

Abstract
We evaluated the effects of the phenothiazine derivative thioridazine on mechanisms of mitochondria potentially implicated in apoptosis, such as those involving reactive oxygen species (ROS) and cytochrome c release, as well as the involvement of drug interaction with mitochondrial membrane in these effects.
Within the 0 – 100 μM range thioridazine did not reduce the free radical 1,1-diphenyl-2-picryl-hydrazyl (DPPH) nor did it chelate iron.
However, at 10 μM thioridazine showed important antioxidant activity on mitochondria, characterized by inhibition of accumulation of mitochondria-generated O2•−, assayed as lucigenin-derived chemiluminescence, inhibition of Fe2+/citrate-mediated lipid peroxidation of the mitochondrial membrane (LPO), assayed as malondialdehyde generation, and inhibition of Ca2+/t-butyl hydroperoxide (t-BOOH)-induced mitochondrial permeability transition (MPT)/protein-thiol oxidation, assayed as mitochondrial swelling.
Thioridazine respectively increased and decreased the fluorescence responses of mitochondria labelled with 1-aniline-8-naphthalene sulfonate (ANS) and 1-(4-trimethylammonium phenyl)-6 phenyl 1,3,5-hexatriene (TMA-DPH).
The inhibition of LPO and MPT onset correlated well with the inhibition of cytochrome c release from mitochondria.
We conclude that thioridazine interacts with the inner membrane of mitochondria, more likely close to its surface, acquiring antioxidant activity toward processes with potential implications in apoptosis such as O2•− accumulation, as well as LPO, MPT and associated release of cytochrome c.
Keywords: Phenothiazines, thioridazine, trifluoperazine, mitochondrial respiration, lipid peroxidation, mitochondrial permeability transition, permeability transition pore, interaction with membranes, cytochrome c release, apoptosis
Introduction
Thioridazine is a derivative of the phenothiazines, a class of compounds with antipsychotic activity including trifluoperazine (TFP), a drug widely used in the investigation of several aspects of drug-membrane interactions. Phenothiazines have a three-ring structure in which two benzene rings are linked by a sulphur and a nitrogen atom. TFP has a piperazine ring and a −CF3 group in the side chain, whereas thioridazine has a piperidine ring and a −SCH3 group (Baldessarini, 1995). They are amphiphilic cations whose main feature is the ability to intercalate into biological membranes (Malheiros et al., 1998; Pavlov & Glaser, 1998).
A fraction of O2 available to the respiratory chain in the inner membrane of mitochondria undergoes incomplete reduction generating the superoxide radical (O2•−), which is readily dismutated to H2O2 (Gonzalez-Flecha & Boveris, 1995; Boveris & Chance, 1973). Increased O2•− generation or decreased mitochondrial antioxidant defences in the presence of Fe2+ give rise, via the Fenton/HaberWeiss reaction, to the highly reactive hydroxyl radical (•OH), which causes oxidative damage to biological molecules, namely, peroxidation of the membrane lipids (LPO) (Halliwell & Gutteridge, 1999). The mitochondrial membrane may also undergo a Ca2+-dependent, CsA-sensitive permeability transition process (MPT) mediated by opening of a non-specific channel, referred to as permeability transition pore (PTP), triggered by different agents including pro-oxidants such as tert-butyl hydroperoxide (t-BOOH) and assessed as mitochondrial swelling (Hunter & Haworth, 1979; Crompton et al., 1988; Zoratti & Szabò, 1995; Bernardi, 1999); membrane protein-thiol cross-linkage subsequent to the thiol oxidation by •OH has been proposed as an underlying mechanism (Kowaltowski et al., 2001). LPO and mainly MPT onset imply the release of cytochrome c from mitochondria into the cytosol, a process now recognized to be closely involved in cell death by apoptotic pathways (Kroemer et al., 1998; Skulachev, 2000; Petronilli et al., 2001). In this regard, TFP, a classical MPT inhibitor (Pereira et al., 1992), was recently reported to inhibit also the HIV-associated apoptosis (Pan et al., 1998).
Within this context, in the present study we evaluated the effects of the phenothiazine derivative thioridazine on mechanisms of mitochondria potentially implicated in apoptosis, such as those involving reactive oxygen species (ROS) and cytochrome c release, as well as the involvement of drug interaction with the mitochondrial membrane in these effects.
Methods
Chemicals
Thioridazine was purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). All other reagents were of the highest commercially available grade. The drug was solubilized in water. All stock solutions were prepared using glass-distilled deionized water.
