

Abstract
Human carboxylesterase 1 (hCE1) is a drug- and endobiotic-processing serine hydrolase that exhibits relatively broad substrate specificity. It has been implicated in a variety of endogenous cholesterol metabolism pathways including the following apparently disparate reactions: cholesterol ester hydrolysis (CEH), fatty acyl Coenzyme A hydrolysis (FACoAH), acyl-CoenzymeA:cholesterol acyltransfer (ACAT), and fatty acyl ethyl ester synthesis (FAEES). The structural basis for the ability of hCE1 to perform these catalytic actions involving large substrates and products has remained unclear. Here we present four crystal structures of the hCE1 glycoprotein in various complexes with endogenous substrates or substrate analogues: Coenzyme A, the fatty acid palmitate, and the bile acids cholate and taurocholate. While the active site of hCE1 was known to be promiscuous and capable of interacting with a variety of chemically-distinct ligands, these structures reveal that the enzyme contains two additional ligand binding sites and that each site also exhibits relatively non-specific ligand binding properties. Using this multisite promiscuity, hCE1 appears structurally capable of assembling several catalytic events depending, apparently, on the physiological state of the cellular environment. These results expand our understanding of enzyme promiscuity and indicate that, in the case of hCE1, multiple non-specific sites are employed to perform distinct catalytic actions.
Keywords: cholesterol metabolism, cholesterol esters, foam cells, atherosclerosis, heart diseases
INTRODUCTION
Several mammalian enzyme systems are involved in the processing and detoxification of both endobiotic and xenobiotic compounds. Central among these are the microsomal drug metabolizing enzymes, including the cytochrome P450s (CYP), the UDP-glucuronosyltransferases (UGT), and the non-specific serine hydrolases, including the human carboxylesterase isoforms 1 and 2. Each of these enzymes or enzyme families exhibits promiscuity in substrate recognition and act on abundant, potentially toxic compounds from endogenous or exogenous sources. In many cases, the nature of enzymatic promiscuity remains mysterious, as it is not clear how one enzyme can act on structurally-distinct substrates with acceptable levels of catalytic efficiency.
Human carboxylesterase 1 (hCE1) is a broad spectrum serine hydrolase involved in metabolizing a variety of clinical and elicit drugs, including cocaine, heroin, meperidine, lidocaine, ACE inhibitors, as well as organophosphate compounds.1–9 The enzyme utilizes a standard two-step hydrolase mechanism in which the catalytic serine is activated for nucleophilic attack by adjacent histidine and glutamic acid residues in a catalytic triad. The acyl-enzyme intermediate formed between the serine and a portion of the substrate is released in the second step of the reaction either by an attacking water molecule (hydrolysis) or by an abundant alcohol (transesterification).1,10 In addition to its role in drug and xenobiotic metabolism, hCE1 is implicated in processing a variety of endobiotics associated with cholesterol and fatty acid homeostasis.1,3,4 The enzyme is expressed in numerous tissues associated with such protective roles, including liver, intestine, testis, kidney, lung, heart, and macrophage/monocytes.1,11 hCE1 has been reported to catalyze cholesteryl ester hydrolysis (CEH) actions, in which a cholesterol-fatty acid conjugate is hydrolyzed to cholesterol and a free fatty acid (Figure 1A).12,13 It has also been shown to perform a similar reaction with a fatty acyl Coenzyme A substrate, generating a free fatty acid and Coenzyme A.14,15 The considerable size of each of these substrates suggests that the enzyme must contain two pores into its active site, which had been hypothesized for mammalian CEs10 and is supported by the structural data reported here.
Figure 1. Endogenous Substrate Processing by hCE1.
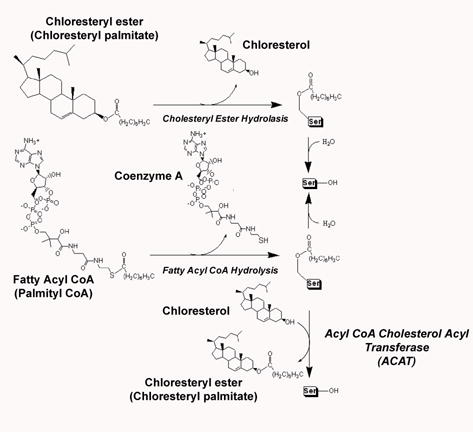
A) Cholesteryl ester hydrolysis (CEH), fatty acyl CoA hydrolysis (FACoAH), and acyl CoA cholesterol acyl transferase (ACAT) reactions.
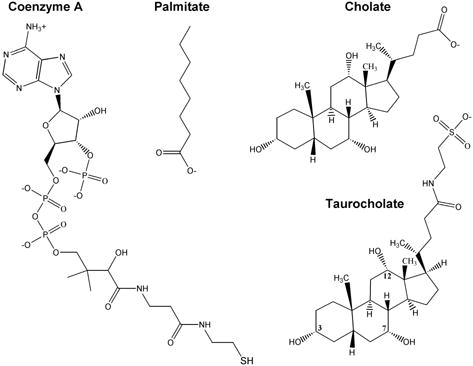
B) Endogenous ligands present in the crystal structures described here.
While the fatty acid acyl intermediate is covalently attached to hCE1, however, several transesterification reactions can occur rather than a simple hydrolysis, and these transesterications are likely driven by the presence of an abundant alcohol-containing compound (Figure 1A). For example, hCE1 is known to act as a fatty acyl ethyl ester synthase, in which an ethanol is transesterified to a fatty acid. When free cholesterol is in abundance, hCE1 has been reported to catalyze acyl-Coenzyme A: acyl transferase (ACAT) action that generates cholesteryl esters.16 Unraveling the structural bases of these varied catalytic actions is the focus of this paper.
Monocytes and macrophages contribute to the development of atherosclerosis, the cause of coronary heart disease (CHD).17 Fatty steaks, the primary atherosclerotic lesion, are initiated by the recruitment and differentiation of monocytes into macrophages at the endothelium of arterial walls. Macrophages then invade the arterial wall and take up significant quantities of oxidized LDL, forming excess cholesterol and fatty acids. These substrates then undergo re-esterification by acyl Coenzyme A cholesterol acyl transferase (ACAT) (Figure 1A). Excess cholesterol esters accumulate in cells, leading to the transformation of macrophages into foam cells forming atherosclerotic plaques, which can block coronary arteries causing a variety of coronary heart problems. hCE1, acting as a cholesteryl ester hydrolase (CEH), functions against ACAT by catalyzing the hydrolysis of cholesteryl esters into free cholesterol and fatty acids (Figure 1A).12,13 Overexpression of hCE1 increases intracellular free cholesterol, essential in extracellular transportation, and prevents foam cell transformation.12,13,17–20
hCE1 homologues have also been shown to play a crucial role in fat and cholesterol metabolism and transportation. In mouse liver, when serum cholesterol is in excess (as in the case of ApoE knockout mice) the expression of mouse hepatic CE significantly increases .21 In adipose tissues and intestine, these enzymes process the hydrolysis of triacylglycerol, the first step towards transporting free fatty acids and glycerol out of adipocytes via VLDL.22–26 These enzymes also play an important role in the hydrolysis of long chain acyl CoA; such as palmitoyl CoA, in liver and kidney. 1,27
Crystal structures of hCE1 have been reported in complexes with cocaine and heroin analogues, as well as with tacrine, mevastatin, tamoxifen and the potent dione inhibitor benzil. 10,28–30 These structures have revealed that the enzyme contains three ligand binding sites: the active site, which is located the bottom of a catalytic gorge, a “side door” secondary pore that leads into the active site from the surface of the enzyme, and the “Z-site” that controls the enzyme’s trimer-hexamer equilibrium. The enzyme uses the Z-site surface to form a hexamer; thus, the Z-site is only available to bind to ligands when the enzyme is in its trimeric form.28,29 While the role of the catalytic gorge in substrate binding is clear, the putative roles played by the side door and Z-site in shuttling substrates to the active site had not yet been firmly established structurally. Here, we examine the structural basis of hCE1’s role in endobiotic metabolism. Four crystal structures are presented to hCE1 in complexes with Coenzyme A, the fatty acid palmitate, and the bile acids cholate and taurocholate (Figure 1B). Based on the various snapshots of ligand binding obtained from these structures, we present models for the manner in which hCE1 can act as a CEH, FACoAH, FAEEH and ACAT. These structures reveal that each of the ligand binding site on hCE1 is promiscuous, and the enzyme appears to harness this promiscuity to assemble a variety of ostensively distinct catalytic functions.
