

Abstract
Background
Common polymorphisms of the transcription factor 7–like 2 gene (TCF7L2) have recently been associated with type 2 diabetes. We examined whether the two most strongly associated variants (rs12255372 and rs7903146) predict the progression to diabetes in persons with impaired glucose tolerance who were enrolled in the Diabetes Prevention Program, in which lifestyle intervention or treatment with metformin was compared with placebo.
Methods
We genotyped these variants in 3548 participants and performed Cox regression analysis using genotype, intervention, and their interactions as predictors. We assessed the effect of genotype on measures of insulin secretion and insulin sensitivity at baseline and at one year.
Results
Over an average period of three years, participants with the risk-conferring TT genotype at rs7903146 were more likely to have progression from impaired glucose tolerance to diabetes than were CC homozygotes (hazard ratio, 1.55; 95 percent confidence interval, 1.20 to 2.01; P<0.001). The effect of genotype was stronger in the placebo group (hazard ratio, 1.81; 95 percent confidence interval, 1.21 to 2.70; P = 0.004) than in the metformin and lifestyle-intervention groups (hazard ratios, 1.62 and 1.15, respectively; P for the interaction between genotype and intervention not significant). The TT genotype was associated with decreased insulin secretion but not increased insulin resistance at baseline. Similar results were obtained for rs12255372.
Conclusions
Common variants in TCF7L2 seem to be associated with an increased risk of diabetes among persons with impaired glucose tolerance. The risk-conferring genotypes in TCF7L2 are associated with impaired beta-cell function but not with insulin resistance. (ClinicalTrials.gov number, NCT00004992.)
The risk of type 2 diabetes is strongly influenced by inheritance. 1 Genetic susceptibility to the common form of type 2 diabetes appears polygenic — that is, it involves a number of variants, each with a modest effect on the risk of disease in an individual person.2 Despite important advances in understanding the genetic determinants of the relatively rare monogenic forms of diabetes,3 the pace of definitive identification of genes that increase the risk of common type 2 diabetes has been slow.
Recently, Grant and colleagues4 reported on the association of a common microsatellite (DG10S478) within intron 3 of the transcription factor 7–like 2 gene (TCF7L2) with type 2 diabetes in an Icelandic case–control sample and replicated this result in two additional case–control cohorts of white patients. The noncoding single-nucleotide polymorphisms rs12255372 and rs7903146 were in strong linkage disequilibrium with DG10S478 (r2= 0.95 and r2= 0.78, respectively) and showed similarly robust associations with type 2 diabetes (P<10−15). The authors recommended that these two single-nucleotide polymorphisms be genotyped in all attempts at replication.
Reproducibility of reported genetic associations is essential in complex human genetics, especially among populations of different races, ethnic backgrounds, and environmental exposures.5–8 Furthermore, the effect that these polymorphisms have on the risk of type 2 diabetes and on validated preventive interventions has not been prospectively ascertained. Finally, the pathophysiological mechanism by which variation in TCF7L2 might influence glycemic traits is not clear. Therefore, we genotyped the rs12255372 and rs7903146 variants in 3548 participants in the Diabetes Prevention Program (DPP) to try to confirm this association, to assess the effect of these variants on the lifestyle and pharmacologic interventions used in the DPP,9 and to explore the effect of these variants on insulin secretion, insulin sensitivity, or both.
