragment from the two NH2-terminal alternatively spliced cTnT isoforms. The insert table shows relative amounts of cTnT-ND produced. It is worth noting that while the troponin structure preserved the TnT core structure against calpain digestion in contrast to that in free cTnT, the protection was less effective than that in intact myofibrils (Figure 8B). Altogether, the results are consistent with the role of cTnT conformation in determining this selective structural modification under physiological conditions.
Similar NH2-terminal truncation of cardiac and fast skeletal muscle TnT by calpain proteolysis
After Triton X-100 extraction of transgenic mouse cardiac muscle strips containing both cTnT and chicken fast skeletal muscle TnT (38), Western blot using polyclonal antibody RATnT raised against chicken fast TnT and mAb 2C8 recognizing both cardiac and fast TnTs detected the production of both cardiac and fast skeletal TnT-ND fragments (Figure 11). Mouse cTnT-ND and chicken fast TnT-ND have different molecular weights due to their amino acid composition in the core structure. While the blot using low cross-linker SDS-gel (Figure 11A) showed only one TnT-ND band, the blot using high cross-linker SDS-gel resolved two TnT fragments with distinct immunoreactivities to RATnT and 2C8 antibodies indicating their cTnT and fast TnT origins. Although the amino acid sequences of the NH2-terminal regions of mouse cardiac and chicken fast skeletal muscle TnTs flanking the truncation sites are very different, the similar modification of both endogenous cTnT and transgenic-expressed fast TnT upon the Triton extraction-activated μ-calpain cleavage suggests a dependence on myofibril structure rather than the amino acid sequences at the cutting sites.
FIGURE 11.
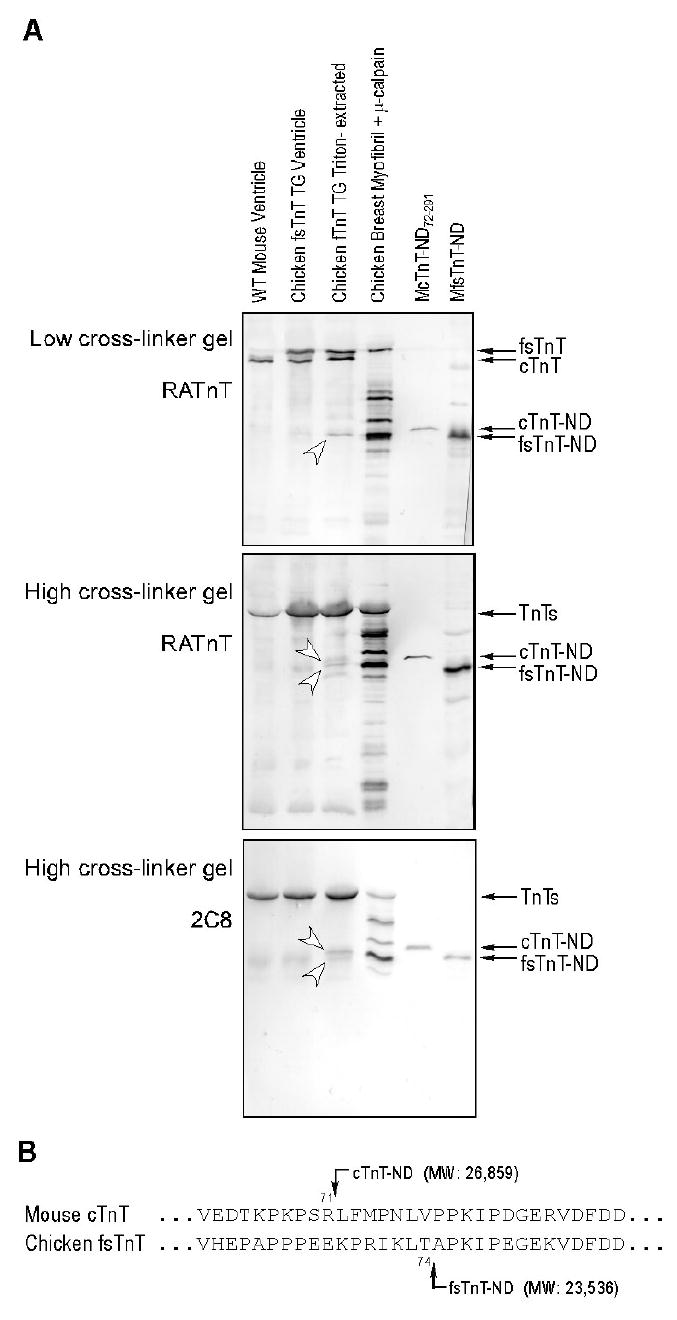
Similar NH2-terminal truncation of cardiac and fast skeletal muscle TnT by μ-calpain modification. A. Western blot using polyclonal antibody RATnT raised against chicken fast TnT and mAb 2C8 recognizing both cardiac and fast TnTs detected TnT-ND fragments (indicated by the arrowheads) in Triton X-100-treated transgenic mouse cardiac muscle strips containing endogenous cTnT and transgenic-expressed chicken fast skeletal muscle TnT (fsTnT) (38). While the blot using low cross-linker SDS-gel (upper panel) showed only one TnT fragment band, the blot using high cross-linker SDS-gel resolved two TnT fragments with distinct immunoreactivities to RATnT and 2C8 antibodies, indicating their cTnT and fast TnT origins. μ-Calpain-treated chicken breast muscle myofibrils, reconstructed mouse cTnT-ND and mouse fast TnT-ND were used as controls. B. Aligned amino acid sequences of the NH2-terminal regions of mouse cardiac and chicken fast skeletal muscle TnTs flanking the truncation sites are shown. The predicted molecular weights of the NH2-terminal truncated cTnT and fast TnT proteins are in agreement with the SDS-gel mobility of the protein fragments detected in A. The observation that cTnT and fast skeletal TnT were modified similarly by μ-calpain cleavage suggests a dependence on myofibril structure rather than the amino acid sequences at the cutting sites.