Isolation of rat liver mitochondria
Mitochondria were isolated by standard differential centrifugation (Pedersen et al., 1978). Male Wistar rats weighing approximately 200 g were sacrificed by cervical dislocation; livers (10 – 15 g) were immediately removed, sliced in 50 ml of medium containing (mM) sucrose 250 EGTA 1 and HEPES – KOH 10, pH 7.2, and homogenized three times for 15 s at 1 min intervals in a Potter-Elvehjem homogenizer. Homogenates were centrifuged at 770×g for 5 min and the resulting supernatant was further centrifuged at 9800×g for 10 min. Pellets were suspended in 10 ml of medium containing (mM) sucrose 250, EGTA 0.3 and HEPES – KOH 10, pH 7.2, and centrifuged at 4500×g for 15 min. The final mitochondrial pellet was suspended in 1 ml of medium containing (mM) sucrose 250 and HEPES – KOH 10, pH 7.2, and used within 3 h. Mitochondrial protein content was determined by the biuret reaction (Cain & Skilleter, 1987).
Assays of thioridazine in mitochondria-free systems
Reduction of DPPH (100 μM) by thioridazine (0 – 100 μM) was monitored from the change in absorbance at 517 nm, 5 min after the drug was incubated with 40 mM sodium acetate, pH 5.5, and 1 ml ethanol (2.5 ml final volume) (Blois, 1958). Iron chelation was monitored through the formation of the FeII(BPS)3 complex (Bolann & Ulvik, 1987). Thioridazine (0 – 100 μM) was added to the standard medium in the presence of 50 μM Fe2+ plus 200 μM BPS and absorbance at 530 nm was measured after 30 min.
Assays with energized mitochondria
The standard incubation medium for all assays was (mM) sucrose 125, KCl 65 and HEPES – KOH 10, pH 7.4. Mitochondria were energized with 5 mM potassium succinate (+2.5 μM rotenone) and the respiration medium included also 0.5 mM EGTA and 10 mM K2HPO4. Mitochondrial respiration was monitored polarographically with an oxygraph equipped with a Clark-type O2 electrode (Gilson Medical Electronics, Middleton, WI, U.S.A.). Mitochondria-generated O2•− was assayed as lucigenin-derived chemiluminescence, monitored with an EG & G Berthold AutoLumat LB 953 apparatus (Bad Wildbad, Germany) (Li et al., 1999). Mitochondrial swelling was estimated from the decrease in absorbance at 540 nm using a Model DU-70 spectrophotometer (Beckman, Coulter Inc., Fullerton, CA, U.S.A.). The electrical transmembrane potential difference (Δψ) was monitored using 0.4 μM rhodamine 123 as an indicator in a F-4500 spectrofluorometer (Hitachi, Tokyo, Japan) with the 505/535 nm excitation/emission wavelength pair (Emaus et al., 1986). The dye rhodamine 123 distributes electrophoretically into the mitochondria in response to Δψ of the inner mitochondrial membrane; the uptake/release of the dye by mitochondria is linearly correlated to the Δψ up to at least 170 mV. The valinomycin-induced K+ diffusion potential was used to perform a calibration curve. Thus, energized mitochondria were incubated with rhodamine 123 in presence of valinomycin and a titration with K+ was performed. The Δψ decay due to the electrogenic influx of the cation, determined by the Nerst equation (Δψ=59 log [K+]in/[K+]out; [K+]in=120 mM), is linearly correlated to the increase in the fluorescence intensity of the dye as it is released from the mitochondria (Åkerman & Wikström, 1976; Emaus et al., 1986).
Fe2+/citrate-mediated lipid peroxidation assay
Lipid peroxidation was assayed as MDA generation. One ml of mitochondrial suspension (1 mg protein) was incubated with thioridazine in the standard medium plus 5 mM succinate, 2.5 μM rotenone, 50 μM (NH4)2Fe(SO)4 and 2 mM sodium citrate for 30 min, at 37°C (1 ml final volume). For MDA determination, 1 ml of 1% TBA (prepared in 50 mM NaOH), 0.1 ml of 10 M NaOH and 0.5 ml of 20% H3PO4 were added, followed by incubation for 20 min at 85°C. The MDA – TBA complex was extracted with 2 ml of n-butanol and absorbance was measured at 535 nm. MDA concentration was calculated from ε=1.56×105 M−1 (Kowaltowski et al., 1996).
Determination of protein-thiol
After 15 min incubation with thioridazine under the swelling assay conditions, mitochondria (0.4 mg protein) were treated with perchloric acid (5% final concentration) and centrifuged at 4500×g for 10 min. The pellet was suspended with 1 ml of medium containing 50 mM EDTA and 100 mM TRIS, pH 8.0. After the addition of 2 mM DTNB, absorbance was determined at 412 nm. The amount of thiol groups was calculated from ε=13 600 M−1 (Jocelyn, 1987).
