

Abstract
Non-random chromosomal translocations are frequently associated with a variety of cancers, especially hematologic malignancies and childhood sarcomas In addition to their diagnostic utility, chromosomal translocations are increasingly being used in the clinic to guide therapeutic decisions. However, the mechanisms which cause these translocations remain poorly understood. Illegitimate V(D)J recombination, class switch recombination, homologous recombination, non-homologous end joining, and genome fragile sites all have potential roles in the production of non-random chromosomal translocations. In addition, mutations in DNA repair pathways have been implicated in the production of chromosomal translocations in humans, mice, and yeast. Although initially quite surprising, the identification of these same oncogenic chromosomal translocations in peripheral blood from healthy individuals strongly suggests that the translocation is not sufficient to induce malignant transformation, and that complementary mutations are required to produce a frank malignancy.
Keywords: Chromosomal translocation, hematologic malignancy, leukemia, lymphoma, non-homologous end-joining, DNA repair
Introduction
Over the past decade, it has become clear that cancer is a genetic disease, that is, a disease caused by gene mutations, which are most commonly acquired, as opposed to inherited. Cytogenetic analysis of some malignancies, especially hematologic malignancies, reveals a sole abnormality, such as a single balanced translocation whereas epithelial tumors will typically display a much more complex pattern of numerical and structural chromosomal aberrations. (Figure 1). (Mitelman Database of Chromosome Aberrations in Cancer, at http://cgap.nci.nih.gov/Chromosomes/Mitelman ). These sole abnormalities serve as a “smoking gun” that identifies gene(s) important for malignant transformation 1. Recent analysis suggests that recurrent balanced chromosomal abnormalities are also common in epithelial tumors, but that these have been difficult to distinguish in a background of numerous structural and numerical chromosomal abnormalities 2 (Box 1). Since the recurrent, balanced translocations have been most intensely studied in the context of hematologic malignancy, this review will focus on the role of recurrent, balanced chromosomal translocations in leukemias and lymphomas (box 2). The more common chromosomal translocations associated with acute and chronic leukemia are listed in Table 1; an exhaustive list of all known recurrent translocation breakpoints can be found at http://cgap.nci.nih.gov/Chromosomes/Mitelman.
Figure 1. Spectral karyotype analysis of malignant cells.
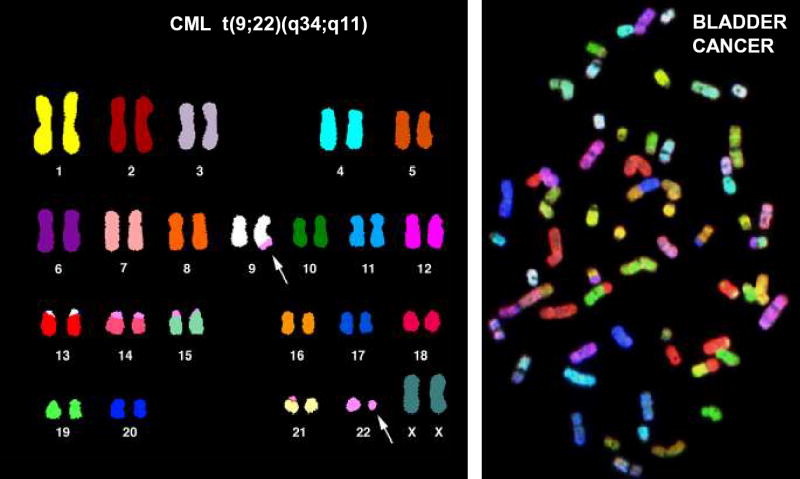
A. Spectral karyotype from a patient with CML, showing a pseudodiploid karyotype with a t(9;22) translocation as the only detectable anomaly. B. Spectral karyotype of a bladder carcinoma, showing numerous numerical and structural chromosomal anomalies. (Both karyotypes courtesy of Drs. Thomas Ried and Hesed Padilla-Nash, NIH, NCI, Bethesda, MD.)
BOX 1 Recurrent, balanced chromosomal translocations in hematologic and non-hematologic tumors.
