

Abstract
Glucocorticoids have been implicated in hypoglycemia-induced autonomic failure but also contribute to normal counterregulation. To determine the influence of normal and hypoglycemia-induced levels of glucocorticoids on counterregulatory responses to acute and repeated hypoglycemia, we compared plasma catecholamines, corticosterone, glucagon, and glucose requirements in male wild-type (WT) and glucocorticoid-deficient, corticotropin-releasing hormone knockout (CRH KO) mice. Conscious, chronically cannulated, unrestrained WT and CRH KO mice underwent a euglycemic (Prior Eu) or hypoglycemic clamp (Prior Hypo) on day 1 followed by a hypoglycemic clamp on day 2 (blood glucose both days, 65 ± 1 mg/dl). Baseline epinephrine and glucagon were similar, and norepinephrine was elevated, in CRH KO vs. WT mice. CRH KO corticosterone was almost undetectable (<1.5 μg/dl) and unresponsive to hypoglycemia. CRH KO glucose requirements were significantly higher during day 1 hypoglycemia despite epinephrine and glucagon responses that were comparable to or greater than those in WT. Hyperinsulinemic euglycemia did not increase hormones or glucose requirements above baseline. On day 2, Prior Hypo WT had significantly higher glucose requirements and significantly lower corticosterone and glucagon responses. Prior Hypo and Prior Eu CRH KO mice had similar day 2 glucose requirements. However, Prior Hypo CRH KO mice had significantly lower day 2 epinephrine and norepinephrine vs. Prior Eu CRH KO and tended to have lower glucagon than on day 1. We conclude that glucocorticoid insufficiency in CRH KO mice correlates with 1) impaired counterregulation during acute hypoglycemia and 2) complex effects after repeated hypoglycemia, neither preventing decreased hormone responses nor worsening glucose requirements.
Keywords: hypoglycemia unawareness, hypoglycemia-associated autonomic failure, catecholamines, adrenal insufficiency
HYPOGLYCEMIA-INDUCED AUTONOMIC FAILURE is an emerging challenge in diabetes management. Although intensive insulin therapy has been identified to reduce long-term complications from diabetes, more aggressive insulin use has also increased the risk of debilitating or even fatal hypoglycemia (9). The ability to decrease insulin, the first physiological defense against falling glucose levels, is absent in type 1 diabetes and in the insulin-dependent stages of advanced type 2 diabetes. Glucagon responses to hypoglycemia are also impaired with increasing duration of insulin-dependent diabetes (8). After glucagon, sympathetic nervous system activation remains the most rapid, and therefore the most effective, defense against decreases in blood glucose (8). However, both the threshold and the absolute magnitude of sympathetic nervous system responses decrease after as few as one or two prior episodes of hypoglycemia (12, 15). Other defenses against hypoglycemia, such as glucocorticoid and growth hormone secretion, exhibit similar impairment after repeated hypoglycemia, although these responses typically have less impact on immediate counterregulation (9). Decrements in sympathetically mediated norepinephrine and epinephrine release, along with the diminution of associated physical symptoms of sympathetic activation, are major contributors to hypoglycemia unawareness, in which diabetic patients lose both the physiological defenses and the subjective warning signs that blood sugar is dangerously low (9).
Glucocorticoids have been shown to inhibit sympathetic activity (4, 31), and Davis et al. (14) have proposed that hypoglycemia-induced increases in glucocorticoids contribute to the inhibition of sympathetic responses to recurrent hypoglycemia. Davis et al. have shown that cortisol infusion mimicked the suppressive effects of prior hypoglycemia on counterregulatory hormone responses (14) and have reported that counterregulatory responses were preserved in primary adrenocortical failure patients (13). These findings seem to contradict the established role of glucocorticoids not only as counterregulatory hormones but also as essential positive regulators of adrenomedullary epinephrine (18, 47), a key counterregulatory defense when glucagon secretion is defective (9).
We have studied counterregulation in the corticotropin-releasing hormone knockout (CRH KO) mouse to develop an animal model to resolve the paradoxical effects of glucocorticoids on adrenomedullary function. The primary metabolic phenotype of the CRH KO mouse is consistent with glucocorticoid deficiency (25, 26, 35). Male CRH KO mice have negligible plasma levels of glucocorticoids under most circumstances, including after hypoglycemia (28, 29, 36). However, CRH KO mice have relatively normal adrenomedullary morphology (34) and marked epinephrine responses to hypoglycemia (28, 29), making them an ideal model to investigate the effects of selective glucocorticoid deficiency on adrenomedullary function. We (28, 29) have previously used bolus insulin injection to test counterregulatory hormone responses to acute and repeated hypoglycemia in CRH KO mice. However, because hypoglycemia induced in this manner is relatively uncontrolled, and because the handling, injection, and blood sampling required by this approach might have been stresses (10, 40) that affected counterregulatory responses (20, 23, 45), we have now assessed counterregulation during glucose clamps in conscious, unrestrained, chronically cannulated CRH KO and wild-type (WT) mice. We hypothesized that, in contrast to WT mice, CRH KO mice would exhibit impaired counterregulation during acute hypoglycemia but would not exhibit counterregulatory deficits after repeated hypoglycemic clamps.
METHODS
Animals
All procedures were approved by the Institutional Animal Care and Use Committees of Albany Medical College and Vanderbilt University. Male CRH WT and KO mice were bred as littermates from heterozygous parents (34). The breeding colony had been maintained on a C57BL/6 background for at least 10 generations. Mice were bred and genotyped at Albany Medical College and were subsequently studied at Vanderbilt University. Mice were 5–8 mo of age at the time of study, representing the time required for weaning, genotyping, quarantine after receipt at Vanderbilt, and surgical preparation. Mice were housed on a 12:12-h light-dark cycle throughout.
Mice were implanted with chronic indwelling carotid artery and jugular vein catheters and recovered for 5–7 days until they had regained their presurgery body weight, as previously described (24). Catheters were filled with heparinized saline (200 U/ml), tunneled under the skin, and exteriorized suprascapularly. Mice were injected subcutaneously with 25 μg/day enoxaparin sodium (Sanofi-Aventis, Bridgewater, NJ) to prevent blood clots.
