

Abstract
This study sought to determine the role of the transcription factor E2F1 in CXCR4-mediated neurotoxicity and HIV neuropathology. We studied the effect of the HIV envelope protein gp120 on the expression of E2F1-dependent apoptotic proteins in human and rodent neurons and examined the expression pattern of E2F1 in the brain of HIV-infected individuals. Our findings suggest that in cultured neurons gp120 increased E2F1 levels in the nucleus, stimulated its transcriptional activity, and enhanced the expression of the E2F1 target proteins Cdc2 and Puma. Studies with neuronal cultures from E2F1 deficient mice demonstrated that the transcription factor is required for gp120-induced neurotoxicity and up-regulation of Cdc2 and Puma. Levels of E2F1 protein were greater in the nucleus of neurons in brains of HIV-infected patients exhibiting dementia when compared to HIV-negative subjects or HIV-positive neurologically normal patients. Overall, these studies indicate that E2F1 is primarily involved in CXCR4-mediated neurotoxicity and HIV neuropathogenesis.
Keywords: neuronal death, Cdc2, cell cycle proteins, chemokines, gp120
Abbreviations: AIDS: Acquired immunodeficiency syndrome, CNS: central nervous system, HAART: highly active antiretroviral therapy, HAD: HIV associated dementia, HIV: human immunodeficiency virus, HIVE: HIV encephalopathy, Rb: retinoblastoma, SIVE: Simian immunodeficiency virus encephalitis
Introduction
The chemokine receptor CXCR4 is virtually ubiquitous and plays important roles in many physiological processes, such as neuronal migration, survival and differentiation; this receptor has also been implicated in the pathogenesis of cancer and HIV – including NeuroAIDS (Tran and Miller, 2003). Analogous to other inflammatory disorders, neuronal injury and death are often found in AIDS patients (Adle-Biassette et al., 1995; Petito and Roberts, 1995; Shi et al., 1996). Cellular alterations not necessarily caused by cell loss, such as dendritic pruning and synaptic impairment, are also thought to contribute to NeuroAIDS (Bellizzi et al., 2005; Everall et al., 1999; Sa et al., 2004). Although highly active antiretroviral therapy (HAART) has greatly reduced HIV mortality and morbidity, the neurological complications of HIV infection significantly affect the clinical management of treated patients, and HIV-dementia still represents a serious concern for patients that have limited access to therapy (reviewed in (Gonzalez-Scarano and Martin-Garcia, 2005; McArthur et al., 2003)). As neurons are not the primary targets of HIV in the brain – the virus mostly infects macrophages and microglia and, to a lesser extent, astroglia - the neuropathogenesis of AIDS is not completely understood. A number of viral and cellular factors produced by infected or activated cells over a long period of time appear to be responsible for the neuronal damage caused by HIV (recent reviews: (Gartner and Liu, 2002; Gonzalez-Scarano and Martin-Garcia, 2005; Jones and Power, 2006; Mattson et al., 2005)). One of these factors is the HIV envelope protein gp120 that binds to chemokine receptors (i.e. CCR5 and CXCR4) on the surface of target cells, including neurons (Gartner and Liu, 2002; Gonzalez-Scarano and Martin-Garcia, 2005; Hesselgesser et al., 1998; Jones and Power, 2006; Mattson et al., 2005)). Several in vitro and in vivo studies have shown the neurotoxic potential of the envelope protein and the involvement of chemokine receptors in this process (Gartner and Liu, 2002; Gonzalez-Scarano and Martin-Garcia, 2005; Jones and Power, 2006; Mattson et al., 2005)). The interaction of the co-receptors with the viral protein only partially resembles the interaction with their natural ligands (Farzan et al., 2002), suggesting that activation of chemokine receptors may lead to cell death. Indeed, both apoptotic and pro-survival actions of chemokines have been reported (Cartier et al., 2005; Hesselgesser et al., 1998; Kaul and Lipton, 1999; Meucci et al., 1998). Thus, chemokine receptors may mediate both survival and apoptotic pathways in neurons.
Brain-derived viruses generally use CCR5 for entry (Gonzalez-Scarano and Martin-Garcia, 2005), but viruses exploiting CXCR4 as co-receptor (or dual tropic viruses) are also found in the CNS and able to infect macrophages (Yi et al., 2003). These viruses are highly neurotoxic in vitro (Gabuzda and Wang, 2000) and generally predominate in the late stages of the disease. The increased permeability of the BBB at these stages of disease contributes to exposure of the brain to peripheral viral envelopes or to other viral proteins and immune/inflammatory mediators, which can interact with neuronal and glial CXCR4 and alter their function. Given the crucial role of CXCR4 in brain physiology (Tran and Miller, 2003), this aspect deserves consideration in spite of the predominant role of CCR5-using viruses in brain infection.
Our previous studies suggest that activation of CXCR4 by its endogenous ligand, the chemokine CXCL12/SDF, promotes expression and activation of the retinoblastoma gene product (Rb) in cultured neurons (Khan et al., 2003). Rb is a major regulator of neuronal survival and its action is primarily due to its ability to repress the transcription factor E2F1, thereby inhibiting the expression of pro-apoptotic genes (DeGregori et al., 1997). Thus, deregulation of the CDK/Rb/E2F1 pathway may be involved in the negative effects of CXCR4 on neuronal survival. Interestingly, aberrant expression of these cell cycle proteins in post-mitotic neurons has been associated with various neuropathological conditions, including HIV/SIV encephalitis (Jordan-Sciutto et al., 2002; Roberts et al., 2003). The aim of this study was to determine whether E2F1 is involved in the neuronal damage caused by HIVgp120. Our in vitro and ex vivo findings suggest that E2F1 is required for the CXCR4-mediated neurotoxicity caused by the HIV envelope protein, and that E2F1 may play an active role in the transcription of neuronal apoptotic genes in HIV-infected brains. As emerging evidence implies that CXCR4/CXCL12 function may be regulated by different cellular and environmental factors (such as crosstalk with neuropeptide pathways and activation of metalloproteinases), these findings also support the hypothesis that alteration of neuronal CXCR4 may be involved in additional neuroinflammatory/neuroimmune disorders.
Material and Methods
Animals
All experiments involving animals were performed according to protocols approved by the Institutional Animal Care and Use Committee of Drexel University College of Medicine. E2F1 deficient mice (strain number 002785, E2F1−/−, Jackson Laboratory, Bar Harbor, ME), obtained by inserting a cassette that disrupted the DNA binding and dimerization domain of the E2F1 gene (Field et al., 1996), and their control mice B6129SF2/J (strain number 101045, E2F1+/+, Jackson Laboratory, Bar Harbor, ME) were bred separately. Genotyping was performed to confirm the absence of E2F1 in the knockout mice by PCR analysis of DNA extracted from mice cerebella as previously described (Field et al., 1996) with minor modifications. Briefly, at the end of each neuronal and glial preparation, each mouse cerebellum was incubated overnight with 300 μg of Proteinase K (Roche, Indianapolis, IN) in Blobel’s solution (50mM Tris, 100mM NaCl, 5mM EDTA, 1% SDS), and DNA was extracted using Phenol/Chloroform/Isoamyl alchohol (Fisher Scientific, Pittsburgh, PA), followed by ethanol precipitation. Three primers (5′-GGATATGATTCTTGGACTTCTTGG-3′, 5′-CTAAATCTGACCACCAAACGC-3′, 5′-CAAGTGCCAGCGGGGCTGCTA AAG-3′) were used to amplify the 172-bp DNA fragment for the wild type (WT) and 227-bp DNA fragment for knockout (KO) mice. PCR samples were run on 3% agarose gel.