Recurrent, balanced chromosomal translocations have been recognized in hematologic malignancies for several decades. As shown in Figure 1, the karyotypes of hematologic malignancies are often quite simple, showing a single balanced translocation, whereas karyotypes of solid tumors often show a complex pattern of numerical and structural abnormalities. These observations have led to the speculation that solid tumors might also have recurrent translocations, but that the recurrent translocations are more difficult to identify in a background of numerous non-specific chromosomal aberrations. It should be noted here that non-hematopoietic solid tumors can be subclassified as carcinomas (typically arising from epithelial tissues), sarcomas, and germ cell tumors; most of the common human malignancies (lung, colon, breast) are epithelial malignancies. Childhood sarcomas and germ cell tumors are solid tumors which are similar to hematologic malignancies in that they often have simple karyotypes that contain a sole abnormality, such as the t(11;22)(q24;q12) associated with Ewing’s sarcoma or the t(X;18(p11;q11) associated with synovial cell sarcoma 72. However, very few recurrent balanced translocations have been cloned from patients with epithelial malignancies. One example is the recently reported MECT-MAML1 fusion caused by the t(11;19) associated with a rare salivary gland tumor (mucoepidermoid carcinoma) 73; additional examples are the chromosomal inversions and translocations leading to RET-PTC1, RET-PTC2, and RET-PTC3 fusions that are associated with papillary thyroid carcinoma 74. All of these translocations can be seen as sole cytogenetic abnormalities or in a background of extensive aneuploidy. Therefore, it is clear that recurrent, balanced translocations resulting in oncogenic fusion genes can occur in epithelial tumors. However, despite the fact that the common epithelial malignancies (lung, colon, breast) are far more frequent than hematologic malignancies, reports of recurrent, balanced translocations that generate oncogenic fusion genes in patients with these common epithelial malignancies are exceedingly rare 75. The reasons for the dearth of balanced, recurrent translocations in epithelial malignancies are not clear, and have been the source of much controversy and speculation 2,76.
BOX 2 Clinical utility of chromosomal translocations.
The clinical importance of recurrent chromosomal translocations in patients with hematologic malignancy has been underscored by the use of these translocations to tailor treatment for leukemia and lymphoma patients. For instance, almost all CML patients with a t(9;22) translocation show dramatic responses to imatinib, a newly developed tyrosine kinase inhibitor 77, and treatment of those acute myeloid leukemia (AML) patients that have a t(15;17) translocation with all trans retinoic acid results in a complete remission in a high percentage of patients 3. These are specific, targeted therapies, directed toward a tyrosine kinase fusion (BCR-ABL) and a retinoic acid receptor fusion (PML-RARA) respectively. Although these specific targeted therapies are only available for a subset of leukemia patients with specific chromosomal abnormalities, it has been noted that patients with certain translocations respond differently to conventional cytotoxic chemotherapy. For instance, AML patients with an inv(16) or t(8;21) respond better to high dose cytarabine than do patients without an inv(16) or t(8;21) 78. As a result of these studies, cytogenetic and molecular genetic findings are now an integral factor in determining chemotherapy regimens for most patients with acute leukemia.
TABLE 1.
Partial list of recurrent chromosomal translocations associated with leukemia
| Disease | Chromosomal abnormality | Activated gene(s) | Mechanism of activation |
|---|---|---|---|
| LYMPHOID | |||
| B-cell ALL | t(8;14)(q24;q32) | MYC | Relocation to IGH locus |
| t(2;8)(p12;q24) | MYC | Relocation to IGK locus | |
| t(8;22)(q24;q11) | MYC | Relocation to IGL locus | |
| B-cell precursor ALL | t(12;21)(p12;q22) | TEL-AML1 fusion | Gene fusion |
| B-cell precursor ALL | t(1;19)(q23;p13) | E2A-PBX1 fusion | Gene fusion |
| B-cell precursor ALL | t(17;19)(q22;p13) | E2A-HLF fusion | Gene fusion |