Glucose clamps
CRH WT and KO mice underwent two hyperinsulinemic glucose clamps separated by 24 h. For each clamp, regular insulin (Humulin-R; Lilly, Indianapolis, IN) was infused intravenously at 20 mU· kg−1 · min−1 for 120 min. The insulin infusion rate was selected to produce a consistent depth and duration of hypoglycemia to define peak and plateau counterregulatory responses (29). On day 1, glucose infusion was adjusted to maintain incoming blood glucose levels (euglycemia) or decreased to ~65 mg/dl (hypoglycemia); on day 2, all mice were subjected to a hypoglycemic clamp. On each day of study, mice were fasted for 6 h before the clamp by being transferred within 1 h of lights-on to 1-liter chambers with bedding but without food or water. Extension lines were attached to the arterial and venous catheters at the time of transfer and exteriorized through the chamber lid. Mice were studied in the postabsorptive state 6 h later (7 h after lights-on). Whenever possible, CRH KO and WT littermates were studied together on the same days.
Arterial blood glucose was measured every 10 min (Hemocue, Lake Forest, CA) during each clamp. The glucose meter coefficient of variation was 1.6–3.5% for glucose values up to 400 mg/dl. Larger arterial blood samples (160 μl) were drawn for hormone analysis at 0 (before insulin infusion), 30, and 120 min on day 1 and at 0, 15, 30, and 120 min on day 2. Blood volume was replaced by donor red blood cells that were washed, resuspended in heparinized saline, warmed, and infused intravenously continuously with the insulin and glucose at 6 μl/min. Between the day 1 and day 2 clamps, euglycemia was restored in hypoglycemic mice by continuing glucose without insulin infusion, and all mice were returned to their home cages overnight with unrestricted access to food and water. After collection of the 120-min blood sample on day 2, mice were immediately killed by pentobarbital sodium overdose (100 mg/kg iv) and decapitation.
Plasma assays
Plasma epinephrine and norepinephrine (detection limit 20 pg/ml) were measured by HPLC according to previously described methods (22). Plasma corticosterone (MP Biomedical, formerly ICN, Irvine, CA), insulin, and glucagon (Linco, St. Louis, MO) were measured with previously described radioimmunoassays (5, 25, 29). All assays had been validated for small volumes of mouse plasma and had inter-assay coefficients of variation no higher than 10%.
Statistics
Data were analyzed by two-way ANOVA with repeated measures across time, with single post hoc comparisons performed by t-test (Statview 5.0; SAS Institute, Cary, NC). Significance was defined as P < 0.05. Data are presented throughout as means ± SE. Where no error bars are visible, the scale of the figure exceeded that of the error.
RESULTS
Day 1: effect of combined CRH and glucocorticoid deficiency on responses to controlled, acute hypoglycemia or euglycemia
WT and CRH KO mice had similar body weights at the time of study (WT vs. CRH KO, 28.1 ± 0.4 vs. 28.9 ± 0.6 g; n = 15 and 9, respectively). Blood glucose levels were similar 7 h after lights-on in 6-h-fasted WT and CRH KO mice (Fig. 1, bottom, and Table 1). During a 2-h hyperinsulinemic hypoglycemic clamp on day 1, blood glucose fell to the same absolute levels and at the same rate in both genotypes (Fig. 1, bottom). Blood glucose averaged 62 ± 1 and 63 ± 1 mg/dl in WT and CRH KO mice, respectively, during the last 60 min of the clamp. The glucose infusion rate required to maintain blood glucose increased gradually over time in both genotypes but was consistently and significantly higher in CRH KO mice during the first 70 min of hypoglycemia (Fig. 1, top). The glucose infusion rates required to maintain euglycemia did not differ between genotypes (Table 1). Plasma insulin was significantly lower in CRH KO than in WT mice at baseline but was similar in all mice after glucose clamps regardless of genotype or target glucose level (Table 2).
Fig. 1.
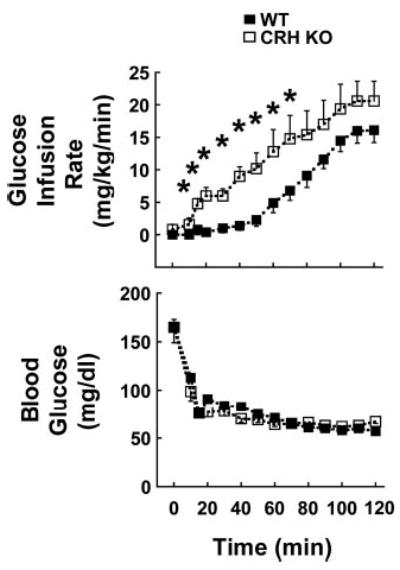
Blood glucose (bottom) and glucose infusion requirements (top) in wild-type (WT, ▪; n = 10) and corticotropin-releasing hormone knockout (CRH KO, □; n = 5) mice exposed to a hyperinsulinemic hypoglycemic clamp on day 1. *P < 0.05 vs. WT.
Table 1.
Day 1 blood glucose and GIR in WT and CRH KO mice subjected to a hyperinsulinemic euglycemic glucose clamp on day 1
| Time, min
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | 0 | 10 | 15 | 20 | 30 | 40 | 50 | 60 | 70 | 80 | 90 | 100 | 110 | 120 | |
| Glucose, mg/dl | WT | 172±5 | 202±31 | 158±33 | 157±18 | 17±316 | 166±9 | 173±7 | 168±11 | 169±8 | 151±8 | 168±12 | 178±17 | 180±12 | 172±14 |
| CRH KO | 152±14 | 181±29 | 158±15 | 137±10 | 147±6 | 150±8 | 161±6 | 165±13 | 171±10 | 155±17 | 147±18 | 154±13 | 151±12 | 156±7 | |
| GIR, mg·kg−1· min−1 | WT | 55 | 61±2 | 67±5 | 68±4 | 68±6 | 72±5 | 71±6 | 74±7 | 75±8 | 79±9 | 79±8 | 77±6 | 76±5 | 76±5 |
| CRH KO | 50 | 50 | 56±2 | 65±4 | 68±4 | 70±5 | 71±5 | 70±4 | 67±4 | 67±2 | 70±2 | 70±2 | 71±2 | 71±2 | |
Values are means ± SE; n = 5 wild-type (WT) and 4 glucocorticoid-deficient, corticotropin-releasing hormone knockout (CRH KO) mice per group. GIR, glucose infusion rate. There were no significant genotype effects on either blood glucose levels or glucose infusion rates.
Table 2.