Selective removal of the NH2-terminal variable region of cTnT preserves the binding of cTnT to TnI and tropomyosin with altered affinities
To examine the effect of the NH2-terminal truncation on cTnT's interactions within the thin filament regulatory system, we compared the binding of intact and the NH2-terminal truncated cTnT to TnI and tropomyosin. The results of ELISA solid phase protein binding experiments in Figure 12A demonstrate that the NH2-terminal truncated cTnT has an increased binding affinity for TnI compared to that of intact cTnT. This is shown by the lower concentration of TnI required for reaching 50% of maximum binding (8.73±1.15 nM for cTnT-ND versus 15.33±1.36 nM for intact cTnT, P<0.005), reflecting a higher Ka in the equilibrium binding step. No significant difference in the maximum binding was observed (Figure 12A insert), indicating no effect on the TnT-TnI coupling strength in the subsequent washing separation steps after the complex formation. The higher Ka in cTnT-ND-TnI binding may facilitate incorporation of the mutant cTnT into the troponin complex and the thin filament as well as affect the allosteric feature of the Ca2+-regulatory system.
FIGURE 12.
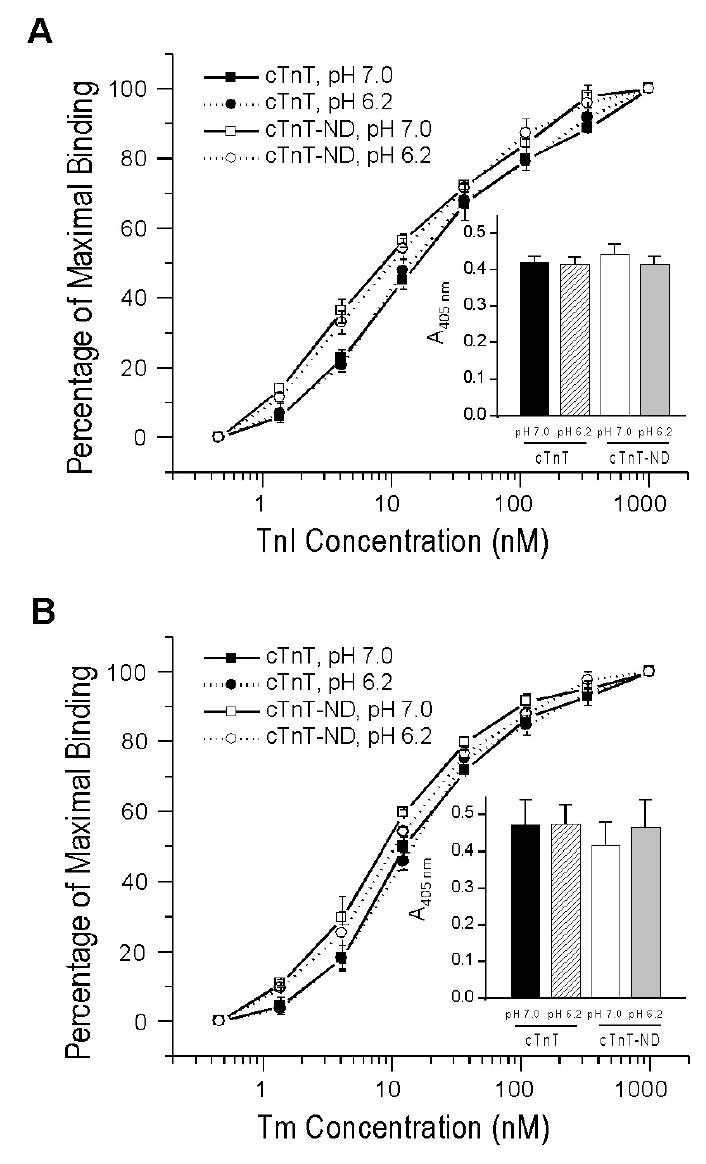
Removal of the NH2-terminal variable region preserves cTnT’s binding to TnI and tropomyosin (Tm) with altered affinities. ELISA protein binding curves demonstrate that the NH2-terminal truncated cTnT has an increased binding affinity for TnI as compared to that of intact cTnT (A). The concentrations of TnI required to reach 50% of maximum binding were 8.73±1.15 nM for cTnT ND and 15.33±1.36 nM for intact cTnT, P < 0.005. No significant difference was seen between the maximum bindings of cTnT-ND and intact cTnT to TnI (Panel A insert). The binding of cTnT-ND to α-tropomyosin also exhibits a higher affinity than that of intact cTnT (B). The concentrations of α-tropomyosin dimer for 50% maximum binding of cTnT ND and intact cTnT were 9.73±0.185 nM and 13.50±1.38 nM, respectively (P < 0.01) The level of maximum binding was not significantly changed (Panel B insert). The decrease of pH from 7.0 to 6.2 did not result in significant change in the binding of both intact and NH2-terminal truncated cTnT to TnI or α-tropomyosin.
The binding of cTnT-ND to tropomyosin also exhibited a higher affinity than that of intact cTnT (Figure 12B). The concentrations of tropomyosin for 50% maximum binding of cTnT-ND and intact cTnT were 9.73±0.185 nM and 13.50±1.38 nM, respectively (P<0.01). The level of maximum binding was not significantly changed (Figure 12B insert). Although no effect on the anchoring strength of TnT on tropomyosin, the altered binding affinity of NH2-terminal truncated cTnT for tropomyosin may affect the allosteric feature of the thin filament regulatory system, contributing to myocardial function during ischemia-reperfusion.