Neuronal cultures
Cortical neurons were obtained from the brains of either mouse or rat embryos and cultured in serum-free medium using the bilaminar co-culture system, as previously described (Khan et al., 2003; Meucci et al., 1998). In this model, pure neuronal cultures are grown in the presence of a separate glial feeder-layer (from the cortex of the same species), which supports their growth and differentiation. Thus, neurons can be separated from glia at any time. Briefly, cortical neurons were plated on poly-L-lysine-coated 15 mm coverslips (35,000 cells) or 60 mm dishes (1 X106 cells). Four hours after plating, the coverslips containing neurons were transferred to a culture dish containing the glial monolayer and co-cultured for at least seven days. Treatments (with or without glia as indicated) usually started at the seventh day in vitro (DIV). When indicated the CXCR4 antagonist, AMD3100 (Sigma-Aldrich, St. Louis, MO), was added to the medium 15 minutes before the gp120.
Human neuroblastoma SH-SY5Y cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in Minimum Essential Medium/F12 medium with 10% Fetal Bovine Serum. Before treatment with gp120IIIB, SH-SY5Y cells were differentiated with retinoic acid (10 μM) in order to induce their neuronal phenotype – as previously described (Pahlman et al., 1984). Both differentiated and undifferentiated cells express CXCR4 and are sensitive to the toxic action of gp120IIIB (Hawkins et al., 1999).
Human brain tissue studies
A total of 23 brain autopsy samples from three groups of patients (Table 1) were obtained from four tissue banks of the National NeuroAIDS Tissue Consortium (NNTC) (Morgello et al., 2001). Specimens included samples from the cerebral cortex and/or hippocampus of: HIV patients with or without neurological impairment (i.e. here defined as HIV/HAD and HIV, respectively) and control patients (HIV-negative). Available information about age, sex, post-mortem interval, viral load in plasma and cerebrospinal fluid (CSF) and CD4+ cell number were provided by NNTC. HIV/HAD patients were diagnosed by HIV RNA levels, CD4 cell counts, and neurological examination - and staged based on the Memorial Sloan-Kettering (MSK) score (Marder et al., 2003). Tissue was fixed in 10% formalin, embedded in paraffin and cut at 6–10 μm for immunohistochemistry. After de-paraffinization and quenching of endogenous peroxidase, sections were incubated at 95°C in Antigen Retrival Solution (Dako, Carpinteria, CA) for 1 h. Sections were then incubated in Tris buffered saline (TBS) with 10% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) for an additional hour, and then overnight with an anti-E2F1 monoclonal antibody (1:100, KH-95, Santa Cruz, CA). This was followed by incubation with a biotin-conjugated donkey anti-mouse antibody (1:250, Jackson Immuno Research, West Grove, PA), and Tyramide Signal Amplification (TSA) from Perkin Elmer (Wellesley, MA) according to the manufacturer’s instructions. Staining was visualized with Vector NovaRed (red-brown; Vector laboratories, Burlingame, CA). When indicated, staining for the cytoplasmic/dendritic neuronal protein, microtubule-associated protein-2 (MAP2a+b), was performed immediately after the E2F1 staining was completed, using the anti-MAP2 monoclonal antibody (1:50, Chemicon, Temecula, CA), followed by incubation with biotinylated donkey anti-mouse secondary antibody (1:250, Jackson ImmunoResearch, West Grove, PA), amplified with Vectastain elite ABC kit (Vector laboratories, Burlingame, CA), and visualized using 3, 3′-diaminobenzidine with Nickel enhancement (blue-purple; Vector Laboratories, Burlingame, CA). Chromogen-based immunostaining was chosen for these experiments to avoid auto-fluorescence of the human tissue. Negative controls (i.e. no primary antibody) were included for each staining. Sections were observed under a Nikon microscope (OPTIPHOT-2) connected to a CCD camera (DP70, Olympus, Melville, NY), and images were taken using the DP software (Olympus, Melville, NY). To determine E2F1 positive nuclei in cortical neurons (identified by MAP2), 10 fields per brain sample were randomly selected and counted using a 20 X objective. The total number of neurons analyzed was about 180/brain. The anti-E2F1 antibodies used in these studies (KH-95) recognize an epitope in the Rb binding domain of E2F1 not accessible when the transcription factor is bound to (and thus inhibited by) Rb (Krek et al., 1993). Therefore, the fraction of E2F1 protein potentially available for transcription (herein defined as “free”) is detected. For cdc2 staining, a monoclonal antibody from Santa Cruz (1:100, sc-54, Santa Cruz, CA) was used and human breast cancer tissue was used as positive control.
Table 1.
Summary of human brain tissue obtained from NNTC.