Plasma insulin in WT and CRH KO mice subjected to hyperinsulinemic euglycemic or hypoglycemic glucose clamps
| Genotype | Group | 0 min | 120 min |
|---|---|---|---|
| WT | Hypoglycemia | 0.9±0.2 | 10.7±0.5 |
| Euglycemia | 2.2±0.5 | 9.7±1.5 | |
| CRH KO | Hypoglycemia | 0.2±0.02* | 14.8±2.5 |
| Euglycemia | 0.1±0.04* | 11.7±0.6 |
Values are means ± SE (pg/ml); n = 10 WT, hypoglycemia, 5 WT, euglycemia, 5 CRH KO, hypoglycemia, and 4 CRH KO, euglycemia. Insulin was measured in plasma drawn either before (0 min) or after (120 min) the 2-h glucose clamp on day 1.
P < 0.05 vs. WT.
On day 1, there was a significant main effect of hypoglycemia on all counterregulatory factors measured except for norepinephrine (Fig. 2; symbols omitted for clarity). Basal plasma epinephrine was similar between WT and CRH KO mice and near the lower limit of detection. Plasma epinephrine increased to similar levels in both genotypes during the hypoglycemic clamp but did not increase during the euglycemic glucose clamp on day 1 (Fig. 2, top). Plasma norepinephrine did not change over time in either genotype, although norepinephrine was significantly higher in CRH KO than in WT mice both basally and after 120 min of hyperinsulinemic euglycemia (Fig. 2, 2nd from top). WT mice exhibited sustained elevations in plasma corticosterone during hypoglycemia, whereas plasma corticosterone was near the lower detection limit and did not change in CRH KO mice (Fig. 2, 3rd on left). In contrast to the response to hypoglycemia, WT mice did not maintain elevated corticosterone levels during euglycemia; corticosterone levels were significantly higher in hypoglycemic than in euglycemic WT mice at 120 min (Fig. 2, 3rd from top; significance symbols omitted for clarity). Basal plasma glucagon was also near the lower detection limit and increased approximately threefold in WT mice during hypoglycemia. However, CRH KO mice exhibited transiently but significantly higher glucagon levels than did WT mice by 30 min of the hypoglycemic clamp (Fig. 2, bottom left). Plasma glucagon was low and did not change at any time during day 1 euglycemia (Fig. 2, bottom right).
Fig. 2.
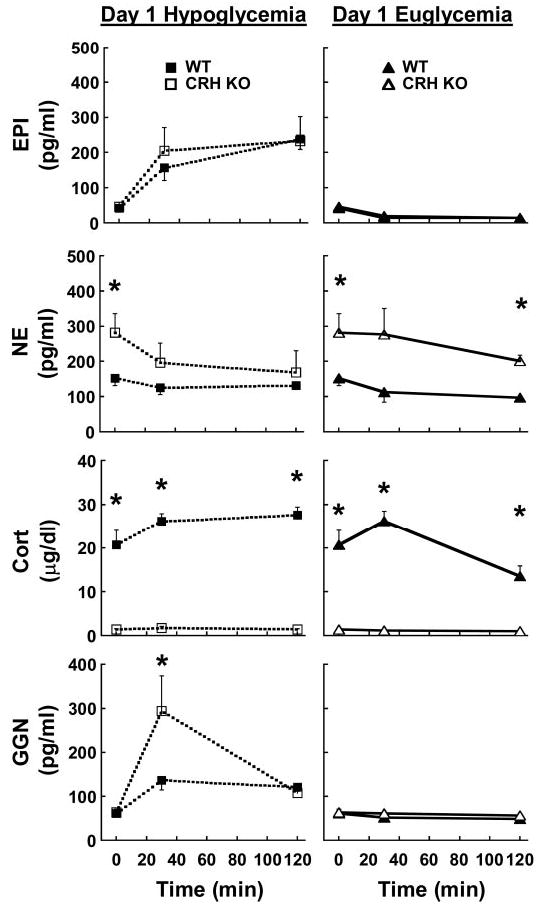
From top to bottom: plasma epinephrine (EPI), norepinephrine (NE), corticosterone (Cort), and glucagon (GGN) in WT (filled symbols) and CRH KO mice (open symbols) during hypoglycemic (squares, left) or euglycemic (triangles, right) hyperinsulinemic glucose clamps on day 1; n = 10 hypoglycemic WT, 5 hypoglycemic CRH KO, 5 euglycemic WT, and 4 euglycemic CRH KO. *P < 0.05 vs. WT in the same clamp group.
Day 2: effect of combined CRH and glucocorticoid deficiency on responses to repeated hypoglycemia
Blood glucose was similar among all mice subjected to a hypoglycemic glucose clamp on day 2 regardless of genotype or prior exposure to hypoglycemia (Fig. 3). Blood glucose levels during day 2 hypoglycemia (65 ± 1 mg/dl during the last 60 min) were also comparable to those during the day 1 hypoglycemic clamp. WT mice exposed to hypoglycemia on day 1 (Prior Hypo) required significantly higher glucose infusion rates to maintain blood glucose during the first 40 min of hypoglycemia on day 2 than did WT mice that had been euglycemic on day 1 (Prior Eu; Fig. 4, left). Prior Hypo CRH KO mice did not exhibit further increases in glucose infusion requirements relative to their Prior Eu controls (Fig. 4, right). Nevertheless, glucose infusion requirements in all CRH KO mice closely resembled those in Prior Hypo WT mice.
Fig. 3.
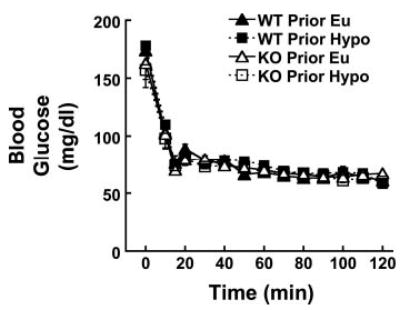
Blood glucose during hyperinsulinemic hypoglycemic clamp in all groups of mice on day 2. There were no significant differences among the groups at any time point. Symbols and groups are as in Fig. 2.
Fig. 4.
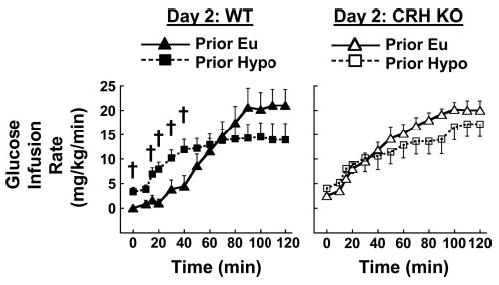
Effects of day 1 euglycemia (Prior Eu, triangles) or hypoglycemia (Prior Hypo, squares) on glucose infusion requirements during hypoglycemic clamp on day 2 in WT (left and filled symbols) and CRH KO mice (right and open symbols). Groups are as in Fig. 2. †P < 0.05 vs. Prior Eu in the same genotype.
