

Abstract
The method of DNA cyclization kinetics reveals special properties of the TATAAACGCC sequence motif found in DNA sequences that have high affinity for core histones. Replacement of 30 bp of generic DNA by three 10-bp repeats of the motif in small cyclization constructs increases cyclization rates by two orders of magnitude. We document a 13° bend in the motif and characterize the direction of curvature. The bending force constant is smaller by nearly 2-fold and there is a 35% decrease in the twist modulus, relative to generic DNA. These features are the likely source of the high affinity for bending around core histones to form nucleosomes. Our results establish a protocol for determination of the ensemble-averaged global solution structure and mechanical properties of any ≈10-bp DNA sequence element of interest, providing information complementary to that from NMR and crystallographic structural studies.
DNA in cells is highly compacted from the unfettered size it would have if free in solution. The first stage of packaging is achieved by wrapping DNA around core histones in a well-defined conformation (1), forming a beads-on-a-string structure that is further compacted by additional proteins into folded chromatin and ultimately into metaphase chromosomes. Interphase chromatin is generally less condensed, but it contains regulatory sites such as promoters at which DNA must be distorted to enable interactions between proteins that bind distant sites on linear DNA. As a relatively stiff polymer, DNA resists the bending required for packaging, which requires free energy assistance from strong interactions with core histones and other proteins.
Variations in DNA sequence can, in principle, modulate the energy cost of bending and packaging. For example, intrinsic curvature of the kind provided by A-tracts (2) should, if in the proper direction, lessen the energy required for bending. Increased local bending flexibility could reduce the energetic cost of packaging, as could an increase in twist flexibility (3), assuming that the helical phasing between distal regulatory sequence elements must be altered to form a protein complex.
The stiffness of DNA (the inverse of its flexibility) is traditionally described by the persistence length P, defined as the average projection of the end-to-end vector of a very long chain on its initial direction. Classical techniques, such as light scattering (4) and rotational diffusion (5), supplemented by DNA cyclization kinetics (6), have led to estimates of around 140–180 bp for P. However, these methods are generally unable to deconvolute rigorously the contributions of curvature and flexibility to apparent persistence length (7), unless special constructs are used (8).
Parameter sets exist for predicting DNA curvature based on roll and tilt values for dinucleotide (9–13) and tetranucleotide (14) steps, but the dinucleotide parameters vary considerably from set to set (15). Nor is there sufficient data to define unambiguously the sequence dependence of the experimental persistence length, except for repeating polymers such as alternating poly[d(A-T)] and poly[d(G-C)], which are found to be relatively more and less flexible, respectively, than genomic DNA (3, 16). Potentially predictive parameters for roll, tilt, and twist flexibilities of individual base pair steps are available from the standard deviation of these parameters in the crystallographic database (11, 12) and accompanying computational approaches (13, 14). De Santis and colleagues (17) have shown that bending flexibility parameters based on thermal stability have predictive power for sequence-dependent nucleosome binding affinity.
The total torsional flexibility of DNA is due to the combined effects of twist and writhe fluctuations, and average values can be determined from study of closed circular molecules (18). Fluorescence polarization anisotropy studies of linear DNA molecules show little dependence of torsional flexibility on average base composition (19). However, local variations of the twist flexibility depending on sequence have not been determined in solution. In summary, there is a clear need for experimentally based parameters that describe the ensemble-averaged solution curvature and local bending and twist flexibility of specific DNA sequences. In addition to providing a test for parameter sets based on crystallography and theory, such a database would allow more stringent test of the intriguing indications that curved and flexible sequences are organized coherently in promoters and other DNA sequences (20–22) and would enable analysis of the properties of the noncoding DNA sequences that constitute ≈95% of the human genome.