RESULTS
hCE1 Structure
We have determined crystal structures of hCE1 in four complexes: CoA, homatropine/palmitate/CoA, palmitate/cholate, and taurocholate (Figure 1B). The cocaine analogue homatropine is known from previous structural studies to stabilize hCE1 and was used as a co-crystallant in some cases to improve crystal quality. The 2.0 Å structure of hCE1-CoA complex is the highest resolution of hCE1 determined to date. The structures of hCE1-homatropine/palmitate/CoA, hCE1-palmitate/cholate and hCE1-taurocholate were refined to 2.8, 3.0 and 3.2 Å, respectively (Table 1). In one asymmetric unit, hCE1 forms a hexamer in complex with CoA, and a trimer in the remaining complexes.
Table 1.
Crystallographic Statistics for Human Carboxylesterase 1 Structures
| Coenzyme A | Homatropine/Palmitate/Coenzme A | Cholate/Palmitate | Taurocholate | |
|---|---|---|---|---|
| Resolution (Å; highest shell) | 30-2.0 (2.0-2.13) | 50-2.8 (3.0-2.8) | 20-3.0 (3.2-3.0) | 31-3.2 (3.4-3.2) |
| Space Group | P21 | P212121 | P212121 | P212121 |
| Asymmetric Unit | One hexamer | One trimer | One trimer | One trimer |
| Cell Constants (Å, °) | a = 88.99 | a=55.56 | a=55.29 | a=55.42 |
| b = 115.37 | b=181.02 | b=179.88 | b=179.95 | |
| c = 175.53 | c=202.56 | c=201.32 | c=201.09 | |
| β = 90.05 | ||||
| Data Collection Facilities | SSRL | SSRL | UNC | SSRL |
| Total Reflections | 1798533 | 238528 | 499077 | 125478 |
| Unique Reflections | 233023 | 50837 | 38284 | 34194 |
| Mean Redundancy | 7.7 | 4.7 | 13.0 | 3.7 |
| Rsym1 (%; highest shell) | 8.2 (33.5) | 12.3 (34.1) | 11.0 (34.1) | 14.4 (40.8) |
| Wilson B factor (Å2) | 21.6 | 48.4 | 47.8 | 45.6 |
| Completeness (%; highest shell) | 97.1 (88.1) | 99.1 (99.7) | 93.0 (83.6) | 98.6 (97.6) |
| Mean I/σ ( highest shell) | 12.7 (2.9) | 8.5 (2.8) | 7.8 (2.4) | 8.5 (3.0) |
| Rcryst2 (%; highest shell) | 18.4 (24.1) | 19.3 (28.9) | 22.6 (30.2)4 | 21.9 (28.6)4 |
| Rfree3 (%; highest shell) | 22.0 (27.4) | 24.4 (35.5) | 27.1 (35.3)4 | 25.5 (31.5)4 |
| Number Protein Atoms5 | 24726 | 12390 | 12390 | 12390 |
| Number Solvent Sites5 | 2715 | 508 | 269 | 253 |
| Number Carbohydrate | 154 | 105 | 105 | 105 |
| Atoms5 | ||||
| Number Ligand Atoms5 | 288 | 120 | 174 | 210 |
| Number Ion Atoms5 | 60 | 33 | 30 | 30 |
| Bound Ligands | One CoA in each active site | One palmitate in two active sites, One CoA in one active site; One homatropine in each Z site; One palmitate in side door | One palmitate in each active site; One cholate in each Z site | One taurocholate in each active site and each Z site |
Rsym = ∑|I−<I>| / ∑I, where I is the observed intensity and <I> is the average intensity of multiple symmetry-related observations of that reflection.
Rcryst = ∑||Fobs|−|Fcalc|| / ∑|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively.
Rfree = ∑||Fobs|−|Fcalc|| / ∑|Fobs| for 7% of the data not used at any stage of structural refinement.
Non-crystallographic symmetry restraints were applied throughout the trimeric complexes except areas (4–6 Å sphere) in contact with ligands.
Number of atoms per asymmetric unit.
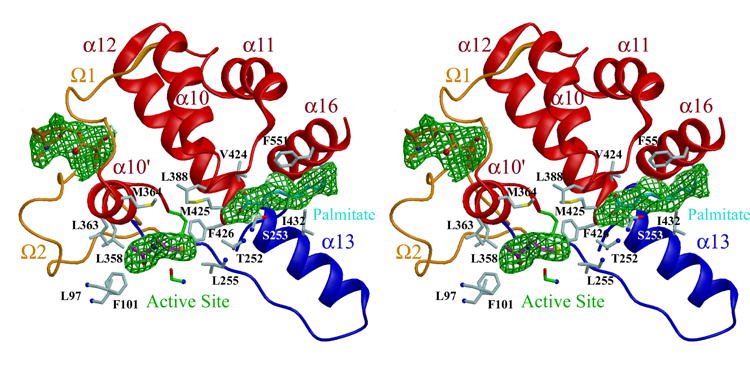
The hCE1 monomer is formed by 17 α-helices and 20 β-strands and is divided into catalytic, αβ, and regulatory domains (Figure 2A). The catalytic domain contains the catalytic triad composed of Ser221, His468 and Glu354, as well as a conserved high-mannose N-linked glycosylation found at Asn79. This carbohydrate moiety is thought to help in protein folding, solubility, and trimer stabilization.29 The αβ domain provides the bulk of the buried surface area in the hCE1 trimer. The regulatory domain is composed of α10–12, α10’ and α16, and two novel Ω loops. This region of the enzyme exhibits relatively higher thermal displacement parameters (crystallographic B-factors), and has been proposed to regulate substrate binding and product release.10,29
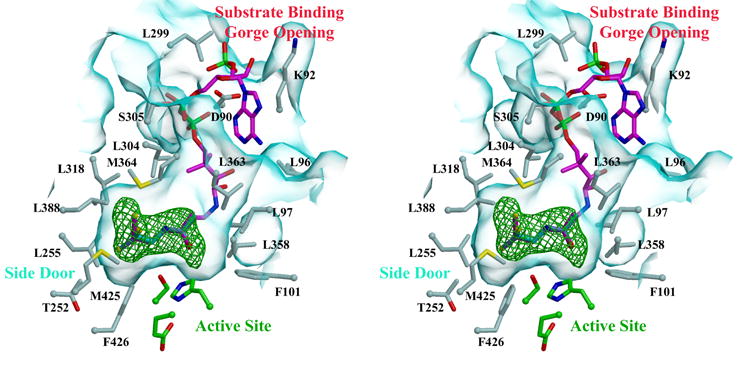
Figure 2. Overall Structure and CoA Binding.