Effect of thioridazine on fluorescence responses of ANS and TMA – DPH-labelled mitochondrial membrane
Mitochondria were incubated at 30°C with 75 μM ANS (2 mg protein) or 1.04 μM TMA – DPH (0.5 mg protein) in the standard incubation medium plus 1 μg ml−1 CCCP before thioridazine was added (2 ml final volume). Fluorescence was measured with a F-4500 spectrofluorometer (Hitachi, Tokyo, Japan) at excitation and emission wavelengths of 380 and 485 nm, respectively, for ANS and of 362 and 432 nm, respectively, for TMA – DPH (Slavík, 1982; Lee et al., 1999).
Determination of cytochrome c
Cytochrome c released from mitochondria was determined by an enzyme immunoassay technique using an ELISA kit (Quantikine M., R&D Systems, Abingdon, U.K.). Mitochondria (0.4 mg protein ml−1) were incubated with thioridazine under the conditions of the specific assays and centrifuged at 16,000×g for 10 min. The supernatant (50 μl) was added to wells and incubated with cytochrome c conjugate for 2 h at 25°C. After five washes, substrate solution was added and the plate was incubated for 30 min. The reaction was stopped and optical density was determined using a microplate reader set at 450 nm with correction at 540 nm. Sample concentrations were determined based on a standard curve within a 0.78 – 25 ng ml−1 concentration range (ε=0,089 ng−1 ml).
Results
Effects of thioridazine on respiration and electrical transmembrane potential difference (Δψ) of isolated rat liver mitochondria
At relatively low concentrations (10 – 75 μM) thioridazine slightly increased the rate of succinate-supported state 4 (resting) respiration of mitochondria (Figure 1A), showing that it is a weak uncoupler of oxidative phosphorylation. However, at higher concentrations thioridazine markedly decreased the rate of succinate-supported state 3 (ADP-stimulated) respiration of mitochondria (Figure 1B, IC50=87.5 μM) in close association with a fall in Δψ (Figure 1C, IC50=89 μM), showing that thioridazine is also a typical inhibitor of oxidative phosphorylation.
Figure 1.
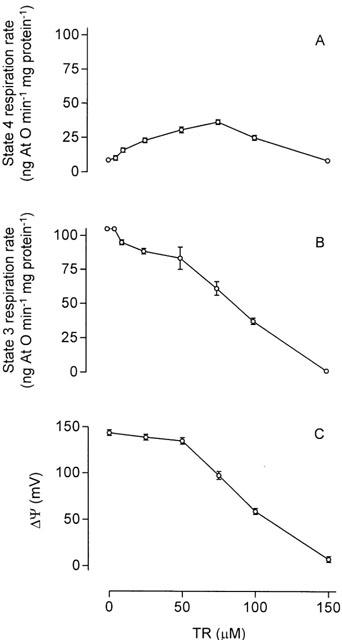
Concentration-response curves for the effects of thioridazine (TR) on state 4 respiration rate (A), state 3 respiration rate (B) and Δψ (C) in isolated rat liver mitochondria. For the respiratory assays, mitochondria (1.5 mg protein) were incubated at 30°C with 5 mM succinate and 2.5 μM rotenone in a standard incubation medium containing 125 mM sucrose, 65 mM KCl and 10 mM HEPES – KOH, pH 7.4, in the presence of 0.5 mM EGTA and 10 mM K2HPO4 (respiration medium) in a final volume of 1.5 ml. State 3 respiration was initiated with 0.4 μmol ADP. For the Δψ assays, mitochondria (2 mg protein) incubated in the standard medium plus 2.5 μM rotenone and 0.4 μM rhodamine 123 in a final volume of 2 ml were energized by the addition of 5 mM succinate. The inhibition of the Δψ response of mitochondria was immediate and remained constant for at least 5 min; its extent was calculated 30 s after drug addition by using a calibration curve, as described in Methods. Data are presented as the mean±s.e.mean of three experiments with different mitochondrial preparations.
Effect of thioridazine on mitochodria-generated O2•−
A fraction of the O2 available to mitochondria during respiration is continuously converted to O2•− which in the presence of its dismutation product H2O2 plus Fe2+ may produce the •OH radical (Boveris & Chance, 1973). As shown in Figure 2, at 10 μM, thioridazine inhibited by about 65% the accumulation of O2•− generated by respiratory chain during succinate-supported resting respiration of isolated rat liver mitochondria, assayed as lucigenin-derived chemiluminescence.
Figure 2.