We have developed a strategy, based on specialized DNA molecules for measurement of cyclization kinetics, that offers a general solution to this problem (23–28). A key feature is inclusion of phased A-tracts, whose intrinsic curvature fixes the mean rotational setting of the circle. Altering the phasing between a test sequence element and the A-tracts has a large effect on cyclization if the test sequence is bent, but not if it is simply altered in isotropic flexibility. The cyclization constructs also provide for variation of the overall DNA length, allowing determination of the bending and twist flexibility and the DNA helical repeat. The constructs are kept small (≈160 bp) as a design principle so that small changes in curvature or flexibility have a large effect on cyclization rate. The method has been used independently by Davis et al. (29) to show that the sequence TATAAAAG is bent (or anisotropically flexible) in a direction opposite to that induced by the TATA box binding protein.
In the work reported here we show that the distinctive curvature and mechanical properties of a small (10 bp) sequence element can be quantitatively determined by this approach. The sequence motif chosen, the TATA tetrad (5′-TATAAACGCC-3′), is found in multiple phased repeats in genomic sequences that are among the strongest known binders of core histones (30). We have repeated the 10-bp sequence element three times to amplify both its curvature and the influence of its flexibility, and inserted it into the cyclization constructs. Molecules in which the 30-bp test nucleosome positioning sequence (NPS) replaces 30 bp of “generic” DNA cyclize approximately 100-fold faster than the parent constructs. Recent improvements in the speed and reliability of the computer simulation of the cyclization process (J.L. and D.M.C., unpublished work) enable us to provide better quantitative interpretation of bend angle and direction, along with the flexibility parameters. The simulations reveal that the accelerated cyclization can be resolved into to a combination of intrinsic (or static) curvature and increased (isotropic) bending flexibility in the NPS, relative to generic DNA.
Materials and Methods
Theoretical Background.
The cyclization kinetic method requires a comparison of the rate constants for unimolecular cyclization and bimolecular ligation (6, 23). The basic assumption is that the rate of each of these processes is limited by the concentration of the substrate for DNA ligase, which consists of molecules in which the complementary DNA ends are transiently hydrogen bonded. It is further required that the ends dissociate rapidly compared with the rate of the ligation step. Under these conditions, the rate constants k1 and k2 for cyclization and bimolecular ligation, respectively, are proportional to the corresponding equilibrium constants K1 and K2 for transient pairing of the substrate ends. The ratio J is given by
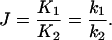 |
The experiments consist of determination of J values for the different DNA constructs by measurement of the reaction kinetics, using gel electrophoresis of 32P-labeled DNA to quantitate the extent of reaction.
Design of the Cyclization Constructs.
The design of our constructs, shown schematically in Fig. 1, is based on earlier work reported from our laboratory (23–28). The molecules contain a set of six phased A-tracts separated from the 30-bp NPS by linkers with variable lengths of 12, 16, and 20 bp. The adaptor region enables variation of the overall length of the constructs.
Figure 1.

A-tract–NPS minicircles. The six phased A-tracts and the 30-bp NPS region are separated by linker regions with variable lengths of 12, 16, of 20 bp. The adaptor region enables variation of the overall length of the constructs. The first number in the nomenclature of our constructs, e.g., 12_154, indicates the length of the phasing linker, and the second number indicates the overall length of the construct.
DNA Sequences.