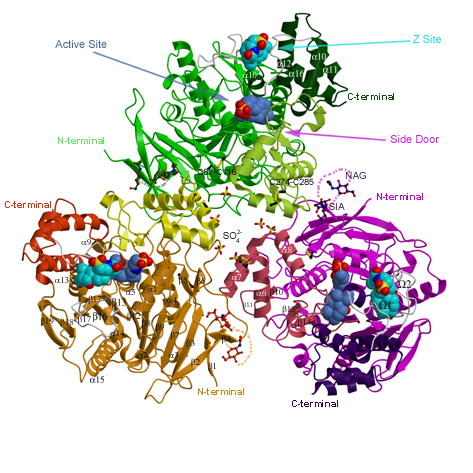
A) The hCE1 trimer colored as follows: the catalytic domains are in green, magenta, and orange; the αβ domains are in light green, pink, and yellow; and the regulatory domains are in dark green, purple, and red. Taurocholate ligands are shown in cornflower blue in the active site and cyan in the Z-site (in between 1 and 2 loops shown in grey). The glycosylation modifications, disulfide linkages, and sulfate ions are also shown.
B) Stereoview of Coenzyme A (CoA) within the active site of hCE1. Note that the thiol tail of CoA ligand is observed in three conformations. The surface of the substrate binding gorge is shown in cyan, while the surrounding residues are rendered in green for the active site and in grey for those lining the gorge. 2.0 Å resolution electron density contoured at 3σ from a simulated-annealing omit map calculated via |Fobs| − |Fcalc|, φcalc (with the ligand and a sphere 1.0 Å around the ligand omitted prior to annealing and map calculation) is shown in green.
hCE1 contains three ligand binding sites: the active site, the side door, and the Z-site (Figure 2A). The active site is located at the base of the catalytic gorge, which in hCE1 is relatively large and conformable to promote the promiscuous binding of substrates. Adjacent to the active site, and separated from the catalytic gorge by a thin wall of amino acid side chains, is the side door. This ligand binding site, first identified in a related mammalian CE structure,10 has been hypothesized to serve as a secondary substrate entrance and/or product release pore. The entrance to the catalytic gorge is flanked by α1 and α10’, a motif common to the active site of other fatty acid ester processing enzymes, including the palmitate protein thioesterases (PPT1 and PPT2).31–33 Adjacent to the gorge opening, Ω1, Ω2, and α10’ form the Z-site, 28,29 a surface ligand-binding site that controls the trimer-hexamer equilibrium of the enzyme.
hCE1-Coenzyme A Complex
The crystal structure of hCE1 crystallized in the presence of Coenzyme A (CoA) was determined and refined to 2.0 Å resolution, the highest resolution of an hCE1 complex reported to date (Table 1). The enzyme packed into a monoclinic space group and is in its hexameric state in this complex, similar to the tacrine and naloxone structures determined previously.28,29 Simulated-annealing omit electron density reveals the thiol tail of CoA bound adjacent to the hCE1 active site and present in 2–3 orientations per protein monomer (Figure 2B). We were unable to place the complete CoA molecule, however, which appeared to be mobile within the trimer-trimer interface of the hexamer. No ligand was observed bound at the side door region of the enzyme and, because of its hexameric state in this structure, the Z-sites of the enzyme were not available to interact with ligands. The position of the bound CoA tails suggests a mechanism by which the enzyme may perform hydrolyze the thioester linkages present in CoA-linked substrates, as discussed below.
hCE1-Cholate-Palmitate Complex
We next co-crystallized the enzyme in complex with cholate (a bile salt and water-soluble cholesterol analog) and palmitoyl CoA, and determined the structure to 3.0 Å resolution (Table 1). In this case, the enzyme packed into an orthorhombic unit cell in its trimeric state, with one trimer per asymmetric unit. In structures of hCE1 determined previously, the enzyme was observed to shift from a hexamer to a trimer when its Z-sites contained bound ligand. Indeed, this complex revealed that cholate molecules were bound in all three Z-sites present in the trimer. The hydroxyl group at position 3 of cholate projects into the Z-site, forming a hydrogen bond with the carbonyl oxygen of Gly356. Intact palmitoyl CoA was used as a co-crystallant in this complex; thus, we were surprised to find upon inspection of the electron density at the active site of the enzyme that only palmitate, the product of palmitoyl CoA hydrolysis, appeared bound in each monomer of the trimer (Figure 3A). This observation suggests that the catalytic activity of the enzyme may have generated this product. In addition, as discussed below, this structure provides insights into the roles that endogenous ligand binding may play in enzyme function, as well as the orientational promiscuity of substrate binding at the enzyme’s active site.
Figure 3. Fatty Acyl CoA Processing.
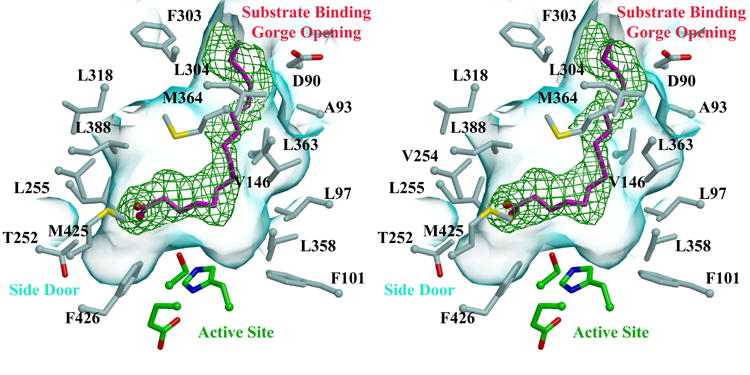
A) Stereoview of palmitate (magenta) within the substrate binding gorge of hCE1. Residues and surfaces are colored as shown in Figure 2B. 3.0 Å resolution electron density contoured at 2σ from a simulated-annealing omit map calculated via |Fobs| − |Fcalc|, φcalc (with the ligand and a sphere 1.0 Å around the ligand omitted prior to annealing and map calculation) is shown in green.
B) Stereoview of palmitate (cyan) at the side door, the thiol tail of CoA (magenta) at the active site and homatropine (orange) at the Z-site. The catalytic and regulatory domains are in cornflower blue and red. Three ligands are found bound in this structure, The catalytic residues are shown in green, while some amino acid side chains within the active site and side door are shown in light blue. 2.8 Å resolution electron density contoured at 2σ from a simulated-annealing omit map calculated via |Fobs| − |Fcalc|, φcalc (with the ligands and a sphere 1.0 Å around the ligands omitted prior to annealing and map calculation) is shown in green.
hCE1-Palmitate-Coenzyme A-Homatropine Complex
The cocaine analog homatropine can be used as a co-crystallant to occupy to the Z-site of hCE1, improving crystallization and diffraction quality.29 Thus, we next co-crystallized hCE1 in the presence of both homatropine and palmitoyl CoA, and determined the structure of the complex to 2.8 Å resolution (Table 1). As expected, homatropine was found at each of the Z-sites in the trimeric asymmetric unit. Similar to hCE1-homatropine complexes previously described,29 homatropine was observed to bind in two conformations at the Z-site of this complex and to form a combination of polar and non-polar contacts. At the active sites, we observed palmitate in two monomers of the asymmetric unit, bound in the orientation observed in the 3.0 Å resolution palmitate/cholate complex described above. In the third active site, we were surprised to find CoA bound as observed in the 2.0 Å resolution structure described above (Figure 3B). We were further surprised to find a palmitate molecule, contacted by Ser253, Val388, and Val424, bound at the side door of this particular monomer (Figure 3B). This is the first observation of a ligand bound to the side door of hCE1; to date, only 4-piperidino piperidine (4PP), a product of CPT-11 activation, has been observed bound to a mammalian CE side door, that of rabbit liver CE.10 There are two features unique to this monomer of hCE1 that may explain why, of the three present in the asymmetric unit of this structure, only it exhibited the binding of the CoA and palmitate products at the active site and side door, respectively. First, this monomer packs more tightly against other symmetrical molecules in the crystal lattice than the other two monomers. Thus, this more constrained environment may facilitate associations with two ligands. Second, the positions of two residues adjacent to the side door, Phe550 and Ser253, appear to accommodate the binding of the palmitate in this monomer relative to the other two (Figure 3B). The distance between these two residues in the palmitate-bound monomer is 6.8 Å, compared to 6.1 Å and 6.4 Å in the other two monomers of the trimer. The structure of this monomer advances our understanding of the potential for functional interactions between the active site and side door in hCE1, as outlined below (see Discussion).