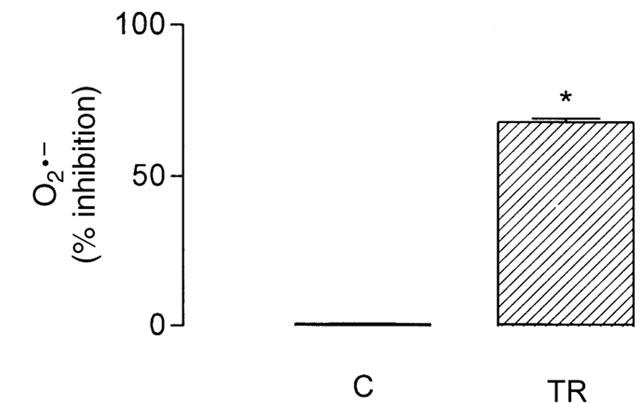
Effects of 10 μM thioridazine (TR) on mitochondria-generated O2•− assayed as lucigenin-derived chemiluminescence. Mitochondria (0.5 mg protein) were incubated with drugs at 30°C in the respiration medium described in the legend to Figure 1, in a final volume of 2 ml. The lucigenin-derived chemiluminesce response was initiated by adding 5 μM lucigenin; data are per cent inhibition of luminescence (integrated area under the curve) in relation to a control in the absence of the drug. The data are presented as the mean±s.e.mean of six experiments with different mitochondrial preparations. Statistical analysis was performed by the Mann – Whitney non-parametric test. *Significantly different from control (P<0.05).
Effect of thioridazine on Fe2+/citrate-mediated lipid peroxidation of the mitochondrial membrane
The •OH radical produced may start a lipid peroxidation process by removing hydrogen atoms from phospholipids and yielding lipid peroxyl radicals (LOO•), thus inducing a sequence of propagation reactions (Halliwell & Gutteridge, 1999). Figure 3(A) shows the effect of thioridazine on Fe2+/citrate-mediated lipid peroxidation of the mitochondrial membrane, assayed as MDA generation, and Figure 3(B) and (C) show, respectively, the effects of the drug on the associated swelling of mitochondria and release of cytochrome c. Thioridazine, at 10 μM, inhibited by about 80% MDA generation, and therefore peroxidation of the mitochondrial membrane lipids, and also inhibited by about 100% the associated swelling of mitochondria and by about 60% the release of cytochrome c induced in mitochondria under the LPO assay condition.
Figure 3.
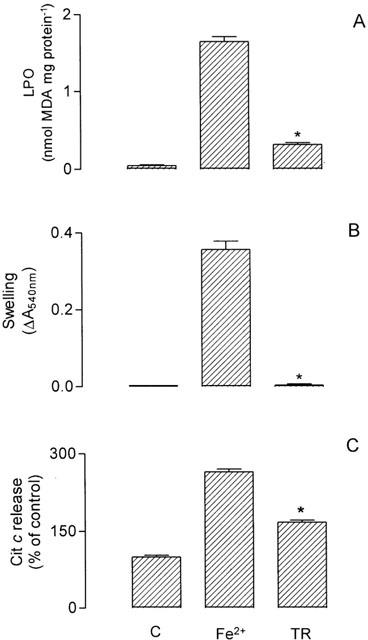
Effects of 10 μM thioridazine (TR) on lipid peroxidation assayed as MDA generation (A) and associated mitochondrial swelling (B) and cytochrome c release (C) induced by Fe2+/citrate in isolated rat liver mitochondria (Fe2+). Mitochondria (1 mg protein) were incubated in the standard medium with 5 mM succinate, 2.5 μM rotenone, 50 μM (NH4)2Fe(SO)4 and 2 mM sodium citrate for 30 min at 37°C (1 ml final volume). See Methods for MDA, mitochondrial swelling and cytochrome c determinations. Data are presented as the mean±s.e.mean of nine (A), nine (B) and six (C) experiments with different mitochondrial preparations. Statistical analysis was performed by Kruskal – Wallis non-parametric analysis of variance (ANOVA) followed by Dunn's multiple comparison test. *Significantly different from Fe2+ (P<0.05 for A and B; P<0.1 for C).
Effect of thioridazine on Ca2+/t-BOOH-induced mitochondrial permeability transition
One of the lines of evidence with regard to the mechanism of Ca2+/t-BOOH-induced MPT is that PTP opening is sensitized by the oxidation of gluthatione and NAD(P)H accumulating ROS such as the •OH radical which, in turn, oxidizes thiol groups of membrane proteins followed by cross-linkage formation (Kowaltowski et al., 2001). In this regard, the phenothiazine TFP, which is a well established MPT inhibitor (Broekemeier et al., 1985), has been proposed to directly interfere with the formation of these membrane protein – thiol cross-linkage (Pereira et al., 1992). Thioridazine, at 10 μM, inhibited by about 80% the Ca2+/t-BOOH-induced swelling of isolated rat liver mitochondria (Figure 4A), and therefore MPT onset, and also inhibited by about 100% the release of cytochrome c induced in mitochondria under the MPT assay condition (Figure 4C). Only about 20% of the mitochondrial membrane protein – thiol groups underwent oxidation under this condition, in agreement with literature data, and thioridazine almost completely protected against it (Figure 4B).