Intracellular acidosis occurs in myocardial ischemic injury with a correlation to the function of troponin (49). Therefore, we compared the response of cTnT-ND to lowered pH with that of intact cTnT. Decrease of pH from 7.0 to 6.2 did not result in significant change in the binary binding affinity of both intact and NH2-terminal truncated cTnT to TnI or tropomyosin (Figure 12). The results suggest that although environmental pH and the NH2-terminal negative charge of TnT is known to affect the bindings of acidic and basic fast skeletal muscle TnT isoforms to skeletal muscle TnI and tropomyosin (50), the cardiac protein isoforms may have a higher resistance to acidosis. The removal of the entire NH2-terminal variable region corresponding to a large amount of negative charges from cTnT did not convey the effect on the core structure and function seen in the fast TnT isoforms in response to the environmental pH. This observation presents an analogy to a feather of cardiac TnI in which proteolytic removal of the entire NH2-terminal region had a functional effect similar to that of protein kinase A phosphorylation of the NH2-terminal region (31).
DISCUSSION
We report in the present study a restricted proteolytic NH2-terminal truncation of cTnT in myocardial ischemia-reperfusion. This structural modification selectively removes the NH2-terminal variable region and preserves the TnT core structure with functional implications. From characterizing its production by μ-calpain cleavage, the following observations suggest the significance of this study.
Regulatory role of the NH2-terminal variable region of TnT
TnT is known as a protein with extended conformation in which the NH2-terminal variable region is a part of the “tail” domain of troponin. The presence of TnI and TnC binding sites in the TnT COOH-terminal domain is confirmed by the X-ray crystallographic three-dimensional structure of partial cardiac (51) and skeletal muscle (52) troponins. The NH2-terminal amino acid sequence is hypervariable among TnT isoforms and regulated by alternative RNA splicing during heart and skeletal muscle development. This region does not contain binding sites for other thin filament proteins but its structural alteration shows fine tuning effects on the Ca2+-regulation of muscle contraction. It has been proposed that the NH2-terminal variable region has its functional effects by modulating the molecular conformation and activity of other regions of TnT (21-23).
The fact that the NH2-terminal region of TnT does not contain binding sites for other thin filament proteins allows for its high sequence variability and forms the foundation for a wide range of modulating effects. The NH2-terminal truncated cTnT produced during myocardial ischemia-reperfusion selectively removes the entire NH2-terminal variable region while retaining the conserved core structure of TnT. This mechanism represents the most extreme modification of TnT in comparison to the developmental (8) and pathological (20) alternative splicing variants. The molecular evolution of the TnT NH2-terminal variable region demonstrates an increase in length and complexity (53). Therefore, the removal of the entire NH2-terminal variable region in cTnT may be a mechanism to resume a fundamental functional state of troponin as a response to stress conditions.