| Bank | Group | HIV | Age/Sex | Race | PMI (h) | Area | CD4 (cells/μl) | CSF VL (copies/ml) | Plasma VL (copies/ml) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | HIV/HAD | + | 43/M | W | <24 | C | 44 | 3,212 | 65 |
| 2 | HIV/HAD | + | 38/M | W | 5.5 | C | 43 | 1,237,903 | -- | |
| 3 | HIV/HAD | + | 32/M | W | 14 | C | 77 | 7,398 | 702,363 | |
| 4 | HIV | + | 34/F | W | 5 | C | 14 | 76 | 15,906 | |
| 5 | HIV | + | 50/M | W | 12.75 | C | 3 | 1,101 | 230,238 | |
| 6 | Control | − | 58/F | H | 7.42 | C | -- | N/A | N/A | |
| 7 | Control | − | 51/F | H | 21.75 | C | -- | N/A | N/A | |
| 8 | Control | − | 66/F | W | 22.8 | C | -- | N/A | N/A | |
| 2 | 9 | HIV/HAD | + | 30/M | B | 6 | C+H | 8 | 14 | 104,300 |
| 10 | HIV/HAD | + | 43/M | H | 31 | C+H | 3 | 65 | 110,493 | |
| 11 | HIV/HAD | + | 43/M | B | 6 | C+H | 10 | 134 | 48,520 | |
| 12 | Control | − | 52/M | W | 17.5 | C+H | N/A | N/A | N/A | |
| 13 | Control | − | 59/F | B | 10 | C+H | N/A | N/A | N/A | |
| 14 | Control | − | 59/M | W | 18 | C+H | N/A | N/A | N/A | |
| 3 | 15 | HIV | + | 39/M | W | 11 | C+H | 13 | N/A | 75,000 |
| 16 | HIV/HAD | + | 58/F | W | 5 | C+H | 29 | N/A | 133,230 | |
| 17 | HIV/HAD | + | 35/M | W | 5.5 | C+H | 26 | N/A | 180,000 | |
| 18 | HIV/HAD | + | 36/M | W | 6 | C+H | 63 | -- | 6,952 | |
| 4 | 19 | HIV/HAD | + | 44/M | W | 12 | C+H | 39 | 1,932 | 198,957 |
| 20 | HIV | + | 50/F | Am In | 10 | C+H | 409 | 11 | 33 | |
| 21 | HIV/HAD | + | 49/M | W | 3 | C+H | 4 | 106 | 109,336 | |
| 22 | HIV/HAD | + | 40/M | W | 10 | C+H | 14 | -- | -- | |
| 23 | Control | − | 75/M | W | 19 | C+H | N/A | N/A | N/A |
HIV/HAD: HIV positive with neurological deficits, HIV: HIV positive without neurological deficits, W: white, H: Hispanic, B: Black, Am In: American Indian, C: cortex, H: hippocampus, PMI: post-mortem interval, VL: viral load, N/A: not applicable, --: not available
Survival assays and fluorescence microscopy
Rat and mouse cortical neurons were treated at 7 DIV with gp120IIIB (200pM); or vehicle (0.1% Bovine Serum Albumin/Phosphate Buffered Saline) for the indicated time (3–24 h). Cells were fixed in 4% paraformaldehyde, and permeabilized with 0.1% Triton-X for 5 min. Cells were incubated with anti-cleaved caspase-3 (polyclonal, 1: 50, #9661, Cell Signaling, Danvers, MA) and anti-E2F1 (1:50, KH-20, Sigma-Aldrich, St. Louis, MO) antibodies overnight, followed by incubation with the appropriate secondary antibody conjugated with Cy-3 (anti-rabbit, 1:250, Jackson ImmunoResearch, West Grove, PA) or Cy-2 (anti-mouse, 1:250, Jackson ImmunoResearch, West Grove, PA), respectively. Hoechst 33342 was used for nuclear staining (0.6 μl/ml). For survival assays, coverslips were observed under an epifluorescent microscope (Olympus IX70, Melville, NY) connected to a CCD camera (Micromax, Trenton, NJ), and images were taken using Metamorph software (Molecular Devices, Sunnyvale, CA). Ten different random fields per coverslip were analyzed (at least 3 coverslips per group); the percentage of apoptotic cells, defined by both cleaved caspase-3 positive staining and condensed/fragmented nucleus, was calculated in a blind manner. Each experiment was repeated at least two times. Furthermore, the intensity of E2F1 staining was calculated in two random fields per treatment by measuring the fluorescence level in the nuclear region of each neuron (defined by Hoechst); these data are shown in figure 2D as average gray levels for each neuron.
Figure 2. Effect of HIV gp120 on E2F1 expression in rat primary neurons.
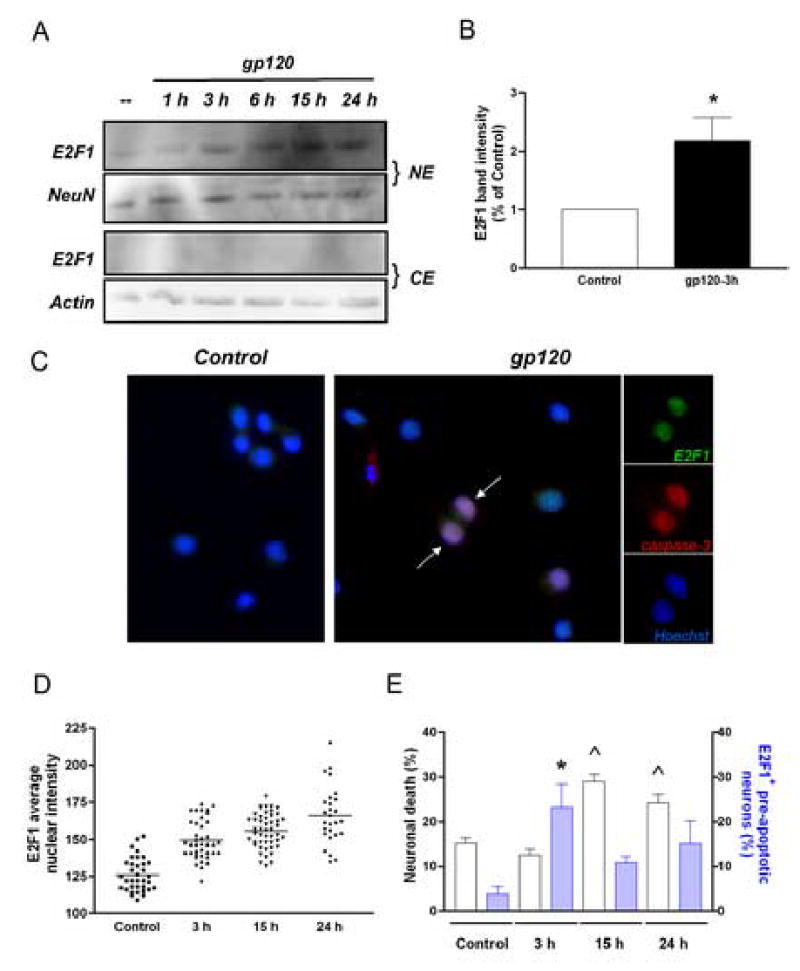
Western blot analysis (A-B) and immunocytochemistry studies (C) indicate that E2F1 is up-regulated in the nucleus of rat primary cortical neurons after treatment with gp120IIIB (200 pM) for the indicated time. The viral protein was added to the co-cultures at 7DIV. Antibodies against the neuronal-specific nuclear protein NeuN or β-actin, were used as markers in the immunoblots with nuclear (NE) and cytosolic (CE) extracts, respectively; Hoechst 33442 was used for nuclear staining in the immunocytochemistry studies. A significant increase in E2F1 protein levels was observed after a few hours of treatment with gp120IIIB (generally 3 h) as shown by the graph in B, which reports the average of three independent experiments using total cell extracts from untreated (control) and gp120-treated neurons (mean ± SEM; *p<0.05). Each scatter plot in graph D shows the average intensity of E2F1 nuclear staining in individual neurons at each time points, as determined by immunostaining (bars at each time represent mean values). E2F1 positive pre-apoptotic neurons, i.e. cleaved caspase-3 positive neurons showing healthy morphology, were also observed after short term treatments with gp120 (arrows in C; blue bars in graph E, ^p<0.001 vs control). Apoptotic neurons, i.e. cleaved caspase-3 positive neurons with condensed/fragmented nuclei, were detected at later time (white bars in graph E; *p<0.001 vs control).