METHODS
THE DIABETES PREVENTION PROGRAM
The DPP Research Group9 conducted a multicenter, randomized clinical trial from 1996 to 2001 at 27 centers in the United States. The institutional review board at each center approved the protocol, and all participants gave written informed consent. The trial was designed to test whether a lifestyle intervention or pharmacologic treatment with metformin would prevent or delay the development of diabetes in persons at increased risk for the disease.9,10 The DPP enrolled 3234 overweight persons in the United States without diabetes who had elevated plasma glucose concentrations after an overnight fast and impaired glucose tolerance. As compared with placebo, the lifestyle interventions and treatment with metformin reduced the incidence of diabetes by 58 percent (95 percent confidence interval, 48 to 66 percent) and 31 percent (95 percent confidence interval, 17 to 43 percent), respectively, over an average follow-up period of approximately three years.9 An additional group of 585 participants was treated with troglitazone, but this treatment was halted during the DPP trial owing to its toxic effects on the liver.10
PARTICIPANTS
The 3548 participants included in this study (92.9 percent of the participants in the DPP trial; 2994 subjects randomly assigned to placebo, lifestyle intervention, or metformin, plus 554 participants initially randomly assigned to troglitazone, in whom only quantitative traits were analyzed) each provided written informed consent for the genetic investigation. Of these participants, 66.8 percent were women, 56.4 percent white, 20.2 percent African American, 16.8 percent Hispanic, 4.3 percent Asian, and 2.4 percent American Indian, according to self-report. The mean (±SD) age was 51±11 years, and the mean body-mass index (BMI) (the weight in kilograms divided by the square of the height in meters) was 34.0±6.7. The development of diabetes was assessed on the basis of semiannual measurements of fasting plasma glucose concentrations and annual oral glucose-tolerance tests (with a 75-g oral glucose load).9,10
GENOTYPING
DNA was extracted from peripheral-blood leukocytes and quantitated with the use of Pico-Green analysis (Molecular Probes). Genotyping was performed by allele-specific primer extension of singleplex amplified products, with detection by matrix-assisted laser desorption–ionization time-of-flight mass spectroscopy on a Sequenom platform.11,12 The genotyping success rate was 99.3 percent, and the consensus rate (on the basis of 222 duplicate genotypes) was 99.1 percent. The allele frequencies for both single-nucleotide polymorphisms in each of the five races or ethnic groups were in Hardy–Weinberg equilibrium (P>0.05).
QUANTITATIVE MEASURES
Data from the baseline oral glucose-tolerance test were used to calculate two measures of insulin secretion13,14 and two measures of insulin sensitivity15,16 as previously described.17 We used glucose and insulin measured in conventional units (milligrams per deciliter and microunits per milliliter, respectively) unless otherwise specified. Measures of insulin secretion included the fasting insulin:glucose ratio, calculated by dividing (insulin at 30 minutes − insulin at 0 minutes) by (glucose at 30 minutes − glucose at 0 minutes), and the corrected insulin response, calculated by means of the following equation: (100 × insulin at 30 minutes) ÷ [glucose at 30 minutes × (glucose at 30 minutes − 70 mg per deciliter)]. Measures of insulin sensitivity included the reciprocal of the fasting insulin level and the insulin-sensitivity index, which is the reciprocal of insulin resistance according to the homeostasis model assessment16 and is calculated by the following equation: 22.5 ÷ [fasting insulin × (fasting glucose ÷ 18.01)]. We have previously shown that both measures of insulin secretion strongly correlate with each other, as do both measures of insulin sensitivity.17
STATISTICAL ANALYSIS
We examined Cox regression models according to genotype, intervention, and interactions between genotype and intervention as the independent variables predicting the incidence of diabetes. To test for interactions, we contrasted the likelihood of the model that included all two-way treatment and genotype interaction terms with the likelihood of the model without any interaction terms; the ratio has a chi-square distribution. If this was significant, we tested each interaction term with the use of the Wald test.18 Models were adjusted for risk factors for diabetes at enrollment. Because there was no evidence of differences between race or ethnic groups in the incidence of diabetes for any of the interventions, initial analyses of the effects of genotype on incidence were performed for all races and ethnic groups combined; they were also repeated only in populations that had similar allele frequencies (whites and African Americans together). No significant interaction between ethnicity and genotype was detected in any of our analyses. The population attributable risk was estimated with data from the placebo group for each ethnic group, calculated as follows: 1 − (1 ÷ [p2HRhom + 2p(1 − p)HRhet + (1 − p)2]), where p is the risk-allele frequency, HRhom is the hazard ratio for homozygotes, and HRhet is the hazard ratio for heterozygotes.
For the quantitative trait analyses, baseline measures in the entire cohort (obtained in 3436 participants, including those randomly assigned to troglitazone) were log-transformed for non-normality and a generalized linear model was performed comparing values according to genotype. In cases in which log transformation did not result in a normal distribution, differences between means were compared with the use of a nonparametric Wilcoxon test. We further obtained an estimate of "composite beta-cell function" by adjusting baseline insulin secretion to insulin sensitivity through linear regression of log-transformed variables.17 For the one-year analyses, we focused on the insulin:glucose ratio as a measure of insulin secretion and the insulin-sensitivity index as a measure of insulin sensitivity, and we used a generalized linear model with interaction terms of genotype and treatment. Means were adjusted for baseline measures. Nominal two-sided P values are reported and were adjusted for multiple comparisons (three genotypic groups within each trait) with the use of the Holm procedure.19 Analyses were done with the use of SAS software, version 8.2 (SAS Institute).