Prior exposure to hypoglycemia on day 1 did not significantly affect basal hormone levels in either genotype the following day (Fig. 5). At the times monitored, we did not detect any differences in epinephrine responses to hypoglycemia between Prior Eu and Prior Hypo WT mice (Fig. 5, top left). However, hypoglycemia-induced epinephrine levels were significantly higher at 15 min in Prior Eu vs. Prior Hypo CRH KO mice (Fig. 5, top right). Plasma norepinephrine did not increase during hypoglycemia in either genotype and did not differ between Prior Eu and Prior Hypo WT mice (Fig. 5, left, 2nd from top). Day 2 norepinephrine levels were initially elevated and declined over time in Prior Eu CRH KO mice (Fig. 5, right, 2nd from top), similar to changes in norepinephrine in hypoglycemic CRH KO mice on day 1. In contrast, Prior Hypo CRH KO mice tended to have lower day 2 norepinephrine levels, which differed significantly from those in Prior Eu CRH KO mice at 15 min (Fig. 5, right, 2nd from top). Prior Hypo WT mice had delayed corticosterone responses to day 2 hypoglycemia, with 15-min corticosterone levels being significantly lower than those in corresponding Prior Eu controls (Fig. 5, left, 3rd from top). Plasma corticosterone remained near the lower limit of detection and was unaffected by acute or repeated hypoglycemia in CRH KO mice (Fig. 5, right, 3rd from top). Glucagon responses to day 2 hypoglycemia were also transiently but significantly reduced in Prior Hypo vs. Prior Eu WT mice (Fig. 5, bottom left). Prior hypoglycemia did not cause similar impairment of the glucagon response in Prior Hypo vs. Prior Eu CRH KO mice (Fig. 5, bottom right). However, peak glucagon levels in CRH KO mice subjected to two hypoglycemic clamps still tended to be lower on day 2 than on day 1, although these differences were not statistically significant.
Fig. 5.
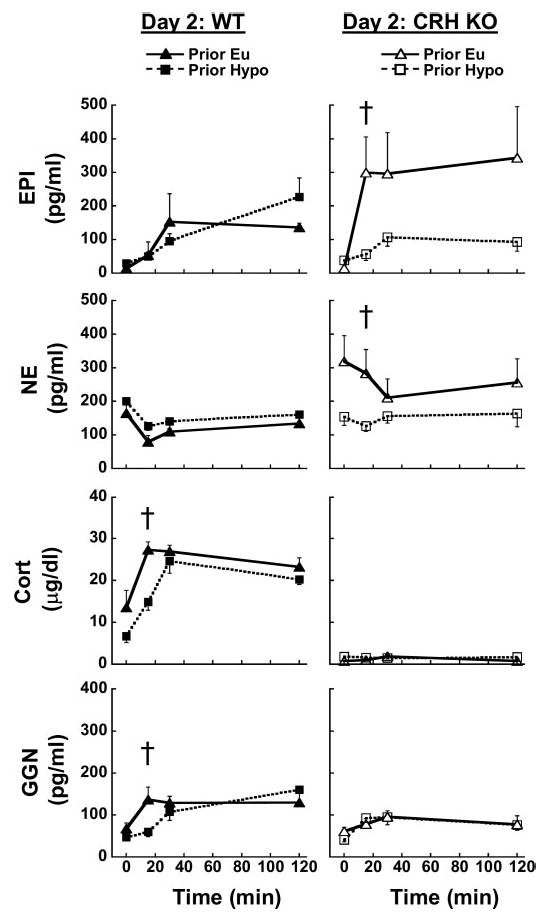
Effects of prior euglycemia or hypoglycemia on (from top to bottom) plasma EPI, NE, Cort, and GGN in WT (left) and CRH KO mice (right) during hypoglycemic clamps on day 2. Symbols and groups are as in Fig. 4. †P < 0.05 vs. Prior Eu in the same genotype.
We also measured food intake overnight between the day 1 and day 2 glucose clamps to determine whether differences in feeding among groups could have influenced our counterregulatory measures. There was a significant main effect of genotype on overnight food consumption (Table 3). However, CRH KO mice actually ate less than WT mice, and differences between WT and CRH KO mice were not significant by post hoc testing in either Prior Eu or Prior Hypo groups.
Table 3.
Food intake between day 1 and day 2 glucose clamps in WT and CRH KO mice
| Genotype | Group | Food Intake, g/g body wt |
|---|---|---|
| WT | Prior Hypo | 0.193.02 |
| Prior Eu | 0.203.03 | |
| CRH KO | Prior Hypo | 0.163.01 |
| Prior Eu | 0.133.01 |
Values are means ± SE; n = 10 WT Prior Hypo, 5 WT Prior Eu, 5 CRH KO Prior Hypo, and 4 CRH KO Prior Eu. There was a significant main effect of genotype, but not of prior hypoglycemia or genotype × hypoglycemia interaction, on overnight food consumption.
DISCUSSION
Using glucose clamp techniques in chronically cannulated, conscious, unrestrained mice, we have shown that combined CRH and glucocorticoid deficiency is associated with defective counterregulation during acute hypoglycemia and does not prevent decreases in counterregulatory hormones after repeated hypoglycemia. These findings are consistent with our prior studies of this model using bolus insulin injection to induce hypoglycemia (29). However, we have found that, unlike in WT mice, decreased counterregulatory hormones after antecedent hypoglycemia are not associated with further increases in glucose requirements in CRH KO mice. Our current and previous data suggest that the role that glucocorticoids play in the impaired counterregulatory response to repeated hypoglycemia is complex. Although activation of the adrenocortical axis is required for protection against hypoglycemia, its prior activation is not essential to explain blunted autonomic responses to subsequent hypoglycemia.
CRH KO mice exhibited impaired counterregulation during acute hypoglycemia (day 1). This impairment was evident from the significantly elevated glucose infusion rates required during the first hour of hypoglycemia. Additional counterregulatory abnormalities were also suggested by the fact that glucose requirements were increased despite relatively normal epinephrine and significantly greater glucagon responses to hypoglycemia in CRH KO vs. WT mice.
Counterregulatory deficits in CRH KO mice during acute hypoglycemia were consistent with their profound glucocorticoid insufficiency. Due to their slower actions, glucocorticoids are not typically considered critical to acute counterregulation (9). However, there is evidence that glucocorticoids contribute acutely to increasing net glucose production during hypoglycemia in normal subjects independently of changes in epinephrine (16). Chronic glucocorticoid insufficiency also increases risks of hypoglycemia; although hypoglycemia risk is most common in children, it is not unheard of in adult patients (21, 43). Both global and hepatic loss of glucocorticoid action in mice causes fasting hypoglycemia (34, 39).