Immunoblotting experiments
Cells were washed with ice cold balanced salt solution and scraped in lysis buffer (25 mM Tris, 150 mM NaCl, 5 mM NaF, 1 mM EDTA, 1 mM DTT, 1% IGEPAL-CA-630, 5 μg each of aprotinin, leupeptin, and pepstatin, 1 mM AEBSF, and 1 mM vanadate) as reported previously (Khan et al., 2003). The protein concentration of cell lysates was determined using the bicinchonic acid assay from Pierce (Rockford, IL). Equal amounts of proteins were loaded in each lane, separated by SDS-PAGE and transferred to PVDF membranes for immunoblotting. The following primary antibodies were used: anti-E2F1 (1:1000, KH-20, Sigma-Aldrich, St. Louis, MO), anti-Cdc2 (1:500, sc-54, Santa Cruz, CA), anti-Puma (1:1000, #4976, Cell Signaling, Danvers, MA), anti-BAD (1:1000, #9292 Cell Signaling, Danvers, MA) and anti-BAD phospho S-128 (1:1000, ab5687, Abcam, Cambridge, MA), anti-CXCR4 (1:1000, H-118, Santa Cruz Biotechnology, Santa Cruz, CA), anti-NeuN (1:1000, MAB377, Chemicon, Temecula, CA), and anti-Actin (1:5000, A2066, Sigma-Aldrich, St. Louis, MO). The last two were used as loading controls for nuclear and cytoplasmic proteins, respectively. Bands were detected by chemiluminescence using Pierce reagents (SuperSignal West Femto maximum sensitivity), according to the manufacture’s instructions, and analyzed using the FluorChem 8900 apparatus from Alpha Innotech (San Leandro, CA).
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed as previously described (Khan et al., 2003). Nuclear extracts (2 μg) were incubated with 3′ biotinylated-oligonucleotides containing a consensus binding site for E2F1 (5′-ATTTAAGTTTCGCGCCCTTTCTCAA-3′) according to the manufacturer’s protocol (LightShift Chemiluminescent EMSA kit, Pierce). After incubation, samples were loaded on a 6% polyacrylamide non-denaturing gel in 1% Tris-acetate-EDTA buffer. After transfer to the membrane, DNA was cross-linked to the membrane by UV light, and DNA-protein complexes were detected by chemiluminescence.
Transfection and luciferase assays
Rat cortical neurons (7 DIV) were transfected with the pGL2AN plasmid (gift from Dr. W Kaelin, Dana-Farber Cancer Institute) containing the luciferase cDNA sequence downstream of an E2F dependent promoter (Neuman et al., 1994), along with the pEGFP, using Lipofectamine 2000 from Invitrogen (2 μg per plasmid in each dish containing 1 million cells). An E2F1 expression plasmid (pMax–E2F1) was transfected with the other plasmids in the positive controls only (2 μg/dish). The Bright-Glo™ (Promega, Madison, WI) reagents were used to detect luciferase activity according to the manufacturer’s protocol, using a spectrophotofluorometer (Wallac 1420 multilabel counter, Perkin Elmer, Wellesley, MA). Expression of pEGFP was measured to determine transfection efficiency and all luciferase measurements were normalized accordingly. Neurons were treated with gp120 (200 pM) 2–3 hours after the transfection.
Statistical Analysis
One-way analysis of variance (ANOVA), followed by Neuman-Keuls multiple comparison procedure, was used for analysis of survival assays, and immunohistochemistry studies. Western blots and Luciferase assays were analyzed using the student t-test. All data are reported as mean ± SEM.
Results
Up-regulation of E2F1 in the nucleus of cortical neurons of HIV/HAD patients
As previously reported (Jordan-Sciutto et al., 2002), widespread up-regulation of E2F1 was observed in both gray and white matter of HIV-infected patients that exhibited neurologic impairment (MSK 1–3; not shown). However, in the present study neuronal E2F1 was mainly expressed in the nucleus (Figure 1), consistent with its traditional role in transcription. In contrast, neurons in the cortex of HIV patients that were neurologically normal (Figure 1A), and of HIV-negative patients (not shown), did not appear to contain detectable levels of E2F1 protein. In order to quantify differences in the expression of E2F1 within the three groups of patients, we calculated the percentage of E2F1 positive neurons in specimens provided by a single tissue bank (bank 1, see Table 1), thus keeping potential variability due to the handling of tissue from different sources to a minimum. These included samples from the frontal cortex of HIV/HAD patients (n=3), of HIV patients without neurological problems (n=2), and of HIV negative control patients (n=3). The postmortem interval for these patients (4 males and 4 females) was less than 24 h and their age ranged from 32 to 66 years. Viral loads in the CSF and blood varied (Table 1). Neurons were identified by double-staining with the cytoplasmic/dendritic neuronal marker MAP2. (Figure 1D). A marked increase in the number of E2F1 positive neurons was observed in HIV/HAD brains, as compared to control or HIV-positive individuals without neurological deficits – suggesting that E2F1 is up-regulated in the neurons of HIV/HAD patients (Figure 1B–E). No significant differences were found between control brains and brains from HIV-positive subjects that were neurologically normal (Figure 1E).
Figure 1. Expression of E2F1 in the nucleus of cortical neuron in HIV/HAD patients.
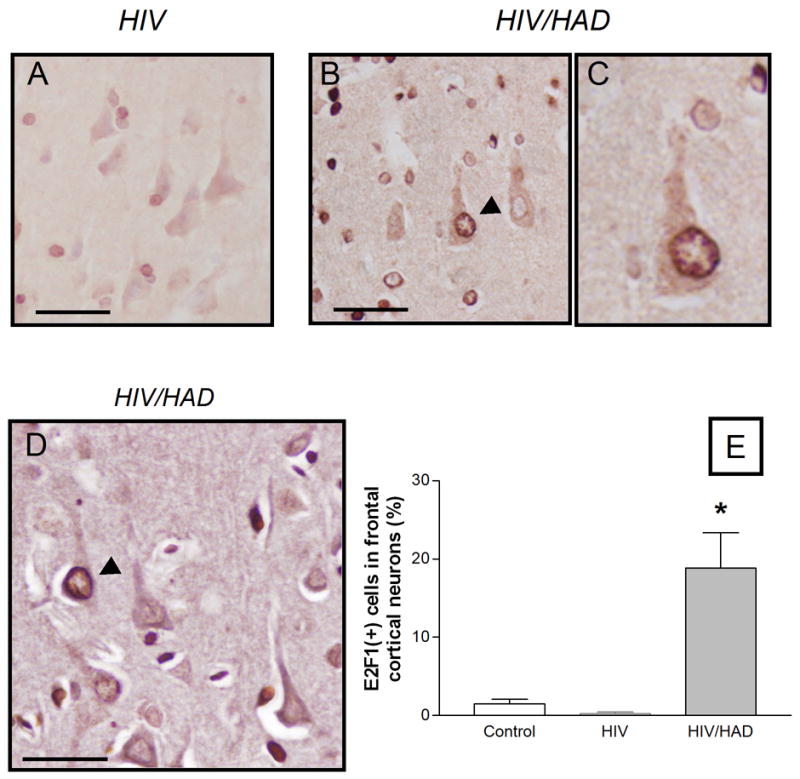
Frontal cortices from HIV-positive patients (with or w/o neurological deficits) and HIV-negative control subjects were used to examine E2F1 expression by immunohistochemistry (as described in detail in the method section). Low levels of E2F1 were detected in the brains of neurologically normal HIV patients (A) or control subjects (graph), while E2F1 was up-regulated in the nucleus of cortical neurons in HIV/HAD brains (arrow head in B; 40X in C). Double-staining with the neuronal marker MAP2 was performed to identify E2F1-positive neurons (D) and quantify differences among the three groups of patients (E, ten fields/brain); *p<0.05 vs HIV; Scale bars 5.0 um.