Figure 4.
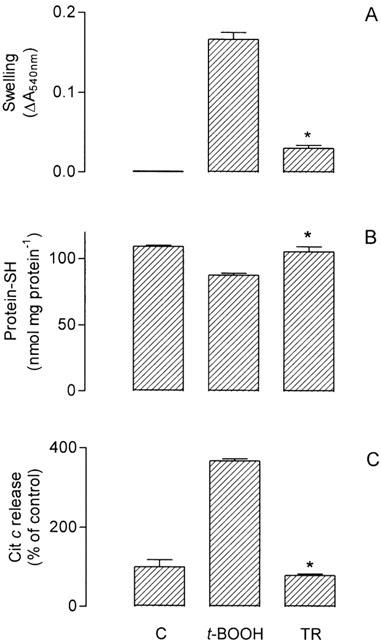
Effects of 10 μM thioridazine (TR) on mitochondrial swelling (A) and associated oxidation of protein-SH (B) and cytochrome c release (C) induced by 10 μM CaCl2+0.5 mM t-BOOH in isolated rat liver mitochondria (t-BOOH). Mitochondria (0.4 mg protein) were incubated in the standard medium with 5 mM succinate and 2.5 μM rotenone, at 30°C (1.5 ml final volume). See also Methods for the determinations. Data are presented as the mean±s.e.mean of 10 (A), six (B) and six (C) experiments with different mitochondrial preparations. Statistical analysis was performed by Kruskal – Wallis non-parametric analysis of variance (ANOVA) followed by Dunn's multiple comparison test. *Significantly different from t-BOOH (P<0.05).
Effects of thioridazine on the fluorescence responses of ANS and TMA – DPH-labelled mitochondria
In order to test the hypothesis that the above effects are mediated by interaction of thioridazine with the mitochondrial membrane, we performed assays with mitochondria labelled with the fluorescent probes ANS and TMA – DPH, which monitor membranes closer to the aqueous interface. ANS is generally assumed to bind to the polar head groups of the phospholipids and to proteins on the membrane surface, with the anionic sulfonate group being the major determinant of binding. The amount of ANS molecules bound to the membranes is highly influenced by the surface charge potential, being inversely proportional to its negative potential (Slavík, 1982) and TMA – DPH is incorporated into the hydrophobic region of membranes oriented parallel to the lipid acyl chain axis, with the cationic trimethylammonium substituent acting as a surface anchor (Lee et al., 1999). As shown in Figure 5, thioridazine respectively increased and decreased the fluorescence responses of ANS and TMA – DPH incubated with the isolated rat liver mitochondria, indicating that the drug interacts with the mitochondrial membrane and that interaction is more likely to occur close to its surface.
Figure 5.
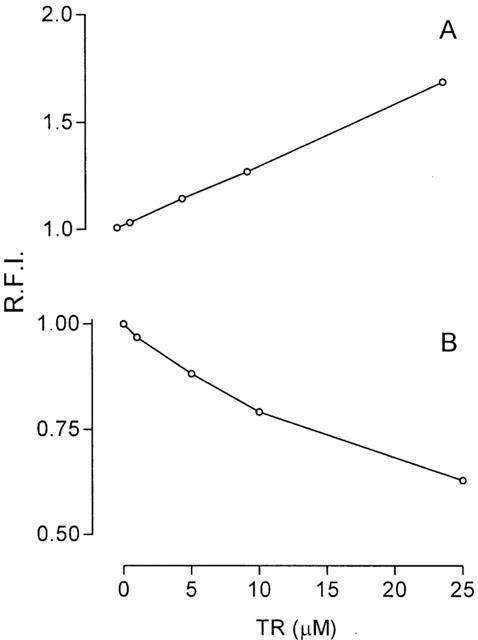
Concentration-response curves for the effects of thioridazine (TR) on membrane-ANS (A) and membrane-TMA – DPH (B) fluorescence response in isolated rat liver mitochondria incubated in the standard medium, as described in Methods. The relative fluorescence intensity (R.F.I.) was measured after drug addition. Data are presented as the mean±s.e.mean of three experiments with different mitochondrial preparations.
Effects of trifluoperazine
The effects of TFP on the mitochondrial parameters/responses above were evaluated in parallel, and, except for potency about 10% lower, they were closely similar to those of thioridazine (results not shown).