Predicted functional consequences of selective deletion of the NH2-terminal variable region of
cTnT. Several previous studies investigating the structure-function relationship of TnT have characterized cTnT model molecules with NH2-terminal deletion similar to cTnT-ND72–291 (Figure 3). For example, a naturally occurring NH2-terminal truncation of rabbit fast skeletal muscle TnT fragment (the TnT 26K fragment, 32) is equivalent to the cTnT-ND72–291. This fragment is able to form a functional troponin complex that exhibits a higher binding strength to tropomyosin compared with that of troponin containing the intact TnT. Reconstituted troponin complex containing the NH2-terminal truncated fast TnT also conferred a decrease in the maximum activation of actomyosin-S1 MgATPase (14). Our results showed that cTnT- ND72–291 also has an increased affinity to tropomyosin (Figure 12B), indicating similar functional effects. Further studies using a similar NH2-terminal deleted cTnT in reconstituted myofilaments demonstrated that the removal of NH2-terminal domain resulted in decreased myofibril force development (16). These data suggest a hypothesis that the NH2-terminal truncation of TnT is not a simple destruction but may function as a regulatory mechanism in both cardiac and skeletal muscles, which may have a role in modulating contractility under physiological and pathological stress conditions.
cTnT-ND72–291 is retained in the myofibrils of ischemia-reperfused cardiac muscle and, therefore, is anticipated to participate in the thin filament regulatory function. A consensus change found in the previous studies due to the presence of NH2-terminal truncated TnT is the decreases in the maximum Ca2+ activated actomyosin ATPase and myofibril force (14–16). Such a decrease of Ca2+ activation by deleting the NH2-terminal domain of cTnT during myocardial ischemia-reperfusion could contribute to the depressed function after ischemia, but may also provide a mechanism against Ca2+ overload-induced contractures (54). The hypothesis that the NH2-terminal truncation of cTnT is not simply destructive is supported by our observation that transgenic mice over-expressing high levels of cTnT-ND72–291 in the cardiac muscle do not show apparent cardiac dysfunction (37). Using the transgenic mouse hearts over-expressing the NH2-terminal truncated cTnT, detailed functional characterization is currently underway.
Myofibril-associated calpain activity as a rapid regulation of myocardial function
Triton X-100 extraction of cardiac muscle fibers induces the production of cTnT-ND, indicating a myofibril-associated proteolytic activity. Exogenous calpain inhibitors suppressed the production of cTnT-ND in Triton-extracted myofibrils. This endogenous calpain activity is independent of the presence or absence of Ca2+ in the incubation buffers (data not shown). Ca2+ concentration in living cardiac muscle cells rises periodically during the systole of each cardiac cycle to reach a level sufficient for μ-calpain activation. Therefore, the myofilament associated μ-calpain may have been Ca2+-primed in the myocytes before skinning. Triton extraction may have removed an endogenous calpain inhibitor or altered the myofilament conformation to activate the specific cleavage of cTnT. Consistent with a regulation by the substrate conformation, μ-calpain treatment of cardiac myofibrils reproduced the cTnT-ND72–291 fragment, in contrast to the treatment of purified cTnT that resulted in non-specific degradation. Although the specific production of cTnT-ND is seen in the μ-calpain treatment of isolated cardiac troponin complex, the protection of TnT core structure was much less effective than that in the intact myofibril (Figures 8 and 10). Therefore, the specific modification of TnT by μ-calpain cleavage is based on the physiological structure of the myofilament. Despite sequence differences, similar NH2-terminal truncation of chicken fast skeletal muscle TnT is produced by endogenous calpain proteolysis in transgenic mouse cardiac muscle (Figure 11), further supporting the myofilament structure-based specific calpain cleavage of the TnT NH2-terminal variable region.
Together with the functional effects of removing the NH2-terminal variable region of TnT, the μ-calpain-mediated proteolytic modification of TnT presents a rapid short term mechanism to adjust muscle function under stress conditions. The cleavage occurs within minutes after myocardial ischemia-reperfusion, which is apparently much faster than regulations by altering TnT isoform gene expression and/or alternative RNA splicing as that seen in the adaptation of skeletal muscle to unloading (24). Since myofilamental TnT only has a half-life of 4–5 days (55), the functional effect would be transient, suitable for an acute response to the usually short-lived ischemia-reperfusion stress conditions.