RESULTS
DISTRIBUTION OF ALLELE FREQUENCIES
Baseline demographic and anthropomorphic characteristics are shown in Table 1. For rs12255372, the frequency of the minor T allele in whites enrolled in the DPP (0.32) was similar to that previously reported in European populations.4 The frequency of minor alleles was similar in African Americans (0.31) but lower in Hispanics (0.23), Asians (0.14), and American Indians (0.05). Similar distributions were noted for rs7903146, with minor allele frequencies of 0.33 in whites, 0.31 in African Americans, 0.24 in Hispanics, 0.17 in Asians, and 0.12 in American Indians. Linkage disequilibrium between both variants was strong in people of European descent (D′ = 0.90, r2 = 0.78) but nearly absent in African Americans (D′ = 0.11, r2 = 0.01). Allele frequencies were similar across treatment groups.
Table 1.
Baseline Characteristics of the Cohort According to Genotype at the rs12255372 and rs7903146 Variants.*
| Characteristic | rs12255372 (N=3509) | P Value† | rs7903146 (N=3537) | P Value | ||||
|---|---|---|---|---|---|---|---|---|
| GG (N = 1783) | GT (N = 1412) | TT (N = 314) | CC (N = 1747) | CT (N = 1454) | TT (N = 336) | |||
| Male sex — no. (%) | 606 (34.0) | 450 (31.9) | 101 (32.2) | 0.43 | 595 (34.1) | 461 (31.7) | 113 (33.6) | 0.36 |
| Age — yr | 50.5±10.7 | 51.0±10.4 | 51.3±10.7 | 0.27 | 50.6±10.8 | 50.7±10.2 | 51.7±11.1 | 0.20 |
| Body-mass index | 34.2±6.7 | 33.8±6.7 | 33.6±6.4 | 0.10 | 34.4±6.7 | 33.8±6.7 | 33.1±6.4 | 0.002 |
| Waist circumference — cm | 106±14.6 | 105±14.6 | 104±13.4 | 0.03 | 106±15.0 | 104±14.1 | 103±13.3 | <0.001 |
Plus–minus values are means ±SD.
P values are based on F tests for continuous variables and chi-square tests for categorical variables.
INCIDENCE OF DIABETES
Grant et al.4 identified the T alleles at both single-nucleotide polymorphisms as the risk variants. In the DPP, participants who were homozygous for the T allele at rs7903146 were more likely to have progression to diabetes than were those who were homozygous for the C allele (hazard ratio, 1.55; 95 percent confidence interval, 1.20 to 2.01; P<0.001). No excess risk was conferred by the heterozygous genotype (Table 2). The effect of the risk-conferring genotype was greatest in the placebo group (hazard ratio, 1.81; 95 percent confidence interval, 1.21 to 2.70; P = 0.004) and less in the metformin group (hazard ratio, 1.62; 95 percent confidence interval, 1.03 to 2.54; P = 0.04) and in the lifestyle-intervention group (hazard ratio, 1.15; 95 percent confidence interval, 0.68 to 1.94; P = 0.60). In the placebo group, the incidence of diabetes for the TT, CT, and CC genotypes at rs7903146 was 18.5, 10.7, and 10.8 per 100 person-years, respectively (Fig. 1). The results were similar for the risk-conferring TT genotype at rs12255372 as compared with the GG genotype, both in the overall group (hazard ratio, 1.53; 95 percent confidence interval, 1.17 to 2.01; P = 0.002) and in the placebo group (hazard ratio, 1.81; 95 percent confidence interval, 1.19 to 2.75; P = 0.005). Results for rs12255372 in the metformin and lifestyle-intervention groups were similar to those for rs7903146 but not significant (hazard ratio, 1.45 and 1.24, respectively; 95 percent confidence intervals, 0.90 to 2.35 and 0.73 to 2.12; P = 0.13 and P = 0.43).
Table 2.