These are derived from the 17A9 molecule described by Sitlani and Crothers (26), with the 30-bp NPS site replacing 30 bp of sequence around the AP-1 site. All molecules have 2-bp 5′ overhanging CG ends, and share the A-tract region of the sequence, which is: (1)CGATGAATTCCTGTACGGATCCGGAAAAAACGGGCAAAAAACGGCAAAAAACGGGCAAAAAACGGCAAAAAACGGGCAAAAAACCGGATC(90). This is followed in the 156-bp B-DNA molecule by (91)CGAATTCTAGACCTAGGTGGATGACTCATTTTTTTTTGCTCGAGCTACAACGTGCTGCCATGGAAT(156) and, in the variable phasing linker sequences, by
12_156
CGAATTATAAACGCCTATAAACGCCTATAAACGCCTTGCTCGAGCTACAACGTGCTGCCATGGAAT
16_156
CGAATTTGCTATAAACGCCTATAAACGCCTATAAACGCCTCGAGCTACAACGTGCTGCCATGGAAT
20_156
CGAATTTGCTCGATATAAACGCCTATAAACGCCTATAAACGCCGCTACAACGTGCTGCCATGGAAT
in which the 30-bp segment is indicated in boldface, and the sequence elements that differ in the phasing linkers are underlined. The molecules with different overall lengths were constructed by using variable bottom-strand PCR primers taken from the set
150 5′-GCAGATATCGATGCAGCACGTTGTAGC
151 GCAGATATCGATCGCAGCACGTTGTAGC
152 GCAGATATCGATTCGCAGCACGTTGTAGC
154 GCAGATATCGATTCCAGCAGCACGTTGTAGC
155 GCAGATATCGATTCCATGCAGCACGTTGTAGC
156 GCAGATATCGATTCCATGGCAGCACGTTGTAGC
157 GCAGATATCGATTCCATGAGCAGCACGTTGTAGC
158 GCAGATATCGATTCCATGGCAAAGCACGTTGTAGC
160 GCAGATATCGATTCTGGACATGGCAGCACGTTGTAGC
161 GCAGATATCGATCTCTGGACATGGCAGCACGTTGTAGC
162 GCAGATATCGATTCTCTGGACATGGCAGCACGTTGTAGC
164 GCAGATATCGATTCTCTCTGGACATGGCAGCACGTTGTAGC
166 GCAGATATCGATGGTCTCTCTGGACATGGCAGCACGTTGTAGC
168 GCAGATATCGATCTGGTCTCTCTGGACATGGCAGCACGTTGTAGC
where the variable sequences are underlined. The ClaI site, indicated in boldface, is used to create the lower strand 2-bp 5′ CG overhang in all molecules. The upper strand PCR primer, 5′-CGCCATGGAATCGATGAATTC, also contained a ClaI site. The molecules were assembled by standard DNA synthesis, ligation, and PCR methods (31). Cyclization substrates were prepared by PCR amplification in the presence of [α-32P]dATP, purified by using the Qiagen (Chatsworth, CA) PCR purification kit, and digested at 37°C overnight with 2.5 units of ClaI (New England Biolabs) per 100 μl. After purification on nondenaturing 10% polyacrylamide gels and electroelution, the radioactivity was monitored by scintillation counting and the concentration was determined by UV absorbance.
Cyclization Kinetics.
Rate constants were measured as described previously (23, 24). Briefly, T4 DNA ligase (New England Biolabs) at 500 units⋅ml−1 was added to a preincubated solution of 2–10 nM DNA in a reaction buffer of 10 mM Tris⋅HCl (pH 7.5), 5 mM MgCl2, 50 mM NaCl, 1 mM EDTA, 50 μg⋅ml−1 BSA, 5% sucrose, 5% (vol/vol) glycerol, 5 mM DTT, and 0.05–0.1% Nonidet P-40 at 21°C. Samples (8 μl) of the reaction mixture were taken at various time points from 0 to 8 min and added to a quenching mixture of 4 μl of 45 mM EDTA, 3 mg⋅ml−1 proteinase K, 15% glycerol, and electrophoresis dyes. Quenched reactions were incubated at 55°C for 10 min and loaded on a native 6% polyacrylamide gel that was prerun for 1 h. The gels were analyzed with a FUJIX Bio-imaging Analyzer System using MacBas software. In most cases the cyclization rate constant k1 and bimolecular rate constant k2 could be obtained directly from the time dependence of the concentration of circular products and linear and circular dimers, respectively (24), related to the approach of Taylor and Hagerman (32). In the case of rapid cyclizers we accessed the bimolecular rate constant either by increasing the DNA concentration to 25 nM or by determining k2 for a standard molecule (17A9) in a parallel reaction.
Computer Simulations.