Proteolytic modifications of cTnT and cTnI have been shown with pathological effects on myocardial contractility (28, 56). A caspase-catalyzed fragmentation of cardiac TnT has been found to reduce force production (29). Ca2+ overload in cardiomyocytes caused by ischemia-reperfusion has been proposed to activate proteolytic cleavage of cTnI at amino acid 192 to remove the COOH terminus (57). The cTnI1–192 fragment reduces the maximal isometric tension of the myocardium and causes a stunning phenotype in the hearts of transgenic mice (28). However the production of cTnT-ND72–291 by μ-calpain modification is most likely a functional regulation rather than a detrimental destruction. The presence of low amounts of cTnT-ND72–291 in normal cardiac muscle also supports the hypothesis that the myofibril associated μ-calpain activity functions in the physiological regulation of contractility.
Tuning thin filament function in myocardial response to stress conditions
The up-regulation of the NH2-terminal truncated cTnT in ischemia-reperfusion indicates that modification of the thin filament function may play a role in the adaptation of cardiac muscle in stress conditions. By decreasing maximum contractile activation, the deletion of the NH2-terminal domain of cTnT may reduce the work of the cardiac muscle during ischemia-reperfusion to prevent permanent damage. This observation suggests that reducing the thin filament Ca2+ activation may be a potential target for the prevention or reduction of myocardial infarction following ischemia-reperfusion. Further studies are needed to investigate this hypothesis.
Posttranslational modification is a rapid mechanism to confer transient functional changes in a protein. Posttranslational regulation of cTnT NH2-terminal structure represents an effective immediate response for myocardial adaptation to functional demands and pathological conditions. To date, this level of myocardial regulation has been mainly studied on phosphorylation modifications (25). Study on restricted proteolytic modification of cTnT represents a new area of research and will provide valuable information to further understand the role of posttranslational regulation in cardiac muscle function and diseases. These studies may also contribute to the development of new preventative and therapeutic strategies for the management of acute coronary inefficiency. Beyond the widely recognized protein destruction in myocardial ischemia-reperfusion, the present study laid a foundation for further investigations into the significance of cTnT modification in ischemic heart disease.
Acknowledgments
We thank Stephen Chong for the construction of NH2-terminal truncated mouse fast TnT, Dr. Jim Lin for the CH1 mAb, and Dr. Jeffrey Robbins for the α-MHC promoter used in the transgenic mouse constructions. ZZ is supported by a postdoctoral fellowship from the American Heart Association Great Midwest Affiliate. BJB participated in this study when this group was at Case Western Reserve University.
Footnotes
This study was supported by grants from the National Institutes of Health (HL078773, AR048816 and HD044824).
Abbreviations: BSA, bovine serum albumin; cTnT, cardiac troponin T; cTnT-ND, NH2-terminal truncated cTnT; ELISA, enzyme-linked immunosorbant assay; mAb, monoclonal antibody; pI, isoelectric point; SDS-PAGE, SDS-polyacrylamide gel electrophoresis; TBS, Trisbuffered saline; TnC, troponin C; TnI, troponin I; TnT, troponin T.
References
- 1.Leavis PC, Gergly J. Thin filament proteins and thin filament-linked regulation of vertebrate muscle contraction. CRC Crit Rev Biochem. 1984;16:235–305. doi: 10.3109/10409238409108717. [DOI] [PubMed] [Google Scholar]
- 2.Zot AS, Potter JD. Structural aspects of troponin-tropomyosin regulation of skeletal muscle contraction. Annu Rev Biophys Biophys Chem. 1987;16:535–559. doi: 10.1146/annurev.bb.16.060187.002535. [DOI] [PubMed] [Google Scholar]
- 3.Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annu Rev Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 4.Perry SV. Troponin T: genetics, properties and function. J Muscle Res Cell Motil. 1998;19:575–602. doi: 10.1023/a:1005397501968. [DOI] [PubMed] [Google Scholar]