hCE1-Taurocholate Complex
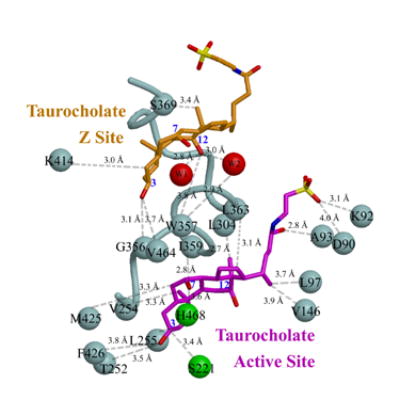
Finally, to focus on the structural basis of cholesteryl ester hydrolysis by hCE1, we co-crystallized the enzyme with another water-soluble cholesterol analog, taurocholate, and determined the structure of this complex to 3.2 Å resolution (Table 1). The enzyme packs into an orthorhombic unit cell common to its trimeric form, with one trimer present per asymmetric unit. Taurocholate molecules are observed bound at each Z-site and active site within the trimer (Figure 4A). Similar to the cholate binding to the Z-site described above, the hydroxyl group at the 3 position of taurocholate forms a hydrogen bond with the main-chain carbonyl oxygen of Gly356 (Figure 4B). In addition to van der Waals contacts between the enzyme and taurocholate, the bile acid is further stabilized by water-mediated interactions between its polar groups at positions 7 and 12 and polar groups on hCE1. At the active site, the cholesterol scaffold of taurocholate is aligned such that the hydroxyl at its 3 position faces down into the substrate binding gorge and the flexible aliphatic sulfate tail unambiguously projects out towards the gorge opening (Figure 4A). Again, a series of polar and non-polar contacts is formed between the enzyme and ligand, including a salt bridge between the sulfate group of the bile acid and the side chain of Lys92 (Figure 4B). Note that the active site and Z-site of the enzyme are separated only by a single short helix, α10’ (Figure 4B). The structural features of this complex suggest how cholesterol-based substrates may be aligned for processing by the enzyme, and how the presence of ligands bound at the Z-site may impact catalytic actions within the substrate binding gorge of the enzyme.
Figure 4. Taurocholate Binding to hCE1.
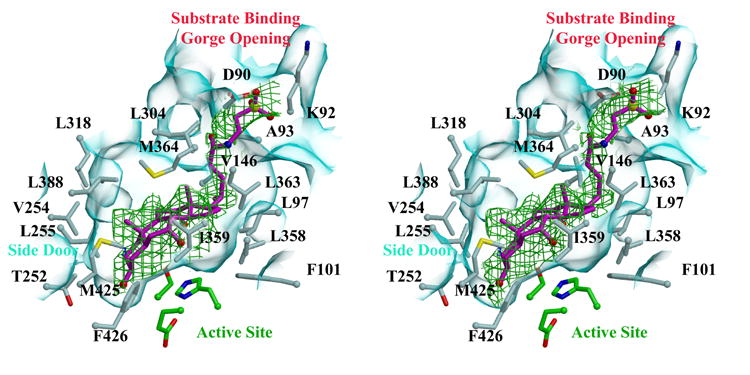
A) Stereoview of taurocholate (magenta) within the substrate binding gorge of hCE1. Residues and surfaces are colored as shown in Figure 2B. 3.2 Å resolution electron density contoured at 2σ from a simulated-annealing omit map calculated via |Fobs| − |Fcalc|, φcalc (with the ligand and a sphere 1.0 Å around the ligand omitted prior to annealing and map calculation) is shown in green.
B) Relationship between taurocholate bound at the active site (magenta) and Z-site (orange) in hCE1. α10’ separates the two sites, with contact residues shown as grey balls, the catalytic residues as green balls, and water molecules in the Z-site as red balls.
DISCUSSION
Z-Site Promiscuity and Allostery
We have proposed previously based on structural and biophysical studies that ligand binding to the hCE1 Z-site shifts the enzyme’s trimer-hexamer equilibrium toward trimer, facilitating the binding of substrates within the active site and promoting catalysis.29,30 The data presented here support this hypothesis. In addition, they further suggest that the Z-site is capable of binding to cholesterol-like molecules, a feature that may play an important role the biological activity of the enzyme in terms of processing cholesteryl esters and other endogenous substrates. The binding of cholesterol-scaffold ligands to the hCE1 Z-site would shift the enzyme from its hexamer to its trimer state, opening up access to its active site and, presumably, promoting catalysis. The Z-site may play a more direct role in the allosteric activation of catalysis. The catalytic triad residue Glu354 lies on a turn just prior in sequence to α10’, the helix that physically separates the active site and Z-site regions of hCE1 and is directly contacted by ligands bound in both sites (Figure 4B). The association of compounds at the Z-site may facilitate the proper positioning of Glu354 for catalysis. Indeed, in the structure of the related rabbit liver CE, in which the region corresponding to α10’ was disordered, the Glu354-equivalent residue (Glu353) was observed in a position not amenable to catalysis.10
These structural observations, as well as recent kinetic data indicating that inhibitors that associate with both the active site and Z-site are mixed-type inhibitors of hCE1,30 strongly suggest that the Z-site functions allosterically in hCE1 activity. In addition, the wide variety of compounds observed to bind to the hCE1 Z-site (e.g., homatropine, tamoxifen, cholate, taurocholate) firmly establish the promiscuity of this region of the enzyme, a feature that mirrors the promiscuity of the active site. If the Z-site functioned allosterically, such relative non-specificity in ligand binding would be expected. Recently, the crystal structure of the human cytochrome P450 CYP3A4 isoform revealed that, similar to the Z-site in hCE1, this drug metabolizing enzyme contains a surface ligand binding site adjacent to its active site.34 This peripheral binding site is proposed to function in substrate screening and allosteric regulation.34 A similar role may exist for the Z-site of hCE1, a conclusion that is supported by the data presented here and by previous structural and functional studies.29,30
hCE1 Side Door Structure and Function
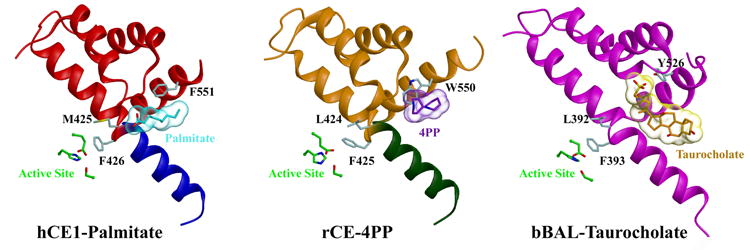
While a product of CPT-11 activation 4PP had previously been observed bound at the side door region of rabbit liver CE (rCE),10 the observation of the fatty acid palmitate bound at hCE1’s side door is the first for this human CE isoform. There are three common features of the side door shown in the crystal structures of hCE1, rCE and a related enzyme-bovine bile salt activated lipase (bBAL).10,35,36 These include (1) a “gate” residue (Met425, Leu424, and Leu392 in hCE1, rCE and bBAL, respectively); (2) a conserved “switch” Phe (Phe426, Phe425, and Phe393 in hCE1, rCE and bBAL, respectively) located in between α12 and α13; and (3) an aromatic “releasing value” residue (Phe551, Trp550, and Tyr526 in hCE1, rCE and bBAL, respectively) located in the terminal helix of each enzyme (Figure 5A–B).