Chronic glucocorticoid insufficiency in CRH KO mice could also increase glucose requirements during insulin-induced hypoglycemia by increasing insulin sensitivity, decreasing substrates for glucose production, or impairing the action of other counterregulatory hormones. The significantly lower baseline insulin levels in CRH KO mice are consistent with increased insulin sensitivity (1, 11, 29). However, glucose requirements during euglycemic hyperinsulinemic clamps on day 1 were similar, suggesting that peripheral insulin responsiveness was normal in CRH KO. Hepatic insulin action could differ between CRH KO and WT mice, but such differences would not have been detected at the insulin infusion rates used in this study. Glucocorticoids also promote the deposition of hepatic glycogen (2, 20), which is the largest and most rapidly accessible source of glucose during acute hypoglycemia (21). Although liver glycogen content is similar in fed WT and CRH KO mice (29), it is possible that CRH KO mice had lower hepatic glycogen levels after the limited fasting in the present studies. In addition, glucocorticoids help to maintain responsiveness of glycogenolytic signaling pathways to glucagon and epinephrine (2, 38); these actions, in combination with possible glycogen depletion, could account for the higher glucose requirements despite normal or enhanced autonomic responses to hypoglycemia in CRH KO mice.
Although glucocorticoids are also required to maintain epinephrine synthesis (18, 46), CRH KO mice did not show any deficits in acute hypoglycemia-induced epinephrine levels. These findings are unexpected in light of clinical and animal evidence that glucocorticoid deficiency impairs adrenomedullary development and function (3, 13, 18, 46), including other studies in CRH KO mice (30). We suspect that the hypoglycemic stimulus in the current experiments was sufficiently limited and controlled as to be within the adrenomedullary capacity of CRH KO mice. In experiments combining the stimuli of retroorbital blood sampling and more severe hypoglycemia induced by bolus insulin injection, we did observe significant deficits in CRH KO epinephrine secretion (29).
WT mice exhibited counterregulatory failure after two episodes of hypoglycemia, evident as elevated glucose infusion requirements and transient but significant decreases in hypoglycemia-evoked glucocorticoid and glucagon levels. Although not the focus of our analyses, counterregulatory hormone responses were similar between day 1 hypoglycemia in Prior Hypo WT mice and day 2 hypoglycemia in Prior Eu WT mice (compare Figs. 2 and 5). Baseline corticosterone levels in WT mice tended to be elevated compared with levels typically reported in rodents (37), but these elevations probably reflect the combined effects of fasting and sampling at a later time in the light cycle (27). WT corticosterone levels were similar to those measured at midday in similarly fasted but otherwise unmanipulated C57BL/6 mice (L. Jacobson, unpublished observations). Both the overall pattern of results and the absolute levels of counterregulatory hormones were entirely consistent with our findings from repeated hypoglycemic glucose clamps in C57BL/6 mice (27). Taken together, our data indicate that controlled, mild hypoglycemia elicits defined, predictable counterregulatory responses in the mouse and that these responses are comparable across a range of ages up to 8 mo.
The absence of an effect of antecedent hypoglycemia on the epinephrine response is probably not attributable to group size or variability. We did not observe significant differences in day 2 plasma epinephrine between Prior Eu and Prior Hypo mice, even after pooling the current data with those from the previous studies in C57BL/6 mice (27), for respective total group sizes of 11 and 16. This pooling was justified by the statistical similarity of hormone levels, as discussed above, and by the genetic similarity from breeding WT mice on a C57BL/6 background (see METHODS). The similarity of epinephrine responses to acute and repeated hypoglycemia most likely reflects either the greater importance of glucagon in the mouse or the inability of our limited sampling to detect transient differences in epinephrine. Our model is not unique in lacking recurrent hypoglycemia-induced suppression of epinephrine. Epinephrine has been found to be unchanged or even increased in humans and rats exhibiting other evidence of counterregulatory failure after repeated hypoglycemia (12, 44).
Although it may also be questioned whether the early and transient decreases in counterregulatory hormones reflect impaired counterregulation in our model, there is also considerable variation in the literature as to whether, or when, counterregulatory responses to hypoglycemia differ after prior euglycemia vs. prior hypoglycemia. The composite picture of counterregulatory failure, in which all counterregulatory hormone responses to hypoglycemia exhibit sustained inhibition, has emerged through intensive investigation and is not illustrated by every study. Studies in humans as well as in rats have found epinephrine (12, 44), norepinephrine (13, 17, 32, 42, 44), glucocorticoids (15, 42, 44), or glucagon (12, 19, 44) to be unaffected even when other counterregulatory hormones were reduced. There are very few studies of counterregulatory hormones during hypoglycemic clamps in mice. As this number expands, we expect the findings will reinforce the validity of our model of counterregulatory failure.
Combined CRH and glucocorticoid deficiency had complex effects on counterregulation after repeated hypoglycemia. Unlike WT mice, Prior Hypo CRH KO mice did not require additional glucose infusion compared with their Prior Eu counterparts. Because of the additional blood volume required, we could not evaluate glucose flux to determine how glucose requirements might have been maintained in Prior Hypo CRH KO mice. However, the lack of change in glucose requirements, despite decreases in counterregulatory hormone levels, suggested that aspects of counterregulation were preserved in CRH KO mice after repeated hypoglycemia.
Contrary to our hypothesis, CRH KO mice had significant decreases in their epinephrine response to a second hypoglycemic exposure. The decline in epinephrine could have been an artifact of slightly higher day 2 epinephrine levels in Prior Eu CRH KO mice. However, epinephrine levels in Prior Hypo CRH KO mice on day 1 were similar to those in Prior Eu CRH KO mice on day 2 and (although also not the focus of our analyses) were still higher than day 2 levels in Prior Hypo CRH KO mice. Elevated levels of norepinephrine in CRH KO mice were also suppressed after antecedent hypoglycemia. These trends are consistent with inhibition of sympathoadrenal activity. Glucagon levels in CRH KO mice did not differ between Prior Eu and Prior Hypo groups during day 2 hypoglycemia, but there was still a tendency for plasma glucagon to decrease between the first and second episodes of hypoglycemia in Prior Hypo CRH KO mice. We cannot exclude the possibility that the apparent decreases in counterregulatory hormones are due to chance variation among the relatively few CRH KO mice studied or that our sampling missed increases in counterregulatory hormones that would have maintained glucose requirements. Nevertheless, our data also do not strongly support the preservation of counterregulatory hormone responses to recurrent hypoglycemia in CRH KO mice. If combined CRH and glucocorticoid deficiency can preserve counterregulation, our current data indicate that this deficiency has more pronounced effects on glucose requirements than on counterregulatory hormone levels. Any decreases in counterregulatory hormones in CRH KO mice might account for why glucose requirements were not lower in CRH KO than in WT mice after repeated hypoglycemia.