- 5.Breitbart RE, Nadal-Ginard B. Complete nucleotide sequence of the fast skeletal troponin T gene. Alternatively spliced exons exhibit unusual interspecies divergence. J Mol Biol. 1986;188:313–324. doi: 10.1016/0022-2836(86)90157-9. [DOI] [PubMed] [Google Scholar]
- 6.Jin JP, Huang QQ, Yeh H-I, Lin JJC. Complete nucleotide sequence and structural organization of rat cardiac troponin T gene. A single gene generates embryonic and adult isoforms via developmentally regulated alternative splicing. J Mol Biol. 1992;227:1269–1276. doi: 10.1016/0022-2836(92)90540-z. [DOI] [PubMed] [Google Scholar]
- 7.Huang QQ, Chen A, Jin JP. Genomic sequence and structural organization of mouse slow skeletal muscle troponin T gene. Gene. 1999;229:1–10. doi: 10.1016/s0378-1119(99)00051-7. [DOI] [PubMed] [Google Scholar]
- 8.Jin JP, Lin JJC. Rapid purification of mammalian cardiac troponin T and its isoform switching in rat hearts during development. J Biol Chem. 1988;263:7309–7315. [PubMed] [Google Scholar]
- 9.Wang J, Jin JP. Primary structure and developmental acidic to basic transition of 13 alternatively spliced mouse fast skeletal muscle troponin T isoforms. Gene. 1997;193:105–114. doi: 10.1016/s0378-1119(97)00100-5. [DOI] [PubMed] [Google Scholar]
- 10.Jin JP, Chen A, Huang QQ. Three alternatively spliced mouse slow skeletal muscle troponin T isoforms: conserved primary structure and regulated expression during postnatal development. Gene. 1998;214:121–129. doi: 10.1016/s0378-1119(98)00214-5. [DOI] [PubMed] [Google Scholar]
- 11.Pearlstone JR, Smillie LB. Binding of troponin-T fragments to several types of tropomyosin. Sensitivity to Ca2+ in the presence of troponin-C. J Biol Chem. 1982;257:10587–10592. [PubMed] [Google Scholar]
- 12.Pearlstone JR, Smillie LB. The interaction of rabbit skeletal muscle troponin-T fragments with troponin-I. Can. J Biochem. 1985;63:212–218. doi: 10.1139/o85-030. [DOI] [PubMed] [Google Scholar]
- 13.Hinkle A, Goranson A, Butters CA, Tobacman LS. Roles for the troponin tail domain in thin filament assembly and regulation. A deletional study of cardiac troponin T. J Biol Chem. 1999;274:7157–7164. doi: 10.1074/jbc.274.11.7157. [DOI] [PubMed] [Google Scholar]
- 14.Pan BS, Gordon AM, Potter JD. Deletion of the first 45 NH2-terminal residues of rabbit skeletal troponin T strengthens binding of troponin to immobilized tropomyosin. J Biol Chem. 1991;266:12432–12438. [PubMed] [Google Scholar]
- 15.Fujita S, Maéda K, Maéda Y. Expression in Escherichia coli and a functional study of a beta-troponin T 25 kDa fragment of rabbit skeletal muscle. J Biochem. 1992;112:306–308. doi: 10.1093/oxfordjournals.jbchem.a123896. [DOI] [PubMed] [Google Scholar]
- 16.Chandra M, Montgomery DE, Kim JJ, Solaro RJ. The N-terminal region of troponin T is essential for the maximal activation of rat cardiac myofilaments. J Mol Cell Cardiol. 1999;31:867–880. doi: 10.1006/jmcc.1999.0928. [DOI] [PubMed] [Google Scholar]
- 17.Gomes AV, Guzman G, Zhao J, Potter JD. Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development. Insights into the role of troponin T isoforms in the heart. J Biol Chem. 2002;277:35341–35349. doi: 10.1074/jbc.M204118200. [DOI] [PubMed] [Google Scholar]
- 18.Anderson PA, Greig A, Mark TA, Malouf NN, Oakeley AE, Ungerleider RM, Allen PD, Kay BK. Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circ Res. 1995;76:681–686. doi: 10.1161/01.res.76.4.681. [DOI] [PubMed] [Google Scholar]
- 19.Biesiadecki BJ, J-P Jin. Exon skipping in cardiac troponin T of turkeys with inherited dilated cardiomyopathy. J Biol Chem. 2002;277:18459–18468. doi: 10.1074/jbc.M200788200. [DOI] [PubMed] [Google Scholar]
- 20.Biesiadecki BJ, Elder BD, Yu ZB, Jin JP. Cardiac troponin T variants produced by aberrant splicing of multiple exons in animals with high instances of dilated cardiomyopathy. J Biol Chem. 2002;277:50275–50285. doi: 10.1074/jbc.M206369200. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Jin JP. Conformational modulation of troponin T by configuration of the NH2-terminal variable region and functional effects. Biochemistry. 1998;37:14519–14528. doi: 10.1021/bi9812322. [DOI] [PubMed] [Google Scholar]