Figure 5. Mechanistic Hypotheses for Endogenous Substrate Processing.
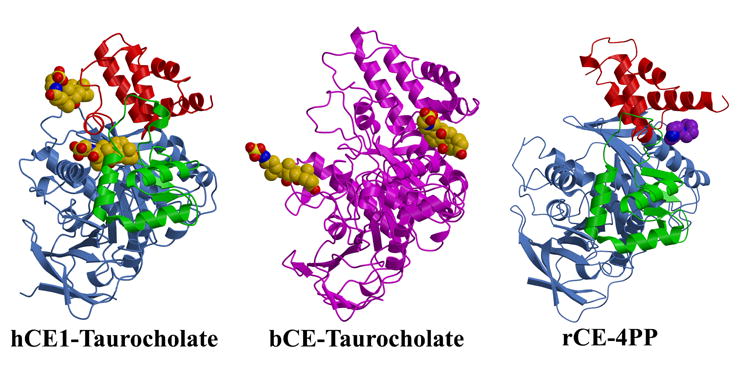
A) The crystal structures of the hCE1-taurocholate complex, the bovine salt-activated lipase (bCE)-taurocholate complex, and the rabbit liver carboxylesterase (rCE)-4-piperidinopiperidine (4PP) complex are shown. The three domains of hCE1 and rCE (catalytic, αβ , and regulatory) are rendered in cornflower blue, green, and red, respectively, while the lipase is shown in magenta. Note that the taurocholate surface binding sites of hCE1 (the Z-site) and the lipase are distinct
B) The active site and side door regions of hCE1, rCE and the bovine salt-activated lipase (bBAL). Note that the ligands, palmitate (in cyan) for hCE1, 4PP (in purple) for rCE, and taurocholate (in orange) for bBAL, are located at an equivalent side door region in each enzyme located in proximity to the active site.
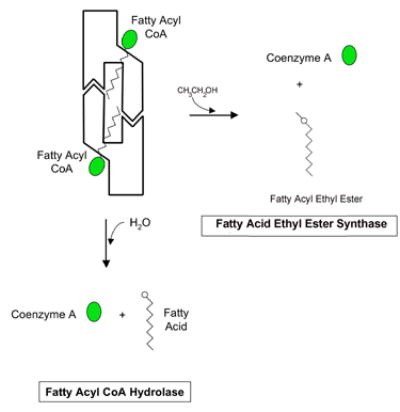
C) Schematic models of fatty acyl CoA hydrolysis and fatty acyl ethyl ester synthesis by the hCE1 hexamer. When the fatty acyl CoA is abundant, it is possible that the hexameric hCE1 would allow the acyl CoA substrate to enter the active site through the side door. See Figure 5D for a definition of the schematic regions of hCE1.
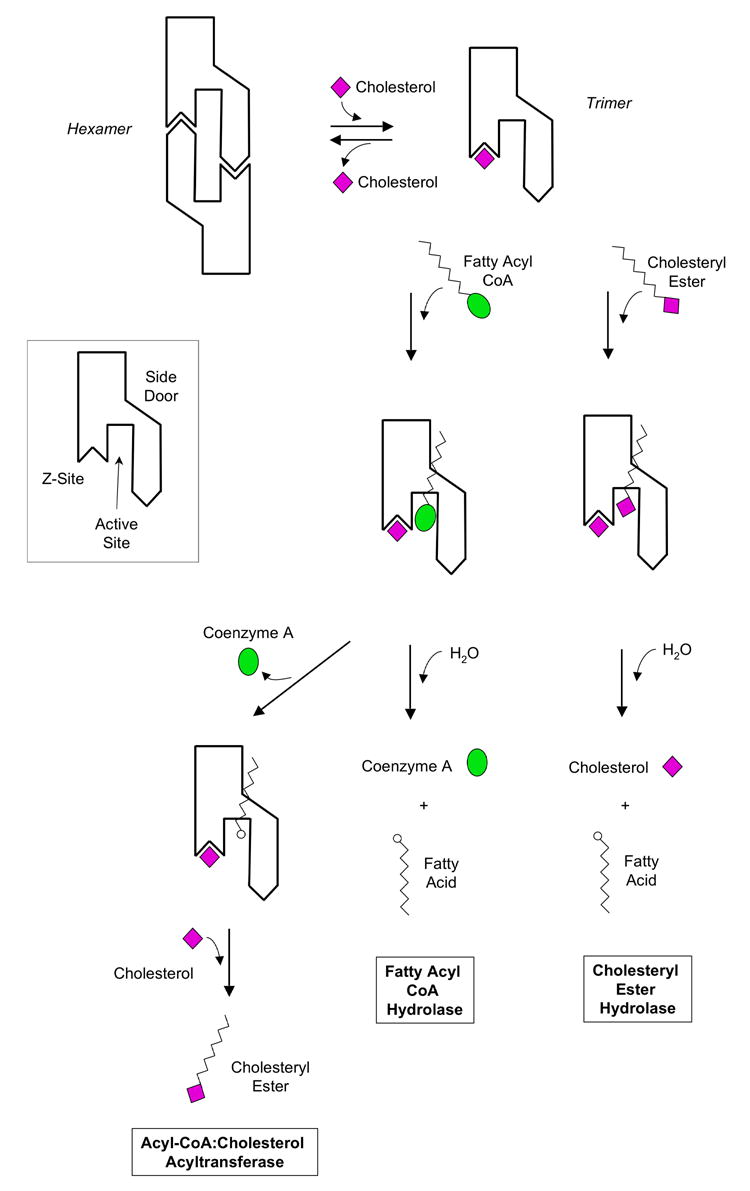
D) Schematic models of cholesteryl ester hydrolysis, fatty acyl CoA hydrolysis, and acyl CoA cholesterol acyl transferase by the hCE1 trimer. First, excess cholesterol could bind the Z-site and shift the trimer-hexamer equilibrium towards trimer. Second, the substrates fatty acyl CoA or a cholesteryl ester could enter the active site through the main substrate binding gorge. For hydrolysis reactions, products (e.g., CoA, cholesterol and fatty acids) could leave through the main substrate binding gorge and the side door. For a transesterification, cholesterol may enter the active site via the substrate binding gorge, producing a cholesteryl ester.
First, the gate residues are highly variable and mutations of these residues alter the activities of the enzyme towards particular substrates.37,38 The binding of palmitate at the side door in the hCE1 structure suggests that Met425 is perhaps the most important residue in the release of the fatty acid after hydrolysis of cholesteryl esters or acyl CoA. This residue corresponds to that described by Wallace et al37, in which a mutation of the equivalent residue in rat homologue of hCE1 alters its substrate preference from carboxylesterase to cholesterol ester hydrolase. In addition, the structures of bBAL suggested that the fatty acid leaving product may have exited the enzyme through a hydrophobic channel analogous to the side door that is guarded by only one tryptophan residue, Trp227.35,36
Second, we have observed a conserved Phe between two connected helices of the catalytic domain and the regulatory domain, immediately after Met425. This residue provides a link between the catalytic and the regulatory domains. Therefore, we hypothesize that when the product exits the active site through the “gate” residues, it would induce a conformational change in the area of these helices. We propose that the conserved Phe functions as a hinge allowing rotation of the regulatory domain, while Met425 functions as a gate allowing the release of product such as fatty acids through the side door.