Monte Carlo simulation of chain configurations was carried out as described (23, 33). Bends at A-tracts were introduced by specific roll and tilt angles at the junctions of the A-tract with adjacent DNA (34). The chain is assumed to be inextensible, and it is assumed that roll, tilt, and twist fluctuations are uncorrelated. Any curvature that results from anisotropic flexibility (29, 35) is represented in our model by a static bend and isotropic flexibility. The program generates libraries of first and second half chains, and screens whole-chain combinations of subsets of these for end-to-end distances below a cut-off value. Chains passing this criterion are further examined for colinear alignment of the helix ends and the twist angle between base pairs at the two ends to calculate J. The original computer algorithm for simulation of DNA cyclization (33) was further developed by Rice (34) and Kahn (36). Most recently, we (J.L. and D.M.C., unpublished work) found that the main source of statistical fluctuations in the results was the excessive importance of a small number of highly bent half chains, particularly in the non-A-tract portion of the molecule. These extreme fluctuations arise from in-phase bend fluctuations, and are less important in the A-tract half, for which the static curvature makes highly bent states more accessible. The new version of the program, available from the authors on request, generates a larger and more diverse population of chains. Rather than combining all first half chains with all second half chains, the program generates a larger (adjustable) number of half chains and combines these in subsets. This approach enables statistical analysis of the J values that result from the individual subsets. The typical standard deviation of simulated values of J ranges from less than 5% for J values greater than 100 nM to about 8% for J values of ≈1 nM. These numbers are significantly smaller than the experimental standard deviation, which is typically 10–25%. The difference between simulated and experimental data sets was characterized by the “absolute error” (36), which is a sum of squares of the difference between theoretical and experimental values of log10 J for all molecules in the set under consideration.
Results and Discussion
Variation of J Values with Phasing Linker Length.
We determined J values for the three different phasing linker lengths of 12, 16, and 20 bp for three different overall lengths, 154, 156, and 158 bp. Fig. 2 shows typical data for the kinetic measurements. The rate of the bimolecular ligation step was determined from the time dependence of the sum of the concentrations of linear dimers and circular multimers (24). As is evident from Fig. 2, the 16-bp linker at 156-bp overall length cyclizes more rapidly than the 12-bp linker. The results are summarized in Table 1, which shows that the highest measured J value is found for the 16-bp linker. Note that whereas the 16-bp linker has an optimum cyclization rate at 156 bp, the rate for the 12-bp linker is greater at 154- than at 156-bp length. Two noncoplanar bends contribute writhe to the molecules, depending on the angle between the bend planes. As a consequence, the optimum number of base pairs needed to yield an integral number of helical turns in the circles depends on the phasing linker length. This rather complicated problem is dealt with automatically by the computer simulations described below, from which we deduce that the ideal linker length required for optimal phasing of the A-tract and test bends would be about 14.5 bp (31). The closest experimental length is 16, which is therefore the most nearly planar molecule among our set.
Figure 2.

Cyclization kinetic measurements on 16_156 and 12_156 constructs. (Upper) Cyclization reactions of 16_156 and 12_156 constructs. A, products of bimolecular ligation; B, cyclization product; and C, linear starting material. (Lower) The corresponding determination of cyclization and bimolecular association rate constants and J factors. □, Circular monomer; ○, products of bimolecular ligation.
Table 1.
Variation of J factors with linker length for the NPS-containing molecules
| DNA length, bp | Linker length, bp | J, nM |
|---|---|---|
| 154 | 12 | 960 (±120) |
| 154 | 16 | 650 (±75) |
| 154 | 20 | 45 (±5) |
| 156 | 12 | 650 (±80) |
| 156 | 16 | 1,650 (±150) |
| 156 | 20 | 30 (±5) |
| 158 | 12 | 70 (±10) |
| 158 | 16 | 500 (±120) |
| 158 | 20 | 25 (±5) |
Numbers in parentheses are errors.
Variation of J Values with Overall Length.
The 16-bp linker was chosen for study of the dependence on overall length. These results are required for a full determination of the bending and twist flexibility and the helical repeat. The constructs differed by 1 or 2 bp in length, for both the NPS-containing molecules and the parent set containing “generic” DNA in the 30-bp test site. [Earlier work showed that there is no appreciable bend in this sequence (26, 27).] The results are summarized in Fig. 3. The expected (18) modulation of the cyclization rate with the ≈10-bp DNA helical periodicity is evident, and there is a striking increase of about two orders of magnitude in the cyclization rate for the NPS-containing molecules compared with the parent set. This dramatic effect must be due at least in part to the curvature of the NPS segment detected in the variation of rate with phasing linker length. However, flexibility changes may also contribute, and it requires a simulation of the process to deconvolute these two variables, as described below.