Incidence of Diabetes According to Genotype at the rs12255372 and rs7903146 Variants.*
| Variable | No. of Participants | Genotype at rs12255372† | Hazard Ratio for GT vs. GG (95% CI) | P Value | GT P Value‡ | Hazard Ratio for TT vs. GG (95% CI) | P Value | TT P Value‡ | PAR | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| GG | GT | TT | |||||||||
| no. of cases of diabetes (cases/100 person-yr) | |||||||||||
| Overall cohort | 2951 | 302 (7.4) | 260 (8.0) | 69 (10.6) | 1.08 (0.92–1.29) | 0.35 | — | 1.53 (1.17–2.01) | 0.002 | — | 0.11 |
| Whites | 1657 | 140 (6.8) | 165 (8.0) | 45 (10.8) | 1.19 (0.94–1.50) | 0.14 | — | 1.67 (1.18–2.37) | 0.004 | — | 0.14 |
| African Americans | 600 | 67 (8.6) | 56 (8.6) | 16 (10.3) | 1.02 (0.71–1.47) | 0.90 | 0.50 | 1.24 (0.70–2.17) | 0.46 | 0.35 | 0.13 |
| Hispanics | 489 | 62 (7.6) | 30 (6.7) | <15 (10.2) | 0.83 (0.53–1.31) | 0.42 | 0.18 | 1.70 (0.75–3.87) | 0.21 | 0.93 | 0.10 |
| Asians | 124 | 20 (8.0) | <15 (7.7) | <15 (13.0) | 0.88 (0.36–2.15) | 0.78 | 0.58 | 1.42 (0.17–11.7) | 0.75 | 0.95 | 0.18 |
| American Indians | 81 | <15 (6.6) | <15 (11.8) | <15 (0.0) | 1.93 (0.39–9.63) | 0.42 | 0.58 | NA | NA | 0.97 | NA |
| Genotype at rs7903146† | Hazard Ratio for CT vs. CC (95%3CI) | P Value | CT P Value‡ | Hazard Ratio for TT vs. CC (95% CI) | P Value | TT P Value‡ | PAR | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CC | CT | TT | |||||||||
| no. of cases of diabetes (cases/100 person-yr) | |||||||||||
| Overall cohort | 2983 | 305 (7.6) | 255 (7.6) | 77 (10.8) | 1.00 (0.84–1.19) | 0.99 | — | 1.55 (1.20–2.01) | <0.001 | — | 0.06 |
| Whites | 1671 | 147 (7.3) | 153 (7.4) | 51 (10.9) | 1.03 (0.81–1.30) | 0.82 | — | 1.62 (1.16–2.25) | 0.004 | — | 0.05 |
| African Americans | 605 | 64 (8.5) | 62 (8.9) | <15 (9.7) | 1.09 (0.76–1.56) | 0.65 | 0.82 | 1.20 (0.66–2.17) | 0.55 | 0.36 | 0.10 |
| Hispanics | 497 | 61 (7.4) | 29 (6.6) | <15 (12.7) | 0.89 (0.56–1.41) | 0.62 | 0.60 | 2.26 (1.14–4.50) | 0.02 | 0.46 | 0.18 |
| Asians | 128 | 22 (9.2) | <15 (6.4) | <15 (8.9) | 0.55 (0.23–1.33) | 0.19 | 0.26 | 0.92 (0.11–7.48) | 0.94 | 0.66 | −0.12 |
| American Indians | 82 | <15 (6.7) | <15 (7.5) | <15 (0.0) | 1.15 (0.35–3.78) | 0.82 | 0.84 | NA | NA | 0.97 | NA |
CI denotes 95 percent confidence interval, PAR population attributable risk (calculated with data from the placebo group), and NA not available because of insufficient data.
In instances where the number of cases is less than 15, exact numbers were not reported to protect the confidentiality of participants, per DPP publication policy.
The P value is for the interaction between the genotype and the race or ethnic group, with the largest groups (whites and those who were homozygous for the major allele) serving as the reference group.
Figure 1. Incidence of Diabetes According to Treatment Group and Genotype at Variant rs7903146.
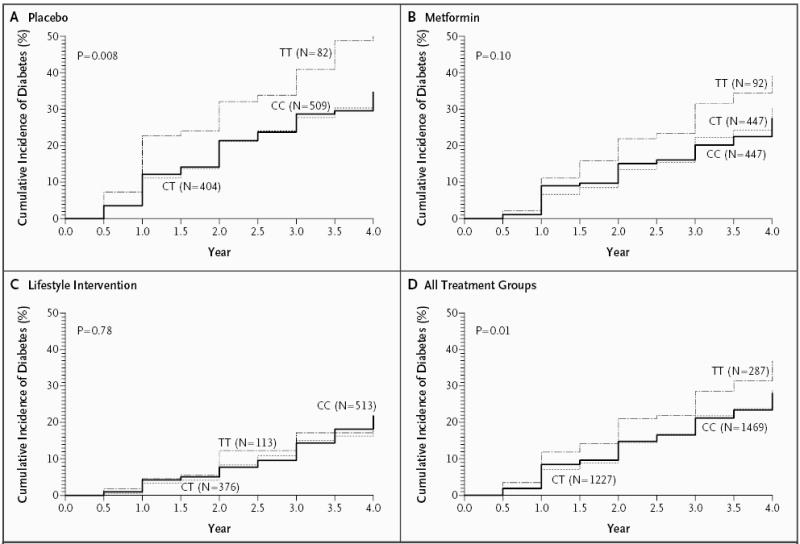
The P values were determined by the log-rank test.