Discussion
Previous literature data have shown that the classical phenothiazine derivative TFP affects the activity of membrane proteins by interacting with hydrophobic sites located in the membrane-embedded domains (Dabbeni-Sala & Palatini, 1990). Also, the presence of antioxidant binding sites in the hydrophobic core of the inner mitochondrial membrane or in the mitochondrial matrix has been previously proposed. These sites may accept a variety of unrelated hydrophobic compounds modulating ROS-associated mitochondrial processes independently of free radical scavenging activities (Gudz et al., 1997; Elimadi et al., 1998). The structure of phenothiazines per se does not suggest free radical scavenging properties. Indeed, within the 0 – 100 μM concentration range thioridazine did not reduce DPPH (data not shown), a stable free radical potentially reactive with all compounds able to donate a hydrogen atom (Blois, 1958). Nevertheless, in the present study thioridazine showed important antioxidant activity on mitochondria, as characterized by the inhibition of accumulation of the O2•− continuously generated during resting respiration, and by inhibition of ROS-associated events, such as Fe2+/citrate-mediated peroxidation of the mitochondrial membrane lipids and Ca2+/t-BOOH-induced mitochondrial permeability transition.
Inhibition of either respiratory chain or ATP synthase activities is the mechanism accounting for the reduced rate of mitochondrial state 3 respiration. We can therefore expect that thioridazine directly and/or indirectly interacts with mitochondrial proteins that are relevant to the control of state 3 respiration, namely, enzymes comprising segments of the respiratory chain and ATP synthase. However, two lines of evidence argue against participation of these mechanisms in the antioxidant activity of thioridazine on mitochondria: (1) at the concentration at which thioridazine exhibited antioxidant activity (10 μM) the drug had no significant effect on mitochondrial respiration; and (2) in general respiratory chain inhibitors rather stimulate than inhibit O2•− generation by mitochondria. The coenzyme Q cycle has been reported to be the main pathway for O2•− generation by the respiratory chain; it accumulates the semiquinone anion in the inner membrane of mitochondria which, in turn, donates electron to O2 (Kowaltowski et al., 2001). Within this context, we believe that the inhibition of accumulation of mitochondria-generated O2•− due to thioridazine comes from the interaction of the drug with the inner mitochondrial membrane, thus impairing the electron donation by the semiquinone anion to the O2.
That thioridazine interacts with the mitochondrial membrane is demonstrated by the increase/decrease in the fluorescence response of mitochondria labelled with ANS/TMA – DPH, respectively. The amount of ANS molecules bound to membranes is inversely proportional to the negative membrane surface potential (Slavík, 1982) and the surface anchor of TMA – DPH is a cationic substituent (Lee et al., 1999). Since thioridazine has a positive charge at the pH of the assays, the increase of the fluorescence response of ANS may reflect a decrease of the negative charge density on the membrane surface by the drug, thereby increasing the binding sites of the probe. On the other hand, the decrease of the fluorescence response of TMA – DPH may reflect a competition between the positive charge of thioridazine and the probe, thereby reducing its anchoring to the membrane. Therefore, the interaction of thioridazine with the inner mitochondrial membrane is more likely to occur close to its surface and may be responsible, at least in part, for the antioxidant activity of the drug on mitochondria in terms of inhibition of accumulation of mitochondria-generated O2•−, and also of LPO and MPT.
The mechanism of LPO inhibition by thioridazine does not involve chelation of the iron required in the Fenton/HaberWeiss reaction, since the drug did not interfere with the formation of the FeII(BPS)3 complex, as evaluated spectrophotometrically at 530 nm (data not shown). Therefore, we believe that LPO inhibition occurs either at the level of initiation of the process by decreasing the availability of O2•−, and therefore of the •OH radical, as discussed above, or at the level of propagation of processes, by slowing down the LPO free radical reactions as the physical state of the mitochondrial membrane is altered by the drug interaction. Concerning MPT inhibition by thioridazine and considering the proposed mechanism discussed above (Kowaltowski et al., 2001), a decrease in the availability of •OH may directly inhibit the protein – thiol oxidation and, if oxidation occurs, a change in the physical state of the mitochondrial membrane would impair the cross-linkage between the oxidized thiols, and therefore the onset of MPT.
The participation of mitochondria in non-apoptotic and apoptotic cell death has been well established. MPT onset can lead to a necrosis-type cell death by ATP depletion and Ca2+ homeostasis disruption, without release of cytochrome c. However, during apoptosis cytochrome c may be released from mitochondria into the cytosol as the mitochondrial membrane becomes permeable due to PTP opening or other processes, and thus acts by triggering the activation of caspases (Susin et al., 1998; Bernadi et al, 2001). In the present study inhibition of cytochrome c release by thioridazine correlated well with inhibition of LPO and MPT onset, indicating that thioridazine may be a useful tool to understand the relationship between mitochondrial mechanisms and induction of apoptosis, playing, in addition, a potential role as a cytoprotective drug in terms of apoptotic cell death.
In general, the present study shows that thioridazine interacts with the inner mitochondrial membrane, more likely close to its surface, acquiring antioxidant activity toward processes with potential implications in apoptosis such as O2•− accumulation, and also peroxidation of mitochondrial membrane lipids, mitochondrial permeability transition and associated release of cytochrome c in mitochondria. Studies on the effects of thioridazine on the mitochondrial mechanisms potentially implicated in apoptosis are now in progress by employing isolated rat liver cells.