Figure 3.

Comparisons of experimental and theoretical log J versus DNA length for constructs with and without the NPS site. The upper sets of curves correspond to constructs having the NPS site with a fixed linker length of 16 bp. The experimental log10 J values are shown as ▵ and the corresponding curve fit is the dashed line. The simulation values are indicated by ▴ and their curve fit is the solid line. The lower sets of curves correspond to the B-DNA molecules containing a generic 30-bp sequence in place of the NPS site. The experimental log10 J values are the ○ with the dashed line indicating the curve fit; the simulation values are the ● with solid line indicating the curve fit. The data are fitted to double Gaussian equations (25).
Verification of the Properties of DNA Lacking the 30-bp Sequence.
In the simulations to determine the properties of the NPS, it is necessary to assign flexibility and helical repeat values to the remainder of the molecule. The values for the A-tract segment come from earlier experiments (34). The experiments for the parent set of molecules reported in Fig. 3 allow determination of average parameters for the rest of the molecule, including the 30-bp segment replacing the NPS site, which we took to be generic B-DNA (27). However, we were concerned that this “generic” 30-bp segment might have properties different from the average for the molecule, so we carried out cyclization kinetic experiments for test molecules in which the 30-bp segment was deleted entirely. Table 2 shows the J values measured, compared with simulation, based on the flexibility and helical repeat parameters determined for the longer B-DNA molecules in Fig. 3. The good correspondence between measured and simulated values for the shortened molecules confirms that the DNA outside the NPS sequence is accurately described by the parameter set. We also infer that the deleted 30-bp segment has properties indistinguishable from those of the rest of the B-DNA portion of the molecule.
Table 2.
Comparison of experimental and simulated J values for molecules lacking the 30-bp insert
| DNA length, bp |
J factor, nM
|
|
|---|---|---|
| Experiment | Simulation* | |
| 124 | 19.8 | 20.8 |
| 126 | 16.0 | 14.8 |
| 128 | 1.2 | 1.2 |
| 134 | 21.5 | 21.0 |
| 136 | 22.5 | 23.4 |
| 138 | 4.0 | 5.4 |
Global Bend Direction Determined by Computer Simulation.
Determination of the properties of DNA molecules by matching simulated and experimental J values for a set of 20 cyclization constructs is a multivariable minimization problem, which we have solved by an iterative series of refinements of the parameters. We took advantage of the dependence, to first order, of some of the molecular properties on single parameters. For example, the variation of J with phasing linker length is primarily a function of the direction of curvature in the NPS sequence, assuming that at least three different phasing linkers have been measured. The distance between successive maxima in the periodic variation of J with overall length in Fig. 3 is determined by the helical repeat, and the peak-to-trough amplitude of the curves is primarily a function of the twist flexibility. However, the overall 100-fold upward shift of the J values for the NPS sequence relative to B-DNA depends on a combination of increased bending flexibility and the magnitude of the curvature, and it must be treated to first order as a two-dimensional minimization problem in which the entire data set, including all phasing linkers, is included.
The cyclization kinetic method can be used to determine the global direction of curvature, but not its structural origin or actual locus. For simplicity we have modeled the bend by roll between the base pairs at either a single base pair step in each NPS 10-bp element, or between adjacent base pair steps (to improve the resolution of global bend direction). The simulations were used to find the position at which a positive roll angle minimizes the disagreement between experiment and calculated log10 J values for the set of phasing linkers. The actual curvature can be due to any combination of roll and tilt angles in the NPS that has the same resultant as our arbitrary roll-bend.