There is considerable evidence that glucose requirements during hypoglycemia can be met in part independently of counterregulatory hormones. Hepatic autoregulation, or hormone-independent changes in hepatic glucose production evoked by changing glucose levels (7, 33), is one of the best characterized examples of this phenomenon. Hepatic autoregulation can be due to autonomic nerve activity or to non-neurally-mediated changes in hepatic responses to hormones and gluconeogenic substrates (33). Although there is disagreement on the importance of hepatic autoregulation to defenses against hypoglycemia, hepatic autoregulation may play a significant role during severe hypoglycemia (33). Hepatic autoregulation could occur in CRH KO mice at less severe hypoglycemia as an adaptation to chronic glucocorticoid insufficiency. If involved, the effects of hepatic autoregulation on glucose requirements in CRH KO mice could be identified by analyzing metabolic responses to repeated hypoglycemia and defined counterregulatory hormone levels. However, initial evaluation of glucose and metabolite kinetics could not be done in the current experiments because of the additional blood volume required.
It may seem puzzling that the effects of repeated hypoglycemia on counterregulatory hormones in CRH KO mice did not resemble those reported by Davis et al. (13) in subjects with primary adrenocortical insufficiency. These subjects were studied after 5 days of basal cortisol infusion; it is possible that CRH KO mice have adapted to chronically low glucocorticoid levels differently than have patients whose replacement was modified 5 days earlier. Our hormone data are consistent with the majority of studies (17, 19, 41, 44) that have not found the inhibitory effects of glucocorticoids on counterregulation reported by Davis et al. (14) and Sandoval et al. (42). Nevertheless, the defense of glucose requirements in repeatedly hypoglycemic CRH KO mice, despite the decreased hormonal responses, suggests that glucocorticoid deficiency might prevent further deficits without actually normalizing counterregulation.
It is unlikely that CRH deficiency rather than glucocorticoid deficiency accounts for the response of CRH KO mice to repeated hypoglycemia. Although findings on the role of CRH in autonomic responses hypoglycemia are contradictory (6, 19), recent studies indicate that CRH may suppress counter-regulatory responses to recurrent hypoglycemia (19). These findings make it doubtful that CRH deficiency counterbalanced or obscured the hypothesized effects of glucocorticoid deficiency in the CRH KO mouse. It is unlikely that glucocorticoid replacement would shed light on this issue, since other counterregulatory responses after repeat hypoglycemia in CRH KO mice were so low and so similar to WT that further changes would have been difficult to detect. We did not find evidence of other processes, such as compensatory food intake, that might have accounted for the suppression of counterregulatory hormones in repeatedly hypoglycemic CRH KO mice. In sum, our data corroborate a significant impact of glucocorticoid deficiency on counterregulatory responses to acute hypoglycemia but do not demonstrate protection against defective counterregulatory hormone secretion after recurrent hypoglycemia.
It remains possible that the low but detectable glucocorticoid levels in CRH KO mice mediated the observed inhibition of counterregulatory hormones after repeated hypoglycemia. This interpretation seems unlikely in view of the number of studies that did not find counterregulatory inhibition even in subjects with normal adrenocortical function given additional glucocorticoids (17, 19, 41, 44). Even if low glucocorticoid levels contributed to counterregulatory hormone suppression after repeated hypoglycemia in CRH KO mice, these low levels also correlated with impaired counterregulation. The contribution of glucocorticoids to counterregulatory failure may be subtle if such a narrow margin exists between glucocorticoid levels that do not permit normal counterregulation during either acute or repeated hypoglycemia.
Acknowledgments
We are grateful to Rebecca Kittell, Charles Harklerode (Albany Medical College), Carlo Malabanan, Wanda Snead, and the Mouse Metabolic Phenotyping Core staff (Vanderbilt University) for technical assistance. We are also grateful to Drs. Louis Muglia (Washington University, St. Louis, MO) and Joseph Majzoub (Children’s Hospital, Boston, MA) for providing mice to maintain a CRH KO mouse breeding colony at Albany Medical College. We are indebted to Dr. David Wasserman (Vanderbilt University) for his continued generous input and helpful suggestions.
Footnotes
GRANTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-62442 (L. Jacobson) and DK-59637 (O. P. McGuinness).