Finally, the conserved aromatic residue in the terminal helix is always seen in association with the product at the side door. It has been proposed that the binding of taurocholate at the equivalent side door in bBAL may help to organize this region of the enzyme for the release of long chain fatty acids.36 In rCE, this stabilization of the side door is provided by a carbohydrate chain that results from post-translational modification of the protein;10 this glycosylation site is missing in hCE1. We hypothesize that this aromatic side chain along with other additional functional domains (e.g. carbohydrate modifications or bile salts) may provide a valve to allow release of products from the side door. In addition, it may function as secondary signaling mechanism for the regulatory domain, and may allow the release of ligands both at the active site and the Z-site.
Catalytic Promiscuity toward Endogenous Substrates
The four structures presented here outline suggest a variety of sometimes conflicting models for the processing of large, endogenous substrates by hCE1. For example, the hCE1-CoA complex indicates that cleavage of a fatty acyl-CoA substrate would require that the long fatty acyl tail extrude from the active site of the enzyme through the side door to allow for the positioning of the scissile thioester linkage at the catalytic residues. In contrast, the orientation of palmitate product observed in the hCE1-cholate-palmitate structure, in which palmitate was apparently generated via cleavage of the palmitoyl CoA compound used in crystallization, suggests the opposite orientation with the CoA at the side door and the palmitate in the catalytic gorge. The static nature and structural features of these two complexes do not allow us to distinguish between these options. However, the snapshot provided by one monomer of the three present in the hCE1-palmitate-CoA-homatropine complex lends support to the mechanism in which a fatty acyl-CoA binds with the CoA in the catalytic gorge and the fatty acid tail extruded via the side door. In this complex, CoA and palmitate were observed bound at the active site and side door regions of the enzyme, respectively, which are the positions expected if the substrate utilized extrusion of the fatty acid tail from the side door. This orientation also suggests that the palmitate product would be released from the catalytic gorge via the side door, as had been proposed previously for 4PP in the rabbit liver CE.10 In terms of cholesteryl ester hydrolysis, the taurocholate-bound hCE1 structure also suggests that productive hydrolysis would require extrusion of the fatty acyl tail from the active site via the side door.
Based on the structural data presented here, two overall scenarios of hCE1 action in the processing of endogenous substrates can be envisioned (Figures 5C–D). In the first scenario, hCE1 is considered in its hexameric state (Figure 5C), in which the binding of a ligand or substrate to the Z-site has not shifted the trimer-hexamer equilibrium toward trimer. For large aliphatic substrates like fatty acyl Coenzyme A, access to the active site of hCE1 may be gained through the side door, which would align the thioester linkage for catalysis with the larger Coenzyme A moiety located outside the enzyme. This arrangement would allow for either hydrolysis for fatty acyl CoA hydrolase activity or for transesterication for fatty acid ethyl ester synthase activity (Figure 5C). The binding of the palmitate fatty acid with its charged carboxylate adjacent to the catalytic residues in palmitate/cholate structure described here (Figure 3A) is consistent with this model. Recall that palmitoyl-CoA was used for co-crystallization in this complex so the palmitate observed was likely generated by fatty acyl CoA hydrolysis.
In the second scenario, hCE1 is considered in its trimeric state, in which the binding of a ligand (e.g., cholesterol as shown schematically) has shifted the trimer-hexamer equilibrium toward trimer (Figure 5D). More open access to the catalytic gorge of the enzyme allows substrates to bind directly to the active site. For both fatty acyl CoA and cholesteryl esters, however, productive binding would require the extrusion of the fatty acyl tail out of the active site through the side door, as shown schematically (Figure 5D). The observed binding of palmitate at the side door in the homatropine/palmitate/CoA structure (Figures 3B) and of taurocholate at the active site in the taurocholate structure (Figures 4A–B) are consistent with this proposal, as are the observed binding of the cholate and taurocholate in the Z-sites of the palmitate/cholate, and taurocholate structures, respectively (Figures 2A, 4B). Such arrangements allow for the enzyme to act as a cholesteryl ester hydrolase, and fatty acyl CoA hydrolase or, via transesterification with cholesterol, as a acyl-CoA:cholesterol acyltransferase (ACAT; Figure 5D). Other enzymes that specifically catalyze palmitoyl ester hydrolysis including palmitoyl-protein thioesterases (PPTs) have obvious alcohol and acyl binding sites.31,40 All PPT structures show a specific binding groove for palmitate. However, our structures suggest that hCE1 has no rigidly-assigned acyl or alcohol binding sites, but rather orients the substrate promiscuously. The promiscuous binding of fatty acid substrates has not been previously reported and may be unique to hCE1 and hCE1-like enzymes. It has been shown that in a rat liver trimeric homolog of hCE1 functions as fatty acid ethyl ester synthase and specific inhibitors can be used in cell culture to prevent the formation of fatty acyl ethyl esters (FAEE).40,41
One dilemma with this model for hCE1 catalysis in its trimeric state is that the largely hydrophobic fatty acid leaving group would be released into the cytoplasm. We propose that this fatty acid product be bound by a lipid cargo protein such as the fatty acid binding protein (FABP). There is evidence that hormone sensitive lipase (HSL), another similar cholesterol ester hydrolase, functions together with FABP. Tissue specific FABP will bind to HSL only when there are fatty acids present, with the absence of fatty acids completely abolishing this interaction.42 FABPs are present in all tissues that express hCE1, and thus FABP may facilitate the departure of the free fatty acid product through the side door.
It is important to note that cholesterol is shown schematically bound to the Z-site of hCE1 in Figure 5D; however, one can envision other ligands, substrates, or substrate analogues bound in that putative allosteric site to shift the enzyme’s oligomeric state toward it more open trimer form. In addition, one can also imagine the hCE1 trimer floating on a lipid droplet with its active sites facing “down” as seen in macrophage foam cell,19,20 and in this way able to draw cholesteryl ester substrates directly into its active site. Thus, the enzyme’s multiple sites (active, Z, side door) and oligomeric state (trimer, hexamer) may respond in various ways to the physiological nature of the cellular environment. During foam cell formation or reverse cholesterol transport, hCE1 may interact with lipid droplets as described and process abundant substrates as a cholesteryl ester hydrolase, for example. Under other conditions, the enzyme may reverse its action and perform fatty acyl CoA hydrolysis or ACAT events (Figure 5D). In all cases, it would be expected that hCE1 acts as an important back-up to other enzyme systems charged with the primary tasks of processing endogenous substrates (e.g., the human ACAT isoforms), and is called into action when abundant compounds are present. Thus, the utilization of an enzyme that contains multiple promiscuous sites and the ability to bind to substrates in various orientations would be expected to be a great asset to cell types like hepatocytes and macrophages that must adjust to significant changes in the levels of key endogenous substrates.
EXPERIMENTAL PROCEDURES
Crystallization
A secreted, 62 kDa form of hCE1 was expressed using baculovirus in Spodoptera frugiperda Sf21 cells and purified as previously described.43,44 hCE1 was concentrated to 3 mg ml−1 in 50mM HEPES pH 7.4, and crystallized in the presence of different ligands; 10 mM Coenzyme A, 10 mM homatropine-10 mM palmitoyl Coenzyme A, 10 mM cholic acid-10 mM palmitoyl Coenzyme A, or 10 mM taurocholate; using sitting drop vapor diffusion at 22°C. Crystals of 200–300 μm in size grew in 8% (w/v) PEG-3350, 0.4 M Li2SO4, 0.1 M NaCl, 0.1 M LiCl, 0.1 M citrate pH 5.5, and 5% glycerol. In the case of homatropine-palmitoyl Coenzyme A complexes, 0.1 mM NaF was also added. The crystals were cryo-protected in 20–40% (w/v) sucrose plus 75–50% (v/v) mother liquor prior to flash cooling in liquid nitrogen.