HIVIIIB-gp120 increases expression of E2F1 in primary cortical neurons
Previous data suggested that activation of neuronal CXCR4 by its natural ligand, the chemokine CXCL12, promoted Rb function (Khan et al., 2003), and that the neuroprotective effect of the chemokine is related to the inhibition of E2F1-mediated apoptosis. Unlike CXCL12, gp120IIIB (an X4-using viral protein) increased Rb phosphorylation in neurons (Khan et al., 2003), resulting in a release of the inhibitory effect of Rb on E2F1 and favoring activation of E2F1. In order to determine whether gp120IIIB also affects E2F1 expression/localization in cultured neurons, protein levels of this transcription factor were measured in nuclear and cytosolic extracts of neurons treated with gp120IIIB. Immunoblot analyses of nuclear and cytolosic fractions revealed that E2F1 was predominantly localized to the nuclear fraction of post-mitotic neurons (Figure 2). No significant changes in the sub-cellular distribution of E2F1 were observed after gp120IIIB treatment, whereas the nuclear levels of the protein were increased by the viral protein (Figure 2A/B). Similar results were obtained from immunocytochemistry-based analysis of E2F1 expression (Figure 2C). These experiments show low levels of E2F1 in control neurons and more intense staining following gp120IIIB treatment (Figure 2C). As indicated in Figure 2D, the average gray level of the E2F1 nuclear staining in individual neurons gradually increased after gp120 treatment. Consistent with previous studies (Khan et al., 2005), gp120 IIIB was found to induce neuronal cell death in these neurons (Figure 2E). These experiments also showed that in gp120-treated neurons E2F1 up-regulation could be detected before the nuclear alterations typical of apoptosis occurred (as assessed by staining with the antibody against active caspase-3 in neurons displaying morphologically normal nuclei) (Figure 2C, arrow). Thus, the increase in E2F1 caused by the viral protein appears to be a relatively early event in the apoptotic process. An increase in the E2F1 protein is indeed already evident after a few hours of gp120 treatment (Figure 2A/B) - when neurons still appear viable. The early increase in E2F1 may indicate a stimulation of E2F1-dependent transcription of E2F1 by the HIV protein (Neuman et al., 1994). Finally, the gp120-induced increase of E2F1 seems to be a direct effect of gp120 on neurons, as similar results were obtained when treatments were performed in the absence of glia (not shown).
HIVIIIB-gp120 stimulates transcription of E2F1-dependent apoptotic genes in neurons
To determine whether gp120IIIB stimulates the transcriptional activity of E2F1, cortical neurons were transfected with a plasmid carrying the sequence of the luciferase gene under the control of an E2F1-dependent promoter (Neuman et al., 1994). A plasmid expressing EGFP was co-transfected to monitor transfection efficiency and for subsequent normalization of luciferase assays, whereas an E2F1 expression vector was used for the positive controls. These experiments showed that treatment with gp120IIIB significantly increased transcriptional activity in transfected neurons (Figure 3A). We additionally tested the effect of gp120IIIB on two endogenous E2F1 transcriptional targets, namely Cdc2 (also known as Cdk1) and Puma, which are known to induce apoptosis in neurons (Hershko and Ginsberg, 2004; Konishi and Bonni, 2003). The neuronal levels of these proteins were enhanced by treatment with gp120IIIB (Figure 3B). This effect was blocked by the CXCR4 antagonist AMD3100 (Figure 3C), indicating that CXCR4 mediates the action of gp120IIIB. Under the same experimental conditions, gp120 also induced a robust increase in the phosphorylation of Bad protein on serine 128 (Figure 3B), which is direct target of Cdc2 (Konishi et al., 2002). Moreover, up-regulation of Cdc2 and Puma (Figure 3D), and Bad phosphorylation (not shown), were also reported in neurons transfected with a plasmid over expressing E2F1 - a condition that mimics gp120IIIB toxicity (DeGregori et al., 1997), and in the absence of glia (not shown). Finally, the ability of gp120 to modulate E2F1 and its targets was also observed in the human SH-SY5Y neuronal cell line (Figure 4A). Electromobility shift assays (EMSA) with SH-SY5Y nuclear extracts demonstrated that gp120IIIB increased the binding of nuclear proteins to E2F-specific oligonucleotide sequences (Figure 4B). Immunoblot analysis showed increased levels of E2F1, Cdc2, and Puma in these cells (Figure 4A). Collectively, these observations demonstrate that the HIV protein stimulated the transcriptional activity of E2F1 in human and rodent neurons, and was able to induce the expression of E2F1-dependent apoptotic genes. This is supported by the observation of expression of Cdc2 in one of the HIV/HAD tissues specimens, (Figure 4D/E).
Figure 3. HIV gp120 up-regulates E2F1 pro-apoptotic targets in rat neurons.

Treatment with gp120IIIB (200 pM) increased transcriptional activity in rat primary neurons as determined by gene reporter assays (A, mean ± SEM of one representative experiment; *p<0.01 vs control). E2F1 over-expression was used as positive control for the assay. Elevated levels of transcriptional targets of E2F1 (Puma and Cdc2) were also found in neuronal extracts from primary rat cultures treated with gp120 (B–C) as well as in neurons transfected with the E2F1 expression plasmid (D). The effect of gp120IIIB was blocked by pre-treatment of cultures with AMD3100 (100 ng/ml) 15 min before the addition of the gp120.
Figure 4. Effect of HIV gp120 on E2F1-dependent proteins in human neurons.

E2F1 pro-apoptotic targets were increased after gp120IIIB (200 pM) treatment in differentiated SH-SY5Y neuroblastoma cells (A). Nuclear extracts from differentiated SH-SY5Y cells treated with the gp120 (200 pM) were used to assess binding activity to E2F1 consensus sequences (B; arrow represents DNA-protein complex). Panels C and D show over-expression of E2F1-dependent proteins, namely Cdc2, in the cytoplasm of frontal cortical neurons in a HIV/HAD patient; higher magnification of one of these neurons is shown in E (Scale bar: 5.0 μm).