- 22.Jin JP, Root DD. Modulation of troponin T molecular conformation and flexibility by metal ion binding to the NH2-terminal variable region. Biochemistry. 2000;39:11702–11713. doi: 10.1021/bi9927437. [DOI] [PubMed] [Google Scholar]
- 23.Jin JP, Chen A, Ogut O, Huang QQ. Conformational modulation of slow skeletal muscle troponin T by an NH(2)-terminal metal-binding extension. Am J Physiol Cell Physiol. 2000;279:C1067–1077. doi: 10.1152/ajpcell.2000.279.4.C1067. [DOI] [PubMed] [Google Scholar]
- 24.Yu ZB, Gao F, Feng H, Jin JP. Role of myofilament protein isofrm regulation in the decreases in contractile force and tolerance to fatigue of slow skeletal muscles after unloading. Biophys J. 2002;82:394a. [Google Scholar]
- 25.Sumandea MP, Burkart EM, Kobayashi T, De Tombe PP, Solaro RJ. Molecular and integrated biology of thin filament protein phosphorylation in heart muscle. Ann NY Acad Sci. 2004;1015:39–52. doi: 10.1196/annals.1302.004. [DOI] [PubMed] [Google Scholar]
- 26.Hirano K, Hirano M, Kanaide H. Regulation of myosin phosphorylation and myofilament Ca2+ sensitivity in vascular smooth muscle. J Smooth Muscle Res. 2004;40:219–236. doi: 10.1540/jsmr.40.219. [DOI] [PubMed] [Google Scholar]
- 27.Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 28.Murphy AM, Kogler H, Georgakopoulos D, McDonough JL, Kass DA, Van Eyk JE, Marban E. Transgenic mouse model of stunned myocardium. Science. 2000;287:488–491. doi: 10.1126/science.287.5452.488. [DOI] [PubMed] [Google Scholar]
- 29.Communal C, Sumandea M, de Tombe P, Narula J, Solaro RJ, Hajjar RJ. Functional consequences of caspase activation in cardiac myocytes. Proc Natl Acad Sci USA. 2002;99:6252–6256. doi: 10.1073/pnas.092022999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu ZB, Zhang LF, Jin JP. A proteolytic NH2-terminal truncation of cardiac troponin I that is up-regulated in simulated microgravity. J Biol Chem. 2001;276:15753–15760. doi: 10.1074/jbc.M011048200. [DOI] [PubMed] [Google Scholar]
- 31.Barbato JC, Huang QQ, Hossain MM, Bond M, Jin JP. Proteolytic N-terminal truncation of cardiac troponin I enhances ventricular diastolic function. J Biol Chem. 2005;280:6602–6609. doi: 10.1074/jbc.M408525200. [DOI] [PubMed] [Google Scholar]
- 32.Ohtsuki I, Shiraishi F, Suenaga N, Miyata T, Tanokura M. A 26K fragment of troponin T from rabbit skeletal muscle. J Biochem. 1984;95:1337–1342. doi: 10.1093/oxfordjournals.jbchem.a134740. [DOI] [PubMed] [Google Scholar]
- 33.Kitamura S, Muroya S, Tanabe S, Okumura T, Chikuni K, Nishimura T. Mechanism of production of troponin T fragments during postmortem aging of porcine muscle. J Agric Food Chem. 2005;53:4178–4181. doi: 10.1021/jf047974l. [DOI] [PubMed] [Google Scholar]
- 34.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 35.Cong JY, Goll DE, Peterson AM, Kapprell HP. The role of autolysis in activity of the Ca2+-dependent proteinases (mu-calpain and m-calpain) J Biol Chem. 1989;264:10096–10103. [PubMed] [Google Scholar]
- 36.Di Lisa F, De Tullio R, Salamino F, Barbato R, Melloni E, Siliprandi N, Schiaffino S, Pontremoli S. Specific degradation of troponin T and I by mu-calpain and its modulation by substrate phosphorylation. Biochem J. 1995;308:57–61. doi: 10.1042/bj3080057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biesiadecki BJ, Jin JP. N-terminal truncation of cardiac troponin T by restricted proteolysis in myocardial ischemia-reperfusion. Biophys J. 2005;88:360a. [Google Scholar]
- 38.Huang QQ, Brozovich FV, Jin JP. Fast skeletal muscle troponin T increases the cooperativity of transgenic mouse cardiac muscle contraction. J Physiol (London) 1999;520:231–242. doi: 10.1111/j.1469-7793.1999.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin JP, Brotto MA, Hossain MM, Huang QQ, Brotto LS, Nosek TM, Morton DH, Crawford TO. Truncation by Glu180 nonsense mutation results in complete loss of slow skeletal muscle troponin T in a lethal nemaline myopathy. J Biol Chem. 2003;278:26159–26165. doi: 10.1074/jbc.M303469200. [DOI] [PubMed] [Google Scholar]