Although the effect of genotype at both single-nucleotide polymorphisms was stronger in the placebo group than in the metformin and lifestyle-intervention groups, there were no significant interactions between genotype and intervention at either locus. Similarly, there were no significant interactions between genotype and race or ethnic group or between genotype and BMI on diabetes incidence (Table 2). When we restricted our analysis to the populations with similar allele frequencies, the effect of the TT genotype, as compared with the CC genotype, at rs7903146 on the risk of diabetes was indistinguishable from the overall result (hazard ratio, 1.63; 95 percent confidence interval, 1.17 to 2.27; P = 0.004 in 2276 whites and African Americans together); we found similar results for rs12255372.
QUANTITATIVE MEASURES
We examined whether the risk allele at either single-nucleotide polymorphism affected quantitative glycemic traits. At baseline, carriers of the T allele at rs7903146 had significantly lower levels of insulin secretion than did CC homozygotes, as measured by both the insulin:glucose ratio and the corrected insulin response (Fig. 2). Similar results were obtained for the T allele at rs12255372.
Figure 2. Effects of Genotype at rs7903146 on Insulin Secretion at Baseline as Measured by the Mean (±SE) Insulin:Glucose Ratio (Panel A) and the Corrected Insulin Response (Panel B).

To convert the insulin:glucose ratio to picomoles per liter ÷ millimoles per liter, multiply by 125.1; to convert the corrected insulin response to picomoles per liter ÷ (millimoles per liter)2, multiply by 2254.9. Statistical comparisons were made on log-transformed values where appropriate. All pairwise comparisons are significant at a P value of less than 0.02; in comparisons between the CC and TT genotypes, the P value is less than 0.001 for both measures of insulin secretion. Although baseline glucose concentrations did not differ significantly across genotypic groups at 0 and 30 minutes, TT homozygotes had significantly lower insulin concentrations at both time points — at 30 minutes, CC, CT, and TT participants had mean insulin concentrations (±SD) of 106.0±69.7, 96.0±56.1, and 89.1±57.2 μU per milliliter, respectively (P<0.001). To convert microunits per milliliter to picomoles per liter, multiply by 6.94.
Surprisingly, two mean baseline measures of insulin sensitivity were significantly higher in the presence of T alleles and in proportion to the number of T alleles (Fig. 3). Composite beta-cell function seemed to be impaired in TT homozygotes, as indicated by a shift in the regression curve downward and to the left (Fig. 3). The greater mean insulin sensitivity in carriers of T alleles at rs7903146 correlated with a concomitant lower mean BMI and waist circumference at baseline and persisted after adjustment for these traits; a similar trend was noted for rs12255372 (Table 1).
Figure 3. Insulin Secretion and Insulin Sensitivity at Baseline for Each Genotype at rs7903146.

Composite beta-cell function is estimated by the relationship between insulin secretion (the insulin:glucose ratio and the corrected insulin response) and insulin sensitivity (insulin-sensitivity index and 1 ÷ fasting insulin). The curves represent the regression line of the logarithm of estimated insulin secretion as a linear function of the logarithm of estimated insulin sensitivity for all participants at baseline, distributed according to genotype at rs7903146. The mean for each group is indicated by the point estimate in each curve. Carriers of the T allele have decreased insulin secretion accompanied by an increase in insulin sensitivity. The shift of the curve downward and to the left in TT homozygotes suggests a defect in composite beta-cell function. To convert insulin-sensitivity index and 1 ÷ fasting insulin to (picomoles per liter × millimoles per liter)−1, multiply by 0.144.
Given these results of quantitative traits, we adjusted our models of the incidence of diabetes for the presence of known risk factors. Initial adjustment for age and BMI at baseline did not alter the results; full adjustment for sex and age, BMI, waist circumference, and the fasting plasma glucose concentration at baseline reduced the hazard ratios slightly as compared with homozygous genotypes, but they remained significant (hazard ratio for the TT genotype as compared with the CC genotype at rs7903146, 1.39; 95 percent confidence interval, 1.06 to 1.82; P = 0.02). When the influence of covariates was assessed with the inclusion of interaction terms in the model, only waist circumference showed a nominally significant interaction with genotype. Including the baseline insulin:glucose ratio in the model as a measure of insulin secretion also minimally decreased the observed effect (hazard ratio for the TT genotype as compared with the CC genotype at rs7903146, 1.41; 95 percent confidence interval, 1.08 to 1.83; P = 0.01). Similar results were obtained for rs12255372.