Acknowledgments
This work was supported by FAPESP and CNPq, Brazil. Results will be presented by Tiago Rodrigues to the Department of Biochemistry, School of Medicine of Ribeirão Preto, University of São Paulo, in partial fulfilment of the requirements for the Doctoral degree.
Abbreviations
- ANS
1-aniline-8-naphthalene sulfonate
- BPS
bathophenanthroline-disulphonic acid
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- CsA
cyclosporin A
- cyt. c
cytochrome c
- DPPH
1,1-diphenyl-2-picryl-hydrazyl
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- EDTA
ethylene diaminetetraacetic acid
- EGTA
ethylene glycol bis (-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
N-(2-hydroxyethyl) piperazine-N′-(2-ethanesulphonic acid)
- MDA
malondialdehyde
- MPT
mitochondrial permeability transition
- PTP
permeability transition pore
- ROS
reactive oxygen species
- TBA
thiobarbituric acid
- t-BOOH
tert-butyl hydroperoxide
- TFP
trifluoperazine
- TMA-DPH
1-(4-trimethylammoniumphenyl)-6 phenyl 1,3,5-hexatriene
- TR
thioridazine
- TRIS
tris(hydroxymethyl)-aminomethane
- Δψ
electrical transmembrane potential difference
References
- ÅKERMAN K.E.O., WIKSTRÖM M.K.F. Safranine as a probe of the mitochondrial membrane potential. FEBS Lett. 1976;68:191–197. doi: 10.1016/0014-5793(76)80434-6. [DOI] [PubMed] [Google Scholar]
- BALDESSARINI R.J.Drugs and the treatment of psychiatric disorders: psychosis and anxiety Goodman & Gilman's The Pharmacological Basis of Therapeutics 1995New York: McGraw-Hill; 399–430.9th edn ed. Hardman, J.G., Gilman, A.G. & Limbird, L.E., pp [Google Scholar]
- BERNARDI P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- BERNARDI P., PETRONILLI V., DI LISA F., FORTE M. A mitochondrial perspective on cell death. Trends Biochem. Sci. 2001;26:112–117. doi: 10.1016/s0968-0004(00)01745-x. [DOI] [PubMed] [Google Scholar]
- BLOIS M.S. Antioxidant determinations by the use of a stable free radical. Nature. 1958;181:1199–1200. [Google Scholar]
- BOLANN B.J., ULVIK R.J. Release of iron from ferritin by xanthine oxidase. Role of the superoxide radical. Biochem. J. 1987;243:55–59. doi: 10.1042/bj2430055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOVERIS A., CHANCE B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROEKEMEIER K.M., SCHMID P.C., SCHMID H.H., PFEIFFER D.R. Effects of phospholipase A2 inhibitors on ruthenium red-induced Ca2+ release from mitochondria. J. Biol. Chem. 1985;260:105–113. [PubMed] [Google Scholar]
- CAIN K., SKILLETER D.N.Preparation and use of mitochondria in toxicological research Biochemical Toxicology 1987Oxford: IRL Press; 217–254.ed. Snell, K. & Mullock B. pp [Google Scholar]
- CROMPTON M., ELLINGER H., COSTI A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- DABBENI-SALA F., PALATINI P. Mechanism of local anesthetic effect. Involvement of F0 in the inhibition of mitochondrial ATP synthase by phenothiazines. Biochim. Biophys. Acta. 1990;1015:248–252. doi: 10.1016/0005-2728(90)90027-2. [DOI] [PubMed] [Google Scholar]
- ELIMADI A., BOUILLOT L., SAPENA R., TILLEMENT J.P., MORIN D. Dose-related inversion of cinnarizine and flunarizine effects on mitochondrial permeability transition. Eur. J. Pharmacol. 1998;348:115–121. doi: 10.1016/s0014-2999(98)00135-6. [DOI] [PubMed] [Google Scholar]
- EMAUS R.K., GRUNWALD R., LEMASTERS J.J. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim. Biophys. Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-x. [DOI] [PubMed] [Google Scholar]
- GONZALEZ-FLECHA B., BOVERIS A. Mitochondrial sites of hydrogen peroxide production in reperfused rat kidney cortex. Biochm. Biophys. Acta. 1995;1243:361–366. doi: 10.1016/0304-4165(94)00160-y. [DOI] [PubMed] [Google Scholar]