Structure Determination and Refinement
Diffraction data were collected at Stanford Synchrotron Radiation Laboratory (SSRL) beamlines 9-1 and 9-2, and at the X-ray Facility at the University of North Carolina-Chapel Hill. The experiments were performed at 100 K using cryo-cooled crystals, and were processed and reduced using DENZO and SCALEPACK, MOSFLM, and HKL2000.45,46 These hCE1 structures were determined by molecular replacement (AMoRe)47 using the structure of hCE1 in complex with tacrine (RBSC code: 1MX1)28 as a search model. Residues 21-553 of the 566 amino acid enzyme were traced for each protein monomer. Structures were refined using torsion angle dynamics in CNS48 with the maximum likelihood function target, and included an overall anisotropic B-factor and a bulk solvent correction. Five to seven percent of the observed data were set aside for cross-validation using free-R prior to any refinement. In the higher resolution structures, hCE1-CoA (2 Å) and hCE1-homatropine-palmitate-CoA (2.8 Å) Complexes, non-crystallographic symmetry (NCS) restraints were employed for each structure at initial refinement stages, and then removed such that each monomer was refined independently. However, for the moderate resolution structures (3.0–3.2 Å) we utilize total NCS restraints in the initial refinement stages and later kept the NCS restraints only in the residues that have no direct contact with the ligands (at least 4 to 6 Å away from the ligands). Manual adjustments were performed using the program O49 and σA-weighted50 electron density maps. Simulated annealing omit and σA-weighted difference density maps were used to position ligands into electron density at the active site, Z-site, or side door. N-linked glycosylation sites were traced in all complexes. Final structures exhibit good geometry with no Ramachandran outliers. Molecular graphic figures were created with MolScript,51 BobScript,52 Raster3D,53 Grasp,54 and Dino (Philippsen, A., www.dino3d.org.).
Acknowledgments
The authors thank C. Chen, J. Chrencik, Y. Xue, E. Ortlund, S. Sakai, K. Park, C. Fleming, B. Hamel, L. Betts, G. Pielak, and D. Erie for discussions and experimental assistance.
References
- 1.Williams FM. Clinical significance ofbesterases in man. Clin Pharmacokinet. 1985;10:392–403. doi: 10.2165/00003088-198510050-00002. [DOI] [PubMed] [Google Scholar]
- 2.Satoh T, Hosokawa M. The mammalian carboxylesterases: from molecules to functions. Annu Rev Pharmacol Toxicol. 1998;38:257–288. doi: 10.1146/annurev.pharmtox.38.1.257. [DOI] [PubMed] [Google Scholar]
- 3.Redinbo MR, Bencharit S, Potter PM. Human carboxylesterase 1: from drug metabolism to drug discovery. Biochem Soc Trans. 2003;31:620–624. doi: 10.1042/bst0310620. [DOI] [PubMed] [Google Scholar]
- 4.Redinbo MR, Potter PM. Mammalian carboxylesterases: from drug targets to protein therapeutics. Drug Discov Today. 2005;10:313–325. doi: 10.1016/S1359-6446(05)03383-0. [DOI] [PubMed] [Google Scholar]
- 5.Kamendulis LM, Brzezinski MR, Pindel EV, Bosron WF, Dean RA. Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J Pharmacol Expt Ther. 1996;279:713–717. [PubMed] [Google Scholar]
- 6.Zhang J, Burnell JC, Dumaual N, Bosron WF. Binding and hydrolysis of meperidine by human liver carboxylesterase hCE-1. J Pharmacol Expt Ther. 1999;290:314–338. [PubMed] [Google Scholar]
- 7.Alexson SE, Diczfalusy M, Halldin M, Swedmark S. Involvement of liver carboxylesterases in the in vitro metabolism of lidocaine. Drug Metab Dispos. 2002;30:643–647. doi: 10.1124/dmd.30.6.643. [DOI] [PubMed] [Google Scholar]
- 8.Geshi E, Kimura T, Yoshimura M, Suzuki H, Koba S, Sakai T, Saito T, Koga AMuramatsu M, Katagiri T. A single nucleotide polymorphism in the carboxylesterase gene is associated with the responsiveness to imidapril medication and the promoter activity. Hypertens Res. 2005;28:719–725. doi: 10.1291/hypres.28.719. [DOI] [PubMed] [Google Scholar]
- 9.Ahmad S, Forgash AJ. Nonoxidative enzymes in the metabolism of insecticides. Drug Metab Rev. 1976;5:141–164. [PubMed] [Google Scholar]
- 10.Bencharit S, Morton CL, Howard-Williams EL, Danks MK, Potter PM, Redinbo MR. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nat Struct Biol. 2002;9:337–342. doi: 10.1038/nsb790. [DOI] [PubMed] [Google Scholar]
- 11.Satoh T, Taylor P, Bosron WF, Sanghani SP, Hosokawa M, La Du BN. Current progress on esterases: from molecular structure to function. Drug Metab Dispos. 2002;30:488–493. doi: 10.1124/dmd.30.5.488. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh S. Cholesteryl ester hydrolase in human monocyte/macrophage: cloning, sequencing, and expression of full-length cDNA. Physiol Genomics. 2000;2:1–8. doi: 10.1152/physiolgenomics.2000.2.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Zhao B, Natarajan R, Ghosh S. Human liver cholesteryl ester hydrolase: cloning, molecular characterization, and role in cellular cholesterol homeostasis. Physiol Genomics. 2005;23:304–310. doi: 10.1152/physiolgenomics.00187.2005. [DOI] [PubMed] [Google Scholar]
- 14.Mukherjee JJ, Jay FT, Choy PC. Purification, characterization and modulation of a microsomal carboxylesterase in rat liver for the hydrolysis of acyl-CoA. Biochem J. 1993;295:81–86. doi: 10.1042/bj2950081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsujita T, Okuda H. Palmitoyl-coenzyme A hydrolyzing activity in rat kidney and its relationship to carboxylesterase. J Lipid Res. 1993;34:1773–1781. [PubMed] [Google Scholar]
- 16.Becker A Bottcher A, Lackner KJ, Fehringer P, Notka F, Aslanidis C, Schmitz G. Purification, cloning, and expression of a human enzyme with acyl coenzyme A: cholesterol acyltransferase activity, which is identical to liver carboxylesterase. Arterioscler Thromb. 1994;14:1346–1355. doi: 10.1161/01.atv.14.8.1346. [DOI] [PubMed] [Google Scholar]
- 17.Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh S, Natarajan R. Cloning of the human cholesteryl ester hydrolase promoter: identification of functional peroxisomal proliferator-activated receptor responsive elements. Biochem Biophys Res Commun. 2001;284:1065–1070. doi: 10.1006/bbrc.2001.5078. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh S, St Clair RW, Rudel LL. Mobilization of cytoplasmic CE droplets by overexpression of human macrophage cholesteryl ester hydrolase. J Lipid Res. 2003;44:1833–1840. doi: 10.1194/jlr.M300162-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Zhao B, Fisher BJ, St Clair RW Rudel LL, Ghosh S. Redistribution of macrophage cholesteryl ester hydrolase from cytoplasm to lipid droplets upon lipid loading. J Lipid Res. 2005;46:2114–2121. doi: 10.1194/jlr.M500207-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Huang GS, Yang SM, Hong MY, Yang PC, Liu YC. Differential gene expression of livers from ApoE deficient mice. Life Sci. 2000;68:19–28. doi: 10.1016/s0024-3205(00)00912-7. [DOI] [PubMed] [Google Scholar]