E2F1 is necessary for the neuronal cell death induced by HIVIIIB-gp120
To confirm the role for E2F1 in gp120IIIB neurotoxicity, we tested the effect of this viral protein in cortical cultures from E2F1-deficient mice (Figure 5). Absence of E2F1 was verified for each neuronal preparation by PCR (Figure 5A). Neuronal cultures from both wild type and knockout animals were treated with 200 pM gp120IIIB for 24 h. The extent of neuronal death caused by gp120IIIB in the wild type mouse cultures was comparable to the neurotoxicity reported in the rat cultures (Figure 5B), though higher levels of basal cell death were generally found in the mouse cultures. However, the effect of the viral protein was abrogated in cultures from E2F1-deficient animals (Figure 5B), even when higher concentration of gp120IIIB (400 pM) were used (data not shown). Caspase-3 activation plus alteration of nuclear morphology were used to evaluate neuronal cell death (Figure 5B/C). As observed in rat neurons, gp120IIIB increased the total levels of the E2F1 protein in mouse cultures within 3 hours of treatment, as determined by Western blot (not shown). Primary neurons from E2F1 knockout mice were resistant to gp120-induced apoptosis and levels of Cdc2 and Puma were not altered by the viral protein in these cultures (Figure 5D). The protein levels of CXCR4 were comparable in extracts from wild type and knockout cultures (Figure 5D). These data suggest that up-regulation of Cdc2 and Puma following gp120IIIB treatment requires E2F1 expression.
Figure 5 . E2F1 is necessary for HIVgp120-induced cell death.
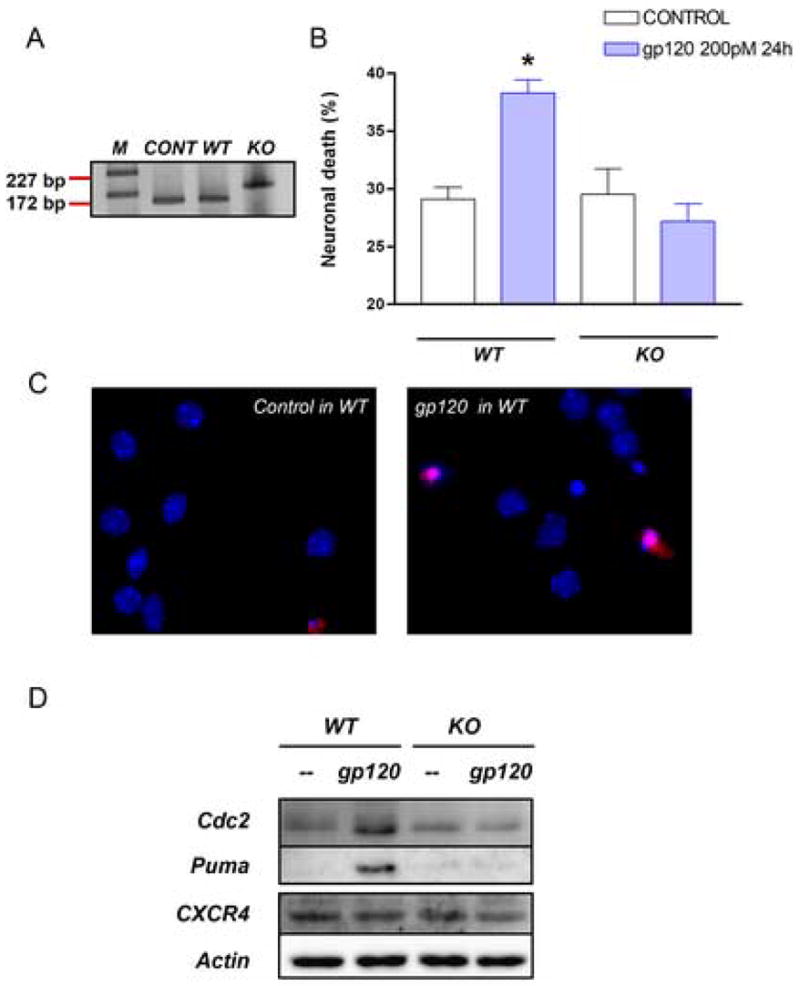
Neuronal cultures from E2F1 KO and wild type mice were used to determine the role of E2F1 in gp120-induced apoptosis. DNA was extracted from the cerebellum of each animal for PCR analysis (A, bands 172 bp and 227 bp correspond to wild type and KO respectively, as expected). Cloned E2F1 was used as positive control. Survival assays performed as reported in the method section, show that KO cultures are resistant to gp120IIIB (B/C, *p<0.0001 vs control in wild type and vs gp120 treatment in KO mice). E2F1 targets, Cdc2 and Puma were up-regulated after gp120 treatment in wild type but not in KO mice - as determined by Western blot analysis with total neuronal cell extracts. No difference in CXCR4 protein levels were observed between WT and KO mice (D).
Discussion
The studies described here demonstrate that activation of CXCR4 by the HIV envelope protein gp120 stimulated the activity of the transcription factor E2F1 in neurons and suggest that the transcription of E2F1-dependent genes may be involved in HIV neuropathology. The experiments on cultured neurons indicate that HIVgp120 up-regulated E2F1 and induced the expression of specific target proteins regulated by this transcription factor. These effects are inhibited by a specific CXCR4 antagonist and do not require the presence of glia, suggesting that the envelope protein acts via neuronal CXCR4. The results with the E2F1 KO cultures revealed that E2F1 is necessary to gp120-induced neurotoxicity and up-regulation of Cdc2 and Puma. Transcriptional up-regulation of apoptotic genes, such as Puma, is also known to be triggered by p53, another major transcription factor involved in HIV apoptosis that is also regulated by CXCR4 (Castedo et al., 2002; Garden et al., 2004; Khan et al., 2005). However, these results suggest that p53 is not sufficient to activate gp120-induced neurotoxic pathways in the absence of E2F1 and may act in concert with (or downstream of) E2F1. The involvement of p53 in HIV apoptosis has been established in both neuronal and non neuronal models (Castedo et al., 2002; Garden et al., 2004) and the two transcription factors can interact in different ways. For instance, E2F1 can induce p53 accumulation via inactivation of the Mdm2 protein, which targets p53 for proteasome degradation (Tao and Levine, 1999). Phosphorylation of specific p53 serine residues by E2F1 is another key step in E2F1-mediated apoptosis (Rogoff et al., 2002). Interestingly, stimulation of CXCR4 by gp120 increases p53 protein levels and phosphorylation in cortical neurons (Khan et al., 2005).
The fact that high levels of free E2F1 (i.e. unbound to Rb) were found in neurons that still appear morphologically normal, but were already primed to death, suggests that E2F1 is recruited in the early stages of gp120-induced neurotoxicity. These data are in agreement with previous observations that gp120 stimulates Rb phosphorylation and promotes the DNA binding activity of E2F1 in neurons, as well as with reports showing increased Rb phosphorylation in the nucleus of neurons of SIVE/HIVE subjects (Jordan-Sciutto et al., 2002; Khan et al., 2003).