At one year from baseline, we detected no significant effects of genotype on the changes in any of the insulin-secretion or insulin-sensitivity indexes associated with the three interventions,17,20 consistent with the absence of interactions between genotype and treatment group.
DISCUSSION
Inconsistent reproducibility has been a vexing problem for genetic association studies in complex diseases.6,7,21 False positive reports of association, false negative attempts at replication, and genetic heterogeneity often complicate the picture, and thus a true genetic association usually emerges only after carefully conducted, large-scale association studies confirm the original report.8
A limited number of common genetic variants meet that high standard in type 2 diabetes.22 In most cases, the genotypic risk is modest (1.15 to 1.25), requiring very large sample sizes for detection. The recent identification of a common allele in the TCF7L2 gene that increases the risk of type 2 diabetes by approximately 1.45 in heterozygotes and 2.41 in homozygotes4 is therefore quite provocative. Despite these significant results in a cross-sectional study,4 it was essential to replicate this genetic association in other cohorts and to do so prospectively. In addition, our evaluation of a potential mechanism by which the risk of diabetes is increased and the determination of whether these variants cause differential responses to validated preventive strategies represent important next steps in exploring the association.
The DPP is a unique study in which to carry out such analyses. The large sample size and the cohort of several races and ethnic groups, reflecting the diversity of the U.S. population with type 2 diabetes, make it possible to test the role of genetic variants in different races or ethnic groups, even if they confer only modest risk. The DPP study is different from other large observational studies23 because of its interventional design and exclusive enrollment of overweight or obese persons with elevated fasting plasma glucose concentrations and impaired glucose tolerance, which indicate a high risk of diabetes at baseline.
Our data indicate that the risk alleles in rs7903146 and rs12255372 predict the risk of diabetes prospectively, beyond that conferred by the clinical risk factors reflected by the DPP eligibility criteria. The genotypic relative risk may differ slightly from the odds ratio documented by Grant et al.4 owing to a different study design, an overestimate of the initial finding,6 population heterogeneity in the DPP, various degrees of linkage disequilibrium between the DG10S478 microsatellite and the single-nucleotide polymorphisms evaluated here, or our limited temporal window (three years on average) for the clinical transition from impaired glucose tolerance to diabetes. The size of the effect seems robust for a sample of this size; given the higher probability conferred by the initial report, our finding is a strong confirmation of the original genetic association.
Grant and coworkers4 exhaustively assessed coding variation in whites by a variety of deep re-sequencing methods in the region, suggesting that other functional variants in this gene are unlikely to have been missed. In addition to rs12255372 and rs7903146, other single-nucleotide polymorphisms in linkage disequilibrium with them were also strongly associated with diabetes. Which of these single-nucleotide polymorphisms is responsible for the observed association requires further study in adequately powered samples. In particular, the absence of linkage disequilibrium between rs7903146 and rs12255372 in African Americans may help distinguish whether one of the two single-nucleotide polymorphisms (or the haplotype formed by the risk alleles at both loci) is the sole source of the association signal; we could not make this distinction after initial exploratory analyses in our data set, perhaps because of inadequate sample size. Given their allele frequencies and assuming an overall genotypic relative risk of 1.54 (Table 2) and diabetes prevalence of 10 percent among African Americans, we estimate that the enrollment of approximately 1400 persons of African ancestry would be necessary for the case–control study to have 80 percent power (with a P value of less than 0.05 considered to indicate statistical significance) to distinguish between rs7903146 and rs12255372 as the source of the association. Sample sizes at least twice as large would be needed if one intended to detect a signal arising solely from the haplotype formed by the minor alleles at both loci.
We studied the TCF7L2 single-nucleotide polymorphisms in nonwhite populations. Subgroup analysis according to race or ethnic group showed similar effect sizes at this locus, but these effect sizes were not individually significant, possibly because of inadequate sample size. However, we cannot rule out effects of genotype on risk that were specific to race or ethnic group; with the results we have obtained here, a cohort of more than 20,000 persons would be needed to detect an interaction between genotype and African-American ethnicity, at a nominal (unadjusted) P value of less than 0.05. Similarly, our sample size may not have been sufficient to detect a significant effect of the heterozygous genotype on the risk of diabetes. Nevertheless, the results of the report by Grant et al.4 and our findings of a specific effect of a single copy of the T allele on quantitative glycemic traits suggest that heterozygosity at this locus may have phenotypic consequences.