Fig. 4a shows the “absolute error” (the sum of squares of the difference between simulated and experimental values of log10 J) for three different linker lengths at a total molecular length of 154 bp. All three curves share a common minimum for a total roll angle, taken here to be 13° for reasons documented below, distributed between nucleotides 8, 9, and 10, in each of the three GCC trinucleotides in the 30-bp sequence. (The kinks at the GC and CC steps were taken to be 3.25° and 9.75°, respectively, so that the scalar sum is 13.) The same bend position was found from fitting the data for the set of 156-bp and 158-bp molecules. If a negative roll angle, indicating a compression of the minor groove, is assumed, the optimum position is shifted by half a helical turn and partitioned over the steps between nucleotides 3, 4, and 5, corresponding to the TAA trinucleotide in the NPS. Taken together, the results are consistent with some combination of a bend toward the minor groove in the A-T-rich section and toward the major groove in the G-C-rich segment of the NPS. This conclusion is consistent with the observed direction of curvature of the NPS when incorporated into nucleosomes, as shown by Widlund et al. (37). Those authors also reported an increase in cyclization rate for molecules containing the NPS, but they did not deconvolute the effects of curvature and flexibility nor establish the direction of curvature of the DNA in the absence of histones.
Figure 4.

Refinement of geometric and flexibility parameters against experiment. A lower value for the absolute error corresponds to a better agreement between experimental and theoretical log10 J factors. (a) Determination of the kink position for molecules with a fixed overall length of 154 bp and varied linker lengths of 12, 16, and 20 bp. Kink angles are centered on a base pair—for example, the second C in CCG has one-fourth the total (scalar) kink angle at the CC step, and three-fourths at CG. (b) Projection of a three-dimensional plot on a two-dimensional xy plane, showing absolute error as a function of kink angle and rms angle fluctuations. The black center corresponds to minimum error (0.4–0.64), with subsequent contours increasing by 0.26. The figure indicates that a kink angle between 11.5° and 14° and roll and tilt fluctuations ranging from 6.25° to 6.75° at each base pair step in the 30-bp NPS site provide the best fit between experimental and simulation values of the log10 J factors. (c) Absolute errors for varying the twist modulus of the 30-bp NPS site. (d) Absolute errors for varying the helical repeat of the 30-bp NPS site.
Determination of Flexibility and Helical Repeat.
The dependence of the absolute error on the twisting modulus and the helical repeat is shown in Fig. 4 c and d. In these simulations, the kink angle and bending flexibility for the NPS segment were set at the optimum values of 13° and rms roll and tilt fluctuations of 6.5° (equivalent to a persistence length of 78 bp), as documented in Fig. 4b. The B-DNA segment has roll and tilt fluctuations of 4.68°, equivalent to a persistence length of 150 bp. The process of iterative refinement of the NPS properties by minimization of the absolute error leads to the parameter set summarized in Table 3. The overall curvature of the 30-bp sequence is about 40° and the average bending flexibility (proportional to the square of the rms angle fluctuations, or to the inverse of the persistence length or bending force constant) is increased by nearly 2-fold from the value for the remainder of the molecule. In independent simulations, we showed that either increased flexibility with zero curvature, or 40° curvature with normal B-DNA flexibility, accounts for about a factor of 10 in the 100-fold increase of J values for the NPS molecules relative to the parent set. There is also a significant increase in twist flexibility, corresponding to the reduction of the twisting modulus from 2.4 to about 1.5 × 10−19 erg⋅cm.
Table 3.
Best-fit parameters derived from the simulations
| DNA | Roll and tilt fluctuations, ° | Torsional modulus, 10−19 erg⋅cm | Kink angle, ° | Helical repeat, bp |
|---|---|---|---|---|
| B-DNA | 4.68 | 2.4 | 0 | 10.45 |
| A-tract | 4.68 | 2.4 | * | 10.33 |
| NPS site | 6.5 | 1.5 | 13 | 10.30 |
The A-tracts are bent by roll and tilt kinks at the junctions (34).
Significance.