In contrast to earlier observations of increased E2F1 expression in the cytosol of neurons in HIVE patients (Jordan-Sciutto et al., 2002), our studies indicate that E2F1 mostly localizes to the nuclei of neurons in both HIV/HAD patients and in neuronal cultures treated with HIVgp120. Though the same anti-E2F1 antibodies were employed in the two studies, the different detection methods and/or tissue preparation might account for this discrepancy. Furthermore, nuclear localization of E2F1 might be restricted to more severe cases of encephalitis, such as those examined in this study. Nonetheless, our in vitro studies (using additional antibodies and alternative approaches) support the link between increased E2F1 transcription in neurons and HIV neuropathogenesis. Stimulation of CXCR4 by the HIV envelope induces up-regulation of E2F1 and E2F1-dependent apoptotic proteins in neurons, including Cdc2/Cdk1 (Castedo et al., 2002; Konishi and Bonni, 2003). This protein is of particular interest as it was found to be specifically associated with E2F1-driven apoptotic pathways in neurons, but not involved in the activation of E2F1-target genes involved in DNA synthesis and replication (Konishi et al., 2002). Increased levels of Cdc2 in neurons were also found in the brain of one HIV/HAD patient. This protein kinase catalyzes the phosphorylation of the BH3-only protein BAD at a distinct site (serine 128), thus inducing BAD-mediated apoptosis in primary neurons and opposing the anti-apoptotic effects of growth factors (Konishi et al., 2002). These data point to the role of E2F1-dependent apoptotic pathways in NeuroAIDS and raise the issue of whether this phenomenon may be restricted to CXCR4 activation. Considering the different impact of the two co-receptors on neuronal physiology, it would not be surprising that CCR5-using envelope proteins behave differently, but it remains to be established. A different behavior of the two co-receptors could explain why only a fraction of HIV-infected individuals develops HAD (even in the absence of HAART), despite the major role of CCR5 in HIV brain infection.
However, other factors also need to be considered. For instance, the ability of different HIV strains to induce syncytia, rather than their co-receptor usage, may be relevant to neuropathogenesis. Indeed, recent studies reported that the presence of dying syncytia in the brain correlates with disease severity (Nardacci et al., 2005). Interestingly, these neuropathology studies also reported induction of Puma in apoptotic neurons of these patients (Nardacci et al., 2005). Syncytia are a common characteristic of HIV-infected tissue, including the brain. Though typically the syncytium-inducing phenotype has been assigned to HIV strains that efficiently induce syncytia in transformed T cells in vitro (i.e. CXCR4-using strains), many R5 HIV-1 strains promote syncytia formation in primary macrophages albeit with variable efficacy, and their different ability in forming syncytia has been suggested to associate with their neuropathogenic potential (Gorry 2002). On the other hand, although brain-derived viruses predominantly use CCR5, viruses that exclusively use CXCR4 for infection of microglia/macrophages have been identified (Gorry et al., 2005) and rarely reported in the CSF (Yi et al., 2003). Thus, both CXCR4 and CCR5-using viruses can potentially induce macrophage syncytia and contribute to HAD, though adaptive viral evolution appears to favor M-tropism of R5 HIV-1 strains (Gorry et al., 2005).
To conclude, we propose that the transcriptional activity of E2F1 and its downstream effectors affect the survival and function of neurons and contribute to the neurological deficits in HIV patients. These apoptotic pathways could also be recruited by the natural CXCR4 ligand (the chemokine CXCL12/SDF) under pathological conditions, such as upon activation of extracellular proteases (Jones and Power, 2006) – extending the clinical relevance of these observations to other neuroinflammatory conditions. Indeed, we and others have previously reported that chemokines affect cell cycle proteins expression in neurons (Khan et al., 2003; Khan et al., 2005). As E2F1 up-regulation appears to be an early event in the neuropathogenesis, interfering with its activity might provide some level of therapeutic intervention. This could be achieved by blocking Rb phosphorylation by cyclin dependent kinase inhibitors (CDKI), which are currently in clinical trial for cancer therapy (Schwartz et al., 2005). Inhibition of CDKs can also prevent syncytia apoptosis of HIV-infected cells (Castedo et al., 2002), increasing the chances of success.
Acknowledgments
Supported in part by NIH grants, DA 19808 and DA15014 (OM), NS 41561 (RR), and by the W.W. Charitable Trust (OM). The NNTC is supported by NIMH and NINDS, contact NIH-N01MH32002, NY: MH59724, UCLA: NS38841, UCSD: MH59745-06, TX: NS45491.
References
- Adle-Biassette H, Levy Y, Colombel M, Poron F, Natchev S, Keohane C, Gray F. Neuronal apoptosis in HIV infection in adults. Neuropathol Appl Neurobiol. 1995;21:218–227. doi: 10.1111/j.1365-2990.1995.tb01053.x. [DOI] [PubMed] [Google Scholar]
- Bellizzi MJ, Lu SM, Masliah E, Gelbard HA. Synaptic activity becomes excitotoxic in neurons exposed to elevated levels of platelet-activating factor. J Clin Invest. 2005;115:3185–3192. doi: 10.1172/JCI25444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Castedo M, Roumier T, Blanco J, Ferri KF, Barretina J, Tintignac LA, Andreau K, Perfettini JL, Amendola A, Nardacci R, et al. Sequential involvement of Cdk1, mTOR and p53 in apoptosis induced by the HIV-1 envelope. Embo J. 2002;21:4070–4080. doi: 10.1093/emboj/cdf391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M, Masliah E. Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol. 1999;9:209–217. doi: 10.1111/j.1750-3639.1999.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzan M, Babcock GJ, Vasilieva N, Wright PL, Kiprilov E, Mirzabekov T, Choe H. The role of post-translational modifications of the CXCR4 amino terminus in stromal-derived factor 1 alpha association and HIV-1 entry. J Biol Chem. 2002;277:29484–29489. doi: 10.1074/jbc.M203361200. [DOI] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG, Jr, Livingston DM, Orkin SH, Greenberg ME. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- Gabuzda D, Wang J. Chemokine receptors and mechanisms of cell death in HIV neuropathogenesis. J Neurovirol. 2000;6(Suppl 1):S24–32. [PubMed] [Google Scholar]