The original study speculated that genetic variation in TCF7L2 might impair the expression of glucagon-like peptide 1 in enteroendocrine cells, possibly by interfering with β-catenin–mediated transcriptional activation of its gene GCG.24 Our finding that insulin secretion is decreased in carriers of the risk-conferring genotype, which is consistent with an increased incidence of diabetes, lends indirect support to this model. However, it is not readily apparent how rs12255372 and rs7903146, which lie in short interspersed repeat elements approximately 41 kb upstream and 9 kb downstream, respectively, of exon 4 in TCF7L2, might affect TCF7L2 expression or the function of its protein product. Fine mapping of the association signal and directed functional studies should help determine the molecular consequences of genetic variation at this locus.
The enhanced insulin sensitivity (and the lower BMI and smaller waist circumference) in carriers of the T allele at both single-nucleotide polymorphisms was unexpected and may be an artifact of our requirement that patients have high-risk characteristics for diabetes at enrollment. Specifically, if the T allele leads to decreased insulin secretion and a higher risk of diabetes, subjects with additional insulin resistance would be more likely to have diabetes at baseline (all other factors being equal), precluding their enrollment in this trial. Conversely, at-risk participants carrying the T allele may not have had diabetes at the time of enrollment because of enhanced insulin sensitivity owing to other genetic or environmental factors. In either case, our results strongly suggest that these variants do not cause insulin resistance in persons with impaired glucose tolerance and support the notion that variants in TCF7L2 lead to diabetes by means of defects in insulin secretion. Like KCNJ11, TCF7L2 seems to be a diabetes-associated gene in which common polymorphisms primarily affect the beta cell.23,25,26
Finally, we did not detect significant interactions between genotypes at either single-nucleotide polymorphism and the DPP interventions. The absence of an effect may not be surprising, since these interventions succeeded primarily by improving insulin sensitivity17 and these variants affect insulin secretion. However, we did not observe any effect of genotype at these loci in the lifestyle-intervention group, raising the possibility that a behavioral intervention can mitigate the risk conferred by genetic background. Conversely, the intervention groups may have been underpowered for these analyses. Whether the response to drugs designed to improve insulin secretion (e.g., sulfonylureas, meglitinides, or incretins) will be affected by these common variants requires specific testing in pharmacogenetic trials.
In summary, our results from this large prospective study confirm and extend the finding that the transcription factor gene TCF7L2 is associated with susceptibility to type 2 diabetes. Further understanding of the mechanisms by which variation in this gene affects glucose homeostasis may provide new insights into the molecular basis of diabetes and opportunities for more targeted interventions for prevention and therapy.
Footnotes
Dr. Shuldiner reports having received consulting fees from Merck, Sanofi-Aventis, and Sensors for Medicine, as well as lecture fees from Sanofi-Aventis. Dr. Altshuler reports having received consulting fees from Rosetta Inpharmatics (a subsidiary of Merck) and serving on the advisory board of Medical Portfolio Management. No other potential conflict of interest relevant to this article was reported.
We are indebted to Santica Marcovina and Greg Strylewicz for their careful processing of the DNA samples; to Mark Daly, Noël Burt, and our colleagues at the Broad Institute Genetic Analysis Platform for their assistance; and to the participants of the DPP for their commitment and dedication.
Supported by the National Institutes of Health (NIH) through the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), the Office of Research on Minority Health, the National Institute of Child Health and Human Development, the Office of Women's Health, and the National Institute on Aging; the Indian Health Service; the Centers for Disease Control and Prevention; the American Diabetes Association; Bristol-Myers Squibb; and Parke-Davis; an NIH grant (R01DK072041-01, to Drs. Altshuler, Florez, Pollin, and Shuldiner); and in part by the Intramural Research Program of the NIDDK. Many of the clinical centers were supported by the General Clinical Research Center Program, National Center for Research Resources. The clinical centers and the coordinating center were supported by the NIDDK through a cooperative agreement, except the Southwestern American Indian Centers, which were supported directly by the NIDDK and the Indian Health Service. Dr. Florez is supported by an NIH Research Career Award (K23 DK65978-03).