Our results establish a protocol for determination of the global structure and flexibility parameters of defined DNA sequence elements containing an approximately integral number of helical turns. The technique provides a view of overall DNA structure for an ensemble of molecules which is complementary to that available from crystallographic and NMR structural studies. The high sensitivity of J values to DNA properties should make it possible to characterize curvature of a few degrees and flexibility changes of ≈10%; examination of a large number of sequences can enable construction of a database that is predictive for the curvature and flexibility of any arbitrary sequence.
At the present time we can specify only the average flexibility and curvature parameters for the NPS, but future mutagenic studies on the sequence should identify specific sequence determinants of the curvature and enhanced flexibility. It seems likely that the bend, enhanced twist and bending flexibility, and reduced helical repeat all contribute to the strong binding affinity of the NPS for core histones.
Acknowledgments
We thank Zippora Shakkked, Wilma Olson, and Jason Kahn for their comments on the manuscript. This work was supported by Grant GM 21966 from the National Institutes of Health.
Abbreviation
- NPS
nucleosome positioning sequence
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.250476297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.250476297
References
- 1.Luger K, Mader A W, Richmond R K, Sargent D F, Richmond T J. Nature (London) 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Wu H-M, Crothers D M. Nature (London) 1984;308:509–513. doi: 10.1038/308509a0. [DOI] [PubMed] [Google Scholar]
- 3.Hogan M E, Austin R H. Nature (London) 1987;329:263–266. doi: 10.1038/329263a0. [DOI] [PubMed] [Google Scholar]
- 4.Harpst J A. Biophys Chem. 1980;11:295–302. doi: 10.1016/0301-4622(80)80032-9. [DOI] [PubMed] [Google Scholar]
- 5.Hagerman P J. Biopolymers. 1981;20:1503–1535. doi: 10.1002/bip.1981.360200710. [DOI] [PubMed] [Google Scholar]
- 6.Shore D, Langowski J, Baldwin R L. Proc Natl Acad Sci USA. 1981;78:4833–4837. doi: 10.1073/pnas.78.8.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trifonov E N. CRC Crit Rev Biochem. 1985;19:89–106. doi: 10.3109/10409238509082540. [DOI] [PubMed] [Google Scholar]
- 8.Levene S D, Wu H-M, Crothers D M. Biochemistry. 1986;25:3988–3995. doi: 10.1021/bi00362a003. [DOI] [PubMed] [Google Scholar]
- 9.Bolshoy A, McNamara P, Harrington R E, Trifonov E N. Proc Natl Acad Sci USA. 1991;88:2312–2316. doi: 10.1073/pnas.88.6.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Santis P, Palleschi A, Savino M, Scipioni A. Biochemistry. 1990;29:9269–9273. doi: 10.1021/bi00491a023. [DOI] [PubMed] [Google Scholar]
- 11.Gorin A A, Zhurkin V B, Olson W K. J Mol Biol. 1995;247:34–48. doi: 10.1006/jmbi.1994.0120. [DOI] [PubMed] [Google Scholar]
- 12.Olson W K, Babcock M S, Gorin A, Liu G, Markey N L, Martino J A, Pederson S C, Srinivasan A R, Tobias I, Westcott T P. Biophys Chem. 1995;55:7–29. doi: 10.1016/0301-4622(94)00139-b. [DOI] [PubMed] [Google Scholar]
- 13.Packer M J, Dauncey M P, Hunter C A. J Mol Biol. 2000;295:71–83. doi: 10.1006/jmbi.1999.3236. [DOI] [PubMed] [Google Scholar]