- 22.Lehner R, Vance DE. Cloning and expression of a cDNA encoding a hepatic microsomal lipase that mobilizes stored triacylglycerol. Biochem J. 1999;343:1–10. [PMC free article] [PubMed] [Google Scholar]
- 23.Lehner R, Cui Z, Vance DE. Subcellullar localization, developmental expression and characterization of a liver triacylglycerol hydrolase. Biochem J. 1999;338:761–768. [PMC free article] [PubMed] [Google Scholar]
- 24.Gilham D, Lehner R. The physiological role of triacylglycerol hydrolase in lipid metabolism. Rev Endocr Metab Disord. 2004;5:303–309. doi: 10.1023/B:REMD.0000045101.42431.c7. [DOI] [PubMed] [Google Scholar]
- 25.Soni KG, Lehner R, Metalnikov P, O'Donnell P, Semache M Gao W, Ashman K, Pshezhetsky AV, Mitchell GA. Carboxylesterase 3 (EC 3.1.1.1) is a major adipocyte lipase. J Biol Chem. 2004;279:40683–40689. doi: 10.1074/jbc.M400541200. [DOI] [PubMed] [Google Scholar]
- 26.Gilham D, Alam M, Gao W, Vance DE, Lehner R. Triacylglycerol hydrolase is localized to the endoplasmic reticulum by an unusual retrieval sequence where it participates in VLDL assembly without utilizing VLDL lipids as substrates. Mol Biol Cell. 2005;16:984–996. doi: 10.1091/mbc.E04-03-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsujita T, Okuda H. Palmitoyl-coenzyme A hydrolyzing activity in rat kidney and its relationship to carboxylesterase. J Lipid Res. 1993;34:1773–1781. [PubMed] [Google Scholar]
- 28.Bencharit S, Morton CL, Hyatt JL, Kuhn P, Danks MK, Potter PM, Redinbo MR. Crystal structure of human carboxylesterase 1 complexed with the Alzheimer's drug tacrine. From binding promiscuity to selective inhibition. Chem Biol. 2003;10:341–349. doi: 10.1016/s1074-5521(03)00071-1. [DOI] [PubMed] [Google Scholar]
- 29.Bencharit S, Morton CL, Xue Y, Potter PM, Redinbo MR. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat Struct Biol. 2003;10:349–356. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- 30.Fleming CD, Bencharit S, Edwards CC, Hyatt JL, Tsurkan L, Bai F, Fraga C, Morton CL, Howard-Williams EL, Potter PM, Redinbo MR. Structural insights into drug processing by human carboxylesterase 1: tamoxifen, mevastatin, and inhibition by benzil. J Mol Biol. 2005;352:165–177. doi: 10.1016/j.jmb.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 31.Bellizzi JJ3rd Widom J, Kemp C, Lu JY, Das AK, Hofmann SL, Clardy J. The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis. Proc Natl Acad Sci. 2000;97:4573–4578. doi: 10.1073/pnas.080508097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das AK, Bellizzi JJ, 3rd, Tandel S, Biehl E, Clardy J, Hofmann SL. Structural basis for the insensitivity of a serine enzyme (palmitoyl-protein thioesterase) to phenylmethylsulfonyl fluoride. J Biol Chem. 2000;275:23847–23851. doi: 10.1074/jbc.M002758200. [DOI] [PubMed] [Google Scholar]
- 33.Calero G, Gupta P, Nonato MC, Tandel S, Biehl ER, Hofmann SL, Clardy J. The crystal structure of palmitoyl protein thioesterase-2 (PPT2) reveals the basis for divergent substrate specificities of the two lysosomal thioesterases, PPT1 and PPT2. J Biol Chem. 2003;278:37957–37964. doi: 10.1074/jbc.M301225200. [DOI] [PubMed] [Google Scholar]
- 34.Williams PA, Cosme J, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Wang CS, Tang J, Dyda F, Zhang XC. The crystal structure of bovine bile salt activated lipase: insights into the bile salt activation mechanism. Structure. 1997;5:1209–1218. doi: 10.1016/s0969-2126(97)00271-2. [DOI] [PubMed] [Google Scholar]
- 36.Terzyan S, Wang CS, Downs D, Hunter B, Zhang XC. Crystal structure of the catalytic domain of human bile salt activated lipase. Protein Sci. 2000;9:1783–1790. doi: 10.1110/ps.9.9.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wallace TJ, Kodsi EM, Langston TB, Gergis MR, Grogan WM. Mutation of residues 423 (Met/Ile), 444 (Thr/Met), and 506 (Asn/Ser) confer cholesteryl esterase activity on rat lung carboxylesterase. Ser-506 is required for activation by cAMP-dependent protein kinase. J Biol Chem. 2001;276:33165–33174. doi: 10.1074/jbc.M105644200. [DOI] [PubMed] [Google Scholar]
- 38.Wierdl M, Morton CL, Nguyen NK, Redinbo MR, Potter PM. Molecular modeling of CPT-11 metabolism by carboxylesterases (CEs): use of pnb CE as a model. Biochemistry. 2004;43:1874–1882. doi: 10.1021/bi035586r. [DOI] [PubMed] [Google Scholar]
- 39.Devedjiev Y, Dauter Z, Kuznetsov SR, Jones TL, Derewenda ZS. Crystal structure of the human acyl protein thioesterase I from a single X-ray data set to 1.5 A. Structure Fold Des. 2000;8:1137–1146. doi: 10.1016/s0969-2126(00)00529-3. [DOI] [PubMed] [Google Scholar]
- 40.Kaphalia BS, Ansari GA. Purification and characterization of rat hepatic microsomal low molecular weight fatty acid ethyl ester synthase and its relationship to carboxylesterases. J Biochem Mol Toxicol. 2001;15:165–171. doi: 10.1002/jbt.14. [DOI] [PubMed] [Google Scholar]
- 41.Kaphalia BS, Mericle KA, Ansari GA. Mechanism of differential inhibition of hepatic and pancreatic fatty acid ethyl ester synthase by inhibitors of serine-esterases: in vitro and cell culture studies. Toxicol Appl Pharmacol. 2004;200:7–15. doi: 10.1016/j.taap.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 42.Jenkins-Kruchten AE, Bennaars-Eiden A, Ross JR, Shen WJ, Kraemer FB, Bernlohr DA. Fatty acid-binding protein-hormone-sensitive lipase interaction. Fatty acid dependence on binding. J Biol Chem. 2003;278:47636–47643. doi: 10.1074/jbc.M307680200. [DOI] [PubMed] [Google Scholar]
- 43.Morton CL, Potter PM. Comparison of Escherichia coli, Saccharomyces cerevisiae, Pichia pastoris, Spodoptera frugiperda, and COS7 cells for recombinant gene expression. Application to a rabbit liver carboxylesterase. Mol Biotechnol. 2000;16:193–202. doi: 10.1385/MB:16:3:193. [DOI] [PubMed] [Google Scholar]
- 44.Danks MK, Morton CL, Krull EJ, Cheshire PJ, Richmond LB, Naeve CW, Pawlik CA, Houghton PJ, Potter PM. Comparison of Activation of CPT-11 by Rabbit and Human Carboxylesterases for Use in Enzyme/Prodrug Therapy. Clin Cancer Res. 1999;5:917–924. [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Daresbury Laboratories; Warrington: 1993. Data collection and processing. [Google Scholar]
- 46.Collaborative Computing Project. Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 47.Navaza J. AmoRe: an automated package for molecular replacement. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 48.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 49.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 50.Read RJ. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr A. 1986;42:140–149. [Google Scholar]
- 51.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystollogr. 1991;24:946–950. [Google Scholar]
- 52.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D. 1999;55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 53.Merritt EA, Murphy MEP. Raster3D version 2.0-a program for photorealistic molecular graphics. Acta Crystallogr D. 1991;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 54.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]