References
- 1.Armstrong L, Bell PM. Addison’s disease presenting as reduced insulin requirement in insulin dependent diabetes. Br Med J. 1996;312:1601–1602. doi: 10.1136/bmj.312.7046.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bollen M, Keppens S, Stalmans W. Specific features of glycogen metabolism in the liver. Biochem J. 1998;336:19–31. doi: 10.1042/bj3360019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bornstein SR, Breidert M, Ehrhart-Bornstein M, Kloos B, Scherbaum WA. Plasma catecholamines in patients with Addison’s disease. Clin Endocrinol. 1995;42:215–218. doi: 10.1111/j.1365-2265.1995.tb01866.x. [DOI] [PubMed] [Google Scholar]
- 4.Bray GA. Reciprocal relation of food intake and sympathetic activity: experimental observations and clinical implications. Int J Obesity Metab Disord. 2000;24(Suppl 2):S8–S17. doi: 10.1038/sj.ijo.0801269. [DOI] [PubMed] [Google Scholar]
- 5.Brissova M, Nicholson WE, Shiota M, Powers AC. Assessment of insulin secretion in the mouse. In: Ozcan S, editor. Methods in Molecular Medicine. Totowa, NJ: Humana; 2003. pp. 23–45. [DOI] [PubMed] [Google Scholar]
- 6.Brown MR, Gray TS, Fisher LA. Corticotropin-releasing factor receptor antagonist: effects on the autonomic nervous system and cardiovascular function. Regul Pept. 1986;16:321–329. doi: 10.1016/0167-0115(86)90032-7. [DOI] [PubMed] [Google Scholar]
- 7.Coker RH, Koyama Y, Denny JC, Camacho RC, Lacy DB, Wasserman DH. Prevention of overt hypoglycemia during exercise: stimulation of endogenous glucose production independent of hepatic catecholamine action and changes in pancreatic hormone concentration. Diabetes. 2002;51:1310–1318. doi: 10.2337/diabetes.51.5.1310. [DOI] [PubMed] [Google Scholar]
- 8.Cryer PE. Diverse causes of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med. 2004;350:2272–2279. doi: 10.1056/NEJMra031354. [DOI] [PubMed] [Google Scholar]
- 9.Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care. 2003;26:1902–1912. doi: 10.2337/diacare.26.6.1902. [DOI] [PubMed] [Google Scholar]
- 10.Dallman MF, Akana SF, Cascio CS, Darlington DN, Jacobson L, Levin N. Regulation of ACTH secretion: variations on a theme of B. Rec Prog Horm Res. 1987;43:113–173. doi: 10.1016/b978-0-12-571143-2.50010-1. [DOI] [PubMed] [Google Scholar]
- 11.Dallman MF, Strack AM, Akana SF, Bradbury MJ, Hanson ES, Scribner KA, Smith M. Feast and famine: critical role of glucocorticoids with insulin in daily energy flow. In: Martini L, Ganong WF, editors. Frontiers in Neuroendocrinology. New York: Raven; 1993. pp. 303–347. [DOI] [PubMed] [Google Scholar]
- 12.Davis MR, Shamoon H. Counterregulatory adaptation to hypoglycemia in normal humans. J Clin Endocrinol Metab. 1991;73:995. doi: 10.1210/jcem-73-5-995. [DOI] [PubMed] [Google Scholar]
- 13.Davis SN, Shavers C, Costa F. Prevention of an increase in plasma cortisol during hypoglycemia preserves subsequent counterregulatory responses. J Clin Invest. 1997;100:429–438. doi: 10.1172/JCI119550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis SN, Shavers C, Costa F, Mosqueda-Garcia R. Role of cortisol in the pathogenesis of deficient counterregulation after antecedent hypoglycemia in normal humans. J Clin Invest. 1996;98:680–691. doi: 10.1172/JCI118839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis SN, Shavers C, Mosqueda-Garcia R, Costa F. Effects of differing antecedent hypoglycemia on subsequent counterregulation in normal humans. Diabetes. 1997;46:1328–1335. doi: 10.2337/diab.46.8.1328. [DOI] [PubMed] [Google Scholar]
- 16.DeFeo P, Perriello G, Torlone E, Ventura MM, Fanelli C, Santeusanio F, Brunetti P, Gerich JE, Bolli GB. Contribution of cortisol to glucose counterregulation in humans. Am J Physiol Endocrinol Metab. 1989;257:E35–E42. doi: 10.1152/ajpendo.1989.257.1.E35. [DOI] [PubMed] [Google Scholar]
- 17.Evans SB, Wilkinson CW, Bentson K, Gronbeck P, Zavosh A, Figlewicz DP. PVN activation is suppressed by repeated hypoglycemia but not antecedent corticosterone in the rat. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1426–R1436. doi: 10.1152/ajpregu.2001.281.5.R1426. [DOI] [PubMed] [Google Scholar]
- 18.Finotto S, Krieglstein M, Schober A, Deimling F, Lindner K, Brühl B, Beier K, Metz J, Garcia-Arraras JE, Roig-López JL, Monaghan P, Schmid W, Cole TJ, Kellendonk C, Tronché F, Schütz G, Unsicker K. Analysis of mice carrying targeted mutations of the glucocorticoid receptor gene argues against an essential role of glucocorticoid signaling for generating adrenal chromaffin cells. Development. 1999;126:2935–2944. doi: 10.1242/dev.126.13.2935. [DOI] [PubMed] [Google Scholar]
- 19.Flanagan DE, Keshavarz T, Evans ML, Flanagan S, Fan X, Jacob RJ, Sherwin RS. Role of corticotropin-releasing hormone in the impairment of counterregulatory responses to hypoglycemia. Diabetes. 2003;52:605–613. doi: 10.2337/diabetes.52.3.605. [DOI] [PubMed] [Google Scholar]
- 20.Fujiwara T, Cherrington AD, Neal DN, McGuinness OP. Role of cortisol in the metabolic response to stress hormone infusion in the conscious dog. Metabolism. 1996;45:571–578. doi: 10.1016/s0026-0495(96)90026-8. [DOI] [PubMed] [Google Scholar]
- 21.Gerich JE. Hypoglycemia. In: De-Groot LJ, Jameson JL, editors. Endocrinology. 5th ed. Philadelphia, PA: Saunders; 2006. pp. 1203–1229. [Google Scholar]
- 22.Goldstein DS, Feuerstein G, Izzo JJL, Kopin IJ, Keiser HR. Validity and reliability of liquid chromatography with electrochemical detection for measuring plasma levels of norepinephrine and epinephrine in man. Life Sci. 1981;28:467–475. doi: 10.1016/0024-3205(81)90139-9. [DOI] [PubMed] [Google Scholar]
- 23.Gustavson SM, Chu CA, Nishizawa M, Farmer B, Neal D, Yang Y, Vaughan S, Donahue EP, Flakoll P, Cherrington AD. Glucagon’s actions are modified by the combination of epinephrine and gluconeogenic precursor infusion. Am J Physiol Endocrinol Metab. 2003;285:E534–E544. doi: 10.1152/ajpendo.00059.2003. [DOI] [PubMed] [Google Scholar]
- 24.Halseth AE, Bracy DP, Wasserman DH. Overexpression of hexokinase II increases insulin- and exercise-stimulated muscle glucose uptake in vivo. Am J Physiol Endocrinol Metab. 1999;276:E70–E77. doi: 10.1152/ajpendo.1999.276.1.E70. [DOI] [PubMed] [Google Scholar]