- GUDZ T., ERIKSSON O., KUSHNAREVA Y., SARIS N.E., NOVGORODOV S. Effect of butylhydroxytoluene and related compounds on permeability of the inner mitochondrial membrane. Arch. Biochem. Biophys. 1997;342:143–156. doi: 10.1006/abbi.1997.0113. [DOI] [PubMed] [Google Scholar]
- HALLIWELL B., GUTTERIDGE J.M. Free Radical in Biology and Medicine. Oxford: University Press; 1999. pp. 36–95. [Google Scholar]
- HUNTER D.R., HAWORTH R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- JOCELYN P.C. Spectrophotometric assay of thiols. Meth. Enzymol. 1987;143:44–67. doi: 10.1016/0076-6879(87)43013-9. [DOI] [PubMed] [Google Scholar]
- KOWALTOWSKI A.J., CASTILHO R.F., GRIJALBA M.T., BECHARA E.J., VERCESI A.E. Effect of inorganic phosphate concentration on the nature of inner mitochondrial membrane alterations mediated by Ca2+ ions. A proposed model for phosphate-stimulated lipid peroxidation. J. Biol. Chem. 1996;271:2929–2934. doi: 10.1074/jbc.271.6.2929. [DOI] [PubMed] [Google Scholar]
- KOWALTOWSKI A.J., CASTILHO R.F., VERCESI A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- KROEMER G., DALLAPORTA B., RESCHE-RIGON M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998;60:610–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- LEE J., YU B.P., HERLIHY J.T. Modulation of cardiac mitochondrial membrane fluidity by age and calorie intake. Free Radic. Biol. Med. 1999;26:260–265. doi: 10.1016/s0891-5849(98)00195-6. [DOI] [PubMed] [Google Scholar]
- LI Y., ZHU H., TRUSH M.A. Detection of mitochondria-derived reactive oxygen species production by the chemilumigenic probes lucigenin and luminol. Biochim. Biophys. Acta. 1999;1428:1–12. doi: 10.1016/s0304-4165(99)00040-9. [DOI] [PubMed] [Google Scholar]
- MALHEIROS S.V., PAULA E., MEIRELLES N.C. Contribution of trifluoperazine/lipid ratio and drug ionization to hemolysis. Biochim. Biophys. Acta. 1998;1373:332–340. doi: 10.1016/s0005-2736(98)00090-x. [DOI] [PubMed] [Google Scholar]
- PAN G., ZHOU T., RADDING W., SAAG M.S., MOUNTZ J.D., MCDONALD J.M. Calmodulin antagonists inhibit apoptosis of CD4+ T-cells from patients with AIDS. Immunopharmacol. 1998;40:91–103. doi: 10.1016/s0162-3109(98)00018-6. [DOI] [PubMed] [Google Scholar]
- PAVLOV P.F., GLASER E. Inhibition of protein import into mitochondria by amphiphilic cations: potential targets and mechanism of action. Biochem. Biophys. Res. Commun. 1998;252:84–91. doi: 10.1006/bbrc.1998.9590. [DOI] [PubMed] [Google Scholar]
- PEDERSEN P.L., GREENAWALT J.W., REYNAFARJE B., HULLIHEN J., DECKER G.L., SOPER J.W., BUSTAMENTE E. Preparation and characterization of mitochondria and submitochondrial particles of rat liver and liver-derived tissues. Meth. Cell. Biol. 1978;20:411–481. doi: 10.1016/s0091-679x(08)62030-0. [DOI] [PubMed] [Google Scholar]
- PEREIRA R.S., BERTOCCHI A.P., VERCESI A.E. Protective effect of trifluoperazine on the mitochondrial damage induced by Ca2+ plus prooxidants. Biochem. Pharmacol. 1992;44:1795–1801. doi: 10.1016/0006-2952(92)90074-s. [DOI] [PubMed] [Google Scholar]
- PETRONILLI V., PENZO D., SCORRANO L., BERNARDI P., DI LISA F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001;276:12030–12034. doi: 10.1074/jbc.M010604200. [DOI] [PubMed] [Google Scholar]
- SKULACHEV V.P. Mitochondria in the programmed death phenomena; a principle of biology: ‘it is better to die than to be wrong'. IUBMB Life. 2000;49:365–373. doi: 10.1080/152165400410209. [DOI] [PubMed] [Google Scholar]
- SLAVÍK J. Anilinonaphthalene sulfonate as a probe of membrane composition and function. Biochim. Biophys. Acta. 1982;694:1–25. doi: 10.1016/0304-4157(82)90012-0. [DOI] [PubMed] [Google Scholar]
- SUSIN S.A., ZAMZAMI N., KROEMER G. Mitochondria as regulators of apoptosis: doubt no more. Biochim. Biophys. Acta. 1998;1366:151–165. doi: 10.1016/s0005-2728(98)00110-8. [DOI] [PubMed] [Google Scholar]
- ZORATTI M., SZABÒ I. The mitochondrial permeability transition. Biochim. Biophys. Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]