- Garden GA, Guo W, Jayadev S, Tun C, Balcaitis S, Choi J, Montine TJ, Moller T, Morrison RS. HIV associated neurodegeneration requires p53 in neurons and microglia. Faseb J. 2004;18:1141–1143. doi: 10.1096/fj.04-1676fje. [DOI] [PubMed] [Google Scholar]
- Gartner S, Liu Y. Insights into the role of immune activation in HIV neuropathogenesis. J Neurovirol. 2002;8:69–75. doi: 10.1080/13550280290049525. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gorry PR, Churchill M, Crowe SM, Cunningham AL, Gabuzda D. Pathogenesis of macrophage tropic HIV-1. Curr HIV Res. 2005;3:53–60. doi: 10.2174/1570162052772951. [DOI] [PubMed] [Google Scholar]
- Hawkins V, Shen Q, Chiueh CC. Kynostatin and 17beta-estradiol prevent the apoptotic death of human neuroblastoma cells exposed to HIV-1 protease. J Biomed Sci. 1999;6:433–438. doi: 10.1007/BF02253675. [DOI] [PubMed] [Google Scholar]
- Hershko T, Ginsberg D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J Biol Chem. 2004;279:8627–8634. doi: 10.1074/jbc.M312866200. [DOI] [PubMed] [Google Scholar]
- Hesselgesser J, Taub D, Baskar P, Greenberg M, Hoxie J, Kolson DL, Horuk R. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol. 1998;8:595–598. doi: 10.1016/s0960-9822(98)70230-1. [DOI] [PubMed] [Google Scholar]
- Jones G, Power C. Regulation of neural cell survival by HIV-1 infection. Neurobiol Dis. 2006;21:1–17. doi: 10.1016/j.nbd.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Jordan-Sciutto KL, Wang G, Murphey-Corb M, Wiley CA. Cell cycle proteins exhibit altered expression patterns in lentiviral-associated encephalitis. J Neurosci. 2002;22:2185–2195. doi: 10.1523/JNEUROSCI.22-06-02185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ, Brandimarti R, Musser BJ, Resue DM, Fatatis A, Meucci O. The chemokine receptor CXCR4 regulates cell-cycle proteins in neurons. J Neurovirol. 2003;9:300–314. doi: 10.1080/13550280390201010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ, Shimizu S, Patel JP, Nelson A, Le MT, Mullen-Przeworski A, Brandimarti R, Fatatis A, Meucci O. Regulation of neuronal P53 activity by CXCR 4. Mol Cell Neurosci. 2005;30:58–66. doi: 10.1016/j.mcn.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Bonni A. The E2F-Cdc2 cell-cycle pathway specifically mediates activity deprivation-induced apoptosis of postmitotic neurons. J Neurosci. 2003;23:1649–1658. doi: 10.1523/JNEUROSCI.23-05-01649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Krek W, Livingston DM, Shirodkar S. Binding to DNA and the retinoblastoma gene product promoted by complex formation of different E2F family members. Science. 1993;262:1557–1560. doi: 10.1126/science.8248803. [DOI] [PubMed] [Google Scholar]
- Marder K, Albert SM, McDermott MP, McArthur JC, Schifitto G, Selnes OA, Sacktor N, Stern Y, Palumbo D, Kieburtz K, et al. Inter-rater reliability of a clinical staging of HIV-associated cognitive impairment. Neurology. 2003;60:1467–1473. doi: 10.1212/01.wnl.0000064172.46685.82. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12(Suppl 1):893–904. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, Sacktor N. Human immunodeficiency virus-associated dementia: an evolving disease. J Neurovirol. 2003;9:205–221. doi: 10.1080/13550280390194109. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgello S, Gelman BB, Kozlowski PB, Vinters HV, Masliah E, Cornford M, Cavert W, Marra C, Grant I, Singer EJ. The National NeuroAIDS Tissue Consortium: a new paradigm in brain banking with an emphasis on infectious disease. Neuropathol Appl Neurobiol. 2001;27:326–335. doi: 10.1046/j.0305-1846.2001.00334.x. [DOI] [PubMed] [Google Scholar]
- Nardacci R, Antinori A, Larocca LM, Arena V, Amendola A, Perfettini JL, Kroemer G, Piacentini M. Characterization of cell death pathways in human immunodeficiency virus-associated encephalitis. Am J Pathol. 2005;167:695–704. doi: 10.1016/S0002-9440(10)62044-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman E, Flemington EK, Sellers WR, Kaelin WG., Jr Transcription of the E2F-1 gene is rendered cell cycle dependent by E2F DNA-binding sites within its promoter. Mol Cell Biol. 1994;14:6607–6615. doi: 10.1128/mcb.14.10.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahlman S, Ruusala AI, Abrahamsson L, Mattsson ME, Esscher T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: a comparison with phorbolester-induced differentiation. Cell Differ. 1984;14:135–144. doi: 10.1016/0045-6039(84)90038-1. [DOI] [PubMed] [Google Scholar]
- Petito CK, Roberts B. Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol. 1995;146:1121–1130. [PMC free article] [PubMed] [Google Scholar]
- Roberts ES, Zandonatti MA, Watry DD, Madden LJ, Henriksen SJ, Taffe MA, Fox HS. Induction of pathogenic sets of genes in macrophages and neurons in NeuroAIDS. Am J Pathol. 2003;162:2041–2057. doi: 10.1016/S0002-9440(10)64336-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogoff HA, Pickering MT, Debatis ME, Jones S, Kowalik TF. E2F1 induces phosphorylation of p53 that is coincident with p53 accumulation and apoptosis. Mol Cell Biol. 2002;22:5308–5318. doi: 10.1128/MCB.22.15.5308-5318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa MJ, Madeira MD, Ruela C, Volk B, Mota-Miranda A, Paula-Barbosa MM. Dendritic changes in the hippocampal formation of AIDS patients: a quantitative Golgi study. Acta Neuropathol (Berl) 2004;107:97–110. doi: 10.1007/s00401-003-0781-3. [DOI] [PubMed] [Google Scholar]
- Schwartz GK, Weitzman A, O’Reilly E, Brail L, de Alwis DP, Cleverly A, Barile-Thiem B, Vinciguerra V, Budman DR. Phase I and pharmacokinetic study of LY293111, an orally bioavailable LTB4 receptor antagonist, in patients with advanced solid tumors. J Clin Oncol. 2005;23:5365–5373. doi: 10.1200/JCO.2005.02.766. [DOI] [PubMed] [Google Scholar]
- Shi B, De Girolami U, He J, Wang S, Lorenzo A, Busciglio J, Gabuzda D. Apoptosis induced by HIV-1 infection of the central nervous system. J Clin Invest. 1996;98:1979–1990. doi: 10.1172/JCI119002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Levine AJ. Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc Natl Acad Sci U S A. 1999;96:3077–3080. doi: 10.1073/pnas.96.6.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PB, Miller RJ. Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci. 2003;4:444–455. doi: 10.1038/nrn1116. [DOI] [PubMed] [Google Scholar]
- Yi Y, Chen W, Frank I, Cutilli J, Singh A, Starr-Spires L, Sulcove J, Kolson DL, Collman RG. An unusual syncytia-inducing human immunodeficiency virus type 1 primary isolate from the central nervous system that is restricted to CXCR4, replicates efficiently in macrophages, and induces neuronal apoptosis. J Neurovirol. 2003;9:432–441. doi: 10.1080/13550280390218706. [DOI] [PubMed] [Google Scholar]