- 40.Jin JP, Wang J, Zhang J. Expression of cDNAs encoding mouse cardiac troponin T isoforms: characterization of a large sample of independent clones. Gene. 1996;168:217–221. doi: 10.1016/0378-1119(95)00803-9. [DOI] [PubMed] [Google Scholar]
- 41.Jin JP. Cloned rat cardiac titin class I and class II motifs. Expression, purification, characterization, and interaction with F-actin. J Biol Chem. 1995;270:6908–6916. [PubMed] [Google Scholar]
- 42.Guttmann RP, Elce JS, Bell PD, Isbell JC, Johnson GVW. Oxidation inhibits substrate proteolysis by calpain I but not autolysis. J Biol Chem. 1997;272:2005–2012. doi: 10.1074/jbc.272.3.2005. [DOI] [PubMed] [Google Scholar]
- 43.Wang KK, Nath R, Posner A, Raser KJ, Buroker-Kilgore M, Hajimohammadreza I, Probert AW, Jr, Marcoux FW, Ye Q, Takano E, Hatanaka M, Maki M, Caner H, Collins JL, Fergus A, Lee KS, Lunney EA, Hays SJ, Yuen P. An alpha-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. Proc Natl Acad Sci USA. 1996;93:6687–6692. doi: 10.1073/pnas.93.13.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smillie LB. Preparation and identification of alpha- and beta-tropomyosins. Methods Enzymol. 1982;85:234–241. doi: 10.1016/0076-6879(82)85023-4. [DOI] [PubMed] [Google Scholar]
- 45.Solaro RJ, Pang DC, Briggs FN. The purification of cardiac myofibrils with Triton X-100. Biochim Biophys Acta. 1971;245:259–262. doi: 10.1016/0005-2728(71)90033-8. [DOI] [PubMed] [Google Scholar]
- 46.Jin JP, Yang FW, Yu ZB, Ruse C, Bond M, Chen A. The highly conserved COOH-terminus of troponin I forms a Ca2+-modulated allosteric domain in the troponin complex. Biochemistry. 2001;40:2623–2631. doi: 10.1021/bi002423j. [DOI] [PubMed] [Google Scholar]
- 47.Lin JJC, Chou CS, Lin JLC. Monoclonal antibodies against chicken tropomyosin isoforms: production, characterization, and application. Hybridoma. 1985;4:223–242. doi: 10.1089/hyb.1985.4.223. [DOI] [PubMed] [Google Scholar]
- 48.Wang Q, Reiter RS, Huang QQ, Jin JP, Lin JJ. Comparative studies on the expression patterns of three troponin T genes during mouse development. Anat Rec. 2001;263:72–84. doi: 10.1002/ar.1078. [DOI] [PubMed] [Google Scholar]
- 49.Murphy AM. Heart failure, myocardial stunning, and troponin: a key regulator of the cardiac myofilament. Congest Heart Fail. 2006;12:32–38. quiz 39–40. doi: 10.1111/j.1527-5299.2006.04320.x. [DOI] [PubMed] [Google Scholar]
- 50.Ogut O, Jin JP. Developmentally regulated, alternative RNA splicing-generated pectoral muscle-specific troponin T isoforms and role of the NH2-terminal hypervariable region in the tolerance to acidosis. J Biol Chem. 1998;273:27858–27866. doi: 10.1074/jbc.273.43.27858. [DOI] [PubMed] [Google Scholar]
- 51.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 52.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Ca(2+)-regulated structural changes in troponin. Proc Natl Acad Sci USA. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin JP, Samanez RA. Evolution of a metal-binding cluster in the NH(2)-terminal variable region of avian fast skeletal muscle troponin T: functional divergence on the basis of tolerance to structural drifting. J Mol Evol. 2001;52:103–116. doi: 10.1007/s002390010139. [DOI] [PubMed] [Google Scholar]
- 54.Piper HM, Meuter K, Schafer C. Cellular mechanisms of ischemia-reperfusion injury. Ann Thorac Surg. 2003;75:S644–S648. doi: 10.1016/s0003-4975(02)04686-6. [DOI] [PubMed] [Google Scholar]
- 55.Martin AF. Turnover of cardiac troponin subunits. Kinetic evidence for a precursor pool of troponin-I. J Biol Chem. 1981;256:964–968. [PubMed] [Google Scholar]
- 56.Colantonio DA, Van Eyk JE, Przyklenk K. Stunned peri-infarct canine myocardium is characterized by degradation of troponin T, not troponin I. Cardiovasc. Res. 2004;63:217–225. doi: 10.1016/j.cardiores.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 57.Foster DB, Noguchi T, VanBuren P, Murphy AM, Van Eyk JE. C-terminal truncation of cardiac troponin I causes divergent effects on ATPase and force: implications for the pathophysiology of myocardial stunning. Circ Res. 2003;93:917–924. doi: 10.1161/01.RES.0000099889.35340.6F. [DOI] [PubMed] [Google Scholar]