- 25.Jacobson L. Glucocorticoid replacement, but not CRH deficiency, prevents adrenalectomy-induced anorexia in mice. Endocrinology. 1999;140:310–317. doi: 10.1210/endo.140.1.6416. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson L. Lower weight loss and food intake in protein-deprived, CRH-deficient mice correlates with glucocorticoid insufficiency. Endocrinology. 1999;140:3543–3551. doi: 10.1210/endo.140.8.6910. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson L, Ansari T, McGuinness OP. Counterregulatory deficits occur within 24 h of a single hypoglycemic episode in conscious, unrestrained, chronically-cannulated mice. Am J Physiol Endocrinol Metab. 2006;290:E678–E684. doi: 10.1152/ajpendo.00383.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobson L, Muglia LJ, Weninger SC, Pacák K, Majzoub JA. CRH deficiency impairs but does not block pituitary-adrenal responses to diverse stressors. Neuroendocrinology. 2000;71:79–87. doi: 10.1159/000054524. [DOI] [PubMed] [Google Scholar]
- 29.Jacobson L, Pacak K. Counterregulatory responses after repeated hypoglycemia are not enhanced by combined corticotropin-releasing hormone and glucocorticoid deficiency in mice. Metabolism. 2005;54:1259–1265. doi: 10.1016/j.metabol.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeong KH, Jacobson L, Pacák K, Widmaier EP, Goldstein DS, Majzoub JA. Impaired basal and restraint-induced epinephrine secretion in corticotropin-releasing hormone-deficient mice. Endocrinology. 2000;141:1142–1150. doi: 10.1210/endo.141.3.7370. [DOI] [PubMed] [Google Scholar]
- 31.Kvetnansky R, Fukuhara K, Pacák K, Cizza G, Goldstein DS, Kopin IJ. Endogenous glucocorticoids restrain catecholamine synthesis and release at rest and during immobilization stress in rats. Endocrinology. 1993;133:1411–1419. doi: 10.1210/endo.133.3.8396019. [DOI] [PubMed] [Google Scholar]
- 32.Lingenfelser T, Renn W, Sommerwerck U, Jung MF, Buettner UW, Zaiser-Kaschel H, Kachel R, Eggstein M, Jakober B. Compromised hormonal counterregulation, symptom awareness, and neurophysiological function after recurrent short-term episodes of insulin-induced hypoglycemia in IDDM patients. Diabetes. 1993;42:610–618. doi: 10.2337/diab.42.4.610. [DOI] [PubMed] [Google Scholar]
- 33.Moore MC, Connolly CC, Cherrington AD. Autoregulation of glucose production. Eur J Endocrinol. 1998;138:240–248. doi: 10.1530/eje.0.1380240. [DOI] [PubMed] [Google Scholar]
- 34.Muglia L, Jacobson L, Dikkes P, Majzoub JA. Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature. 1995;373:427–432. doi: 10.1038/373427a0. [DOI] [PubMed] [Google Scholar]
- 35.Muglia LJ, Jacobson L, Luedke CE, Vogt SK, Schaefer ML, Dikkes P, Fukuda S, Sakai Y, Suda T, Majzoub JA. Corticotropin-releasing hormone links pituitary adrenocorticotropin gene expression and release during adrenal insufficiency. J Clin Invest. 2000;105:1269–1277. doi: 10.1172/JCI5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muglia LJ, Jacobson L, Weninger SC, Karalis KP, Jeong K, Majzoub JA. The physiology of corticotropin-releasing hormone deficiency in mice. Peptides. 2001;22:725–731. doi: 10.1016/s0196-9781(01)00385-0. [DOI] [PubMed] [Google Scholar]
- 37.Muglia LJ, Jacobson L, Weninger SC, Luedke CE, Bae DS, Jeong KH, Majzoub JA. Impaired diurnal adrenal rhythmicity restored by constant infusion of corticotropin-releasing hormone in CRH-deficient mice. J Clin Invest. 1997;99:2923–2929. doi: 10.1172/JCI119487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munck A, Fejes-Toth A. Glucocorticoid action: physiology. In: DeGroot LJ, Jameson JL, editors. Endocrinology. Philadelphia, PA: Saunders; 2001. pp. 1632–1646. [Google Scholar]
- 39.Opherk C, Tronche F, Kellendonk CKD, Schultze A, Schmid W, Schütz G. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol. 2004;18:1346–1353. doi: 10.1210/me.2003-0283. [DOI] [PubMed] [Google Scholar]
- 40.Pacak K, Palkovits M, Yadid G, Kvetnansky R, Kopin IJ, Goldstein DS. Heterogeneous neurochemical responses to different stressors: a test of Selye’s doctrine of nonspecificity. Am J Physiol Regul Integr Comp Physiol. 1998;275:R1247–R1255. doi: 10.1152/ajpregu.1998.275.4.R1247. [DOI] [PubMed] [Google Scholar]
- 41.Raju B, McGregor VP, Cryer PE. Cortisol elevations comparable to those that occur during hypoglycemia do not cause hypoglycemia-associated autonomic failure. Diabetes. 2003;52:2083–2089. doi: 10.2337/diabetes.52.8.2083. [DOI] [PubMed] [Google Scholar]
- 42.Sandoval DA, Ping L, Neill AR, Morrey S, Davis SN. Cortisol acts through central mechanisms to blunt counterregulatory responses to hypoglycemia in conscious rats. Diabetes. 2003;52:2198–2204. doi: 10.2337/diabetes.52.9.2198. [DOI] [PubMed] [Google Scholar]
- 43.Selva KA, LaFranchi SH, Boston B. A novel presentation of familial glucocorticoid deficiency (FGD) and current literature review. J Pediatric Endocrinol Metab. 2004;17:85–92. doi: 10.1515/jpem.2004.17.1.85. [DOI] [PubMed] [Google Scholar]
- 44.Shum K, Inouye K, Chan O, Mathoo J, Bilinski D, Matthews SG, Vranic M. Effects of antecedent hypoglycemia, hyperinsulinemia, and excess corticosterone on hypoglycemic counterregulation. Am J Physiol Endocrinol Metab. 2001;281:E455–E465. doi: 10.1152/ajpendo.2001.281.3.E455. [DOI] [PubMed] [Google Scholar]
- 45.Surwit RS, Schneider MS, Feinglos MN. Stress and diabetes mellitus. Diabetes Care. 1992;15:1413–1422. doi: 10.2337/diacare.15.10.1413. [DOI] [PubMed] [Google Scholar]
- 46.Weise M, Mehlinger SL, Drinkard B, Rawson E, Charmandari E, Hiroi M, Eisendorfer G, Yanovski JA, Chrousos GP, Merke DP. Patients with classic congenital adrenal hyperplasia have decreased epinephrine reserve and defective glucose elevation in response to high-intensity exercise. J Clin Endocrinol Metab. 2004;89:591–597. doi: 10.1210/jc.2003-030634. [DOI] [PubMed] [Google Scholar]
- 47.Wurtman RJ, Axelrod J. Adrenaline synthesis: control by the pituitary gland and adrenal glucocorticoids. Science. 1965;150:1464–1465. doi: 10.1126/science.150.3702.1464. [DOI] [PubMed] [Google Scholar]