- 14.Packer M J, Dauncey M P, Hunter C A. J Mol Biol. 2000;295:85–103. doi: 10.1006/jmbi.1999.3237. [DOI] [PubMed] [Google Scholar]
- 15.Crothers D M. Proc Natl Acad Sci USA. 1998;95:15163–15165. doi: 10.1073/pnas.95.26.15163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas T J, Bloomfield V A. Nucleic Acids Res. 1983;11:1919–1930. doi: 10.1093/nar/11.6.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anselmi C, Bocchinfuso G, De Santis P, Savino M, Scipioni A. J Mol Biol. 1999;286:1293–1301. doi: 10.1006/jmbi.1998.2575. [DOI] [PubMed] [Google Scholar]
- 18.Shore D L, Baldwin R L. J Mol Biol. 1983;170:957–981. doi: 10.1016/s0022-2836(83)80198-3. [DOI] [PubMed] [Google Scholar]
- 19.Fujimoto B S, Schurr J M. Nature (London) 1990;344:175–177. doi: 10.1038/344175a0. [DOI] [PubMed] [Google Scholar]
- 20.Wada-Kiyama Y, Kuwabara K, Sakuma Y, Onishi Y, Trifonov E N, Kiyama R. FEBS Lett. 1999;444:117–124. doi: 10.1016/s0014-5793(99)00041-1. [DOI] [PubMed] [Google Scholar]
- 21.Ozoline O N, Deev A A, Trifonov E N. J Biomol Struct Dyn. 1999;16:825–831. doi: 10.1080/07391102.1999.10508295. [DOI] [PubMed] [Google Scholar]
- 22.Pederson A G, Jensen L J, Brunak S, Stærfeldt H-H, Ussery D W. J Mol Biol. 2000;299:907–930. doi: 10.1006/jmbi.2000.3787. [DOI] [PubMed] [Google Scholar]
- 23.Crothers D M, Drak J, Kahn J D, Levene S D. Methods Enzymol. 1992;212B:3–29. doi: 10.1016/0076-6879(92)12003-9. [DOI] [PubMed] [Google Scholar]
- 24.Kahn J D, Crothers D M. Proc Natl Acad Sci USA. 1992;89:6343–6347. doi: 10.1073/pnas.89.14.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahn, J. D., Yun, E. & Crothers, D. M. (1994) (1994) Nature (London)368, 163–166. [DOI] [PubMed]
- 26.Sitlani A, Crothers D M. Proc Natl Acad Sci USA. 1996;93:3248–3252. doi: 10.1073/pnas.93.8.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hockings S C, Kahn J D, Crothers D M. Proc Natl Acad Sci USA. 1998;95:1410–1415. doi: 10.1073/pnas.95.4.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sitlani A, Crothers D M. Proc Natl Acad Sci USA. 1998;95:1404–1409. doi: 10.1073/pnas.95.4.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis N A, Majee S S, Kahn J D. J Mol Biol. 1999;291:249–265. doi: 10.1006/jmbi.1999.2947. [DOI] [PubMed] [Google Scholar]
- 30.Widlund H R, Cao H, Simonsson S, Magnusson E, Simonsson T, Nielsen P E, Kahn J D, Crothers D M, Kubista M. J Mol Biol. 1997;267:807–817. doi: 10.1006/jmbi.1997.0916. [DOI] [PubMed] [Google Scholar]
- 31.Roychoudhury M. Ph.D. thesis. New Haven, CT: Yale Univ.; 2000. [Google Scholar]
- 32.Taylor W H, Hagerman P J. J Mol Biol. 1990;212:363–376. doi: 10.1016/0022-2836(90)90131-5. [DOI] [PubMed] [Google Scholar]
- 33.Levene S D, Crothers D M. J Biomol Struct Dyn. 1983;1:429–435. doi: 10.1080/07391102.1983.10507452. [DOI] [PubMed] [Google Scholar]
- 34.Koo H-S, Drak J, Rice J A, Crothers D M. Biochemistry. 1990;29:4227–4234. doi: 10.1021/bi00469a027. [DOI] [PubMed] [Google Scholar]
- 35.Olson W K, Zhurkin V B. Curr Opin Struct Biol. 2000;10:286–297. doi: 10.1016/s0959-440x(00)00086-5. [DOI] [PubMed] [Google Scholar]
- 36.Kahn J D, Crothers D M. J Mol Biol. 1998;276:287–309. doi: 10.1006/jmbi.1997.1515. [DOI] [PubMed] [Google Scholar]
- 37.Widlund H R, Kuduvalli P N, Bengtsson M, Cao H, Tullius T D, Kubista M. J Biol Chem. 1999;274:31847–31852. doi: 10.1074/jbc.274.45.31847. [DOI] [PubMed] [Google Scholar]