References
- 1.Barroso I. Genetics of type 2 diabetes. Diabet Med. 2005;22:517–35. doi: 10.1111/j.1464-5491.2005.01550.x. [DOI] [PubMed] [Google Scholar]
- 2.Florez JC, Hirschhorn JN, Altshuler D. The inherited basis of diabetes mellitus: implications for the genetic analysis of complex traits. Annu Rev Genomics Hum Genet. 2003;4:257–91. doi: 10.1146/annurev.genom.4.070802.110436. [DOI] [PubMed] [Google Scholar]
- 3.Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971–80. doi: 10.1056/NEJMra002168. [DOI] [PubMed] [Google Scholar]
- 4.Grant SFA, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–3. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 5.Freely associating. Nat Genet. 1999;22:1–2. doi: 10.1038/8702. [DOI] [PubMed] [Google Scholar]
- 6.Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29:306–9. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- 7.Lohmueller K, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33:177–82. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- 8.Hattersley AT, McCarthy MI. What makes a good genetic association study? Lancet. 2005;366:1315–23. doi: 10.1016/S0140-6736(05)67531-9. [DOI] [PubMed] [Google Scholar]
- 9.Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Idem. The Diabetes Prevention Program: design and methods for a clinical trial in the prevention of type 2 diabetes. Diabetes Care. 1999;22:623–34. doi: 10.2337/diacare.22.4.623. [Erratum, Diabetes Care 1999;22:1389.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 12.Tang K, Fu DJ, Julien D, Braun A, Cantor CR, Koster H. Chip-based genotyping by mass spectrometry. Proc Natl Acad Sci U S A. 1999;96:10016–20. doi: 10.1073/pnas.96.18.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sluiter WJ, Erkelens DW, Reitsma WD, Doorenbos H. Glucose tolerance and insulin release, a mathematical approach. I. Assay of the β-cell response after oral glucose loading. Diabetes. 1976;25:241–4. doi: 10.2337/diab.25.4.241. [DOI] [PubMed] [Google Scholar]
- 14.Phillips DI, Clark PM, Hales CN, Osmond C. Understanding oral glucose tolerance: comparison of glucose or insulin measurements during the oral glucose tolerance test with specific measurements of insulin resistance and insulin secretion. Diabet Med. 1994;11:286–92. doi: 10.1111/j.1464-5491.1994.tb00273.x. [DOI] [PubMed] [Google Scholar]
- 15.Hanson RL, Pratley RE, Bogardus C, et al. Evaluation of simple indices of insulin sensitivity and insulin secretion for use in epidemiologic studies. Am J Epidemiol. 2000;151:190–8. doi: 10.1093/oxfordjournals.aje.a010187. [DOI] [PubMed] [Google Scholar]
- 16.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 17.The Diabetes Prevention Program Research Group. Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the Diabetes Prevention Program: effects of lifestyle intervention and metformin. Diabetes. 2005;54:2404–14. doi: 10.2337/diabetes.54.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenberg RS, Kleinbaum DG. Mathematical modeling strategies for the analysis of epidemiologic research. Annu Rev Public Health. 1985;6:223–45. doi: 10.1146/annurev.pu.06.050185.001255. [DOI] [PubMed] [Google Scholar]
- 19.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- 20.The Diabetes Prevention Program Research Group. Prevention of type 2 diabetes with troglitazone in the Diabetes Prevention Program. Diabetes. 2005;54:1150–6. doi: 10.2337/diabetes.54.4.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 22.O'Rahilly S, Barroso I, Wareham NJ. Genetic factors in type 2 diabetes: the end of the beginning? Science. 2005;307:370–3. doi: 10.1126/science.1104346. [DOI] [PubMed] [Google Scholar]
- 23.Lyssenko V, Almgren P, Anevski D, et al. Genetic prediction of future type 2 diabetes. PLoS Med. 2005;2(12):e345. doi: 10.1371/journal.pmed.0020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yi F, Brubaker PL, Jin T. TCF-4 mediates cell type-specific regulation of proglucagon gene expression by β-catenin and glycogen synthase kinase-3β. J Biol Chem. 2005;280:1457–64. doi: 10.1074/jbc.M411487200. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen E-MD, Hansen L, Carstensen B, et al. The E23K variant of Kir6.2 associates with impaired post-OGTT serum insulin response and increased risk of type 2 diabetes. Diabetes. 2003;52:573–7. doi: 10.2337/diabetes.52.2.573. [DOI] [PubMed] [Google Scholar]
- 26.Florez JC, Burtt N, de Bakker PIW, et al. Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. Diabetes. 2004;53:1360–8. doi: 10.2337/diabetes.53.5.1360. [DOI] [PubMed] [Google Scholar]