| t(4;11)(q21;q23) | MLL-AF4 fusion | Gene fusion | |
| pre-T LBL | t(8;14)(q24;q11) | MYC | Relocation to TCRA/D locus |
| t(7;19)(q35;p13) | LYL1 | Relocation to TCRB locus | |
| t(1;14)(p32;q11) | SCL | Relocation to TCRA/D locus | |
| t(14;21)(q11;q22) | OLIG2 | Relocation to TCRA/D locus | |
| t(11;14)(p15;q11 ) | LMO1(RBTN1) | Relocation to TCRA/D locus | |
| t(11;14)(p13;q11) | LMO2(RBTN2) | Relocation to TCRA/D locus | |
| t(10;14)(q24;q11) | HOX11 | Relocation to TCRA/D locus | |
| t(5;14)(q35;q32) | HOX11L2 | ? | |
| t(10;11)(p13;q21) | CALM-AF10 fusion | Gene fusion | |
| t(4;11)(q21;p15) | NUP98-RAP1GDS1 fusion | Gene fusion | |
| MYELOID | |||
| APL | t(15;17)(q21;q21) | PML-RARA fusion | Gene fusion |
| t(11;17)(q23;q21) | PLZF-RARA fusion | Gene fusion | |
| AML | #t(11;v)(q23;v) | MLL | Gene fusion |
| AML, CMML | #t(12;v)(p13;v) | ETV6 | Gene fusion |
| AML | #t(11;v)(p15;v) | NUP98 | Gene fusion |
| AML | t(8;21)(q22;q22) | AML1-ETO fusion | Gene fusion |
| AML | inv(16)(p13;q22) | CBFB-MYH11 fusion | Gene fusion |
| AML | t(16;21)(p11;q22) | FUS-ERG fusion | Gene fusion |
| AML | t(5;14)(q33;q32) | CEV14-PDGFRB fusion | Gene fusion |
| AML | t(8;22)(p11;q13) | P300-MOZ fusion | Gene fusion |
| AML | t(6;9)(p23;q34) | DEK, NUP214 fusion | Gene fusion |
| AMKL | t(1;22)(p13;q13) | RBM15, MKL fusion | Gene fusion |
| AML | t(3;21)(q26;q22) | AML1-EV11 fusion | Gene fusion |
| CML | t(9;22)(q34;q11) | BCR, ABL fusion | Gene fusion |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; pre-T LBL, pre-T cell lymphoblastic leukemia/lymphoma; APL, acute promyelocytic leukemia; AMKL, acute megakaryoblastic leukemia; CML, chronic myelogenous leukemia; CMML, chronic myelo-monocytic leukemia
# “v” represents any of more than 10 chromosomal loci
A large number of recurrent, balanced chromosomal translocations associated with hematologic malignancy have been cloned and characterized, and several recurrent themes have been identified. First, specific chromosomal translocations are often associated with specific subtypes of leukemia or lymphoma. For example, the t(15;17) is found only in the leukemic cells of patients with acute promyelocytic leukemia (APL) and not other forms of leukemia 3, and the t(1;19) is found only in the leukemic cells of patients with B-cell precursor acute lymphoblastic leukemia (ALL) 4. However, there are numerous exceptions to this rule. For instance, the t(4;11) translocation occurs in patients with both acute myelocytic leukemia (AML) and ALL 5 and the t(9;22) is associated with both chronic myelocytic leukemia (CML) and ALL. Nonetheless, the consistent association of specific translocations with specific leukemic phenotypes strongly supports the hypothesis that these non-random chromosomal translocations are causal events during leukemic transformation.
Analysis of the genes involved in chromosomal translocations reveals a second theme. In general, the translocations produce one of two events at a molecular level (Figure 2). Two distinct normal genes, such as BCR and ABL, may be joined and generate a chimeric BCR-ABL fusion protein. Alternately, unscheduled expression of a normal gene product, such as SCL, may result from replacement of normal SCL regulatory elements with those derived from the translocation partner, such as the T-cell receptor δ (TCRD) enhancer 6.
Figure 2. Molecular consequences of chromosomal translocation.
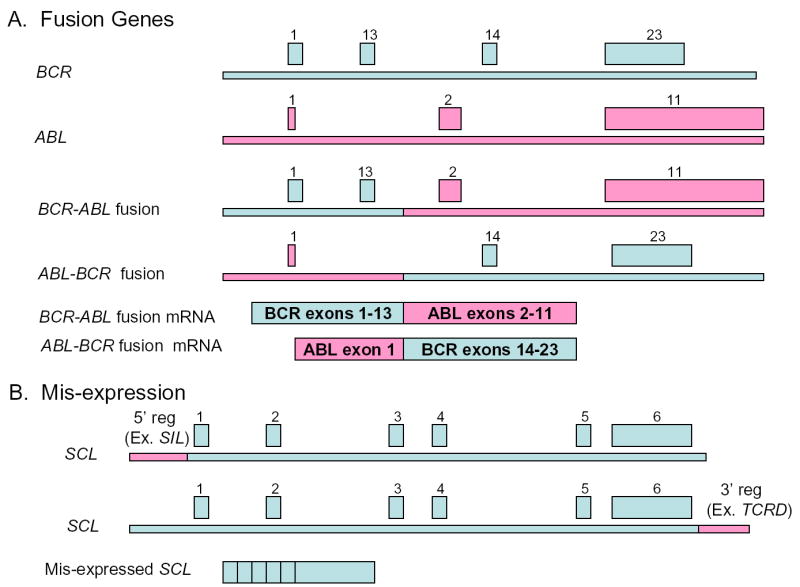
A. Production of a chimeric fusion gene by chromosomal translocation. In this example, the translocation breakpoints occur between exons 13 and 14 of BCR, and exons 1 and 2 of ABL. Two reciprocal fusion genes are formed, with exon 13 of BCR fused to exon 2 of ABL (BCR-ABL fusion), and exon 1 of ABL fused to exon 14 of BCR (ABL-BCR fusion). One of the two reciprocal transcripts is often not expressed. Exons 2–12 and 15–22 of BCR, and exons 3–10 of ABL are not shown for the sake of clarity. B. Mis-expression of a normal gene. Either 5′ or 3′ regulatory sequences (for example, 5′ SIL regulatory sequences or 3′ TCRD enhancer sequences respectively) can be fused to the coding sequence of a growth-regulating gene such as SCL. SCL expression is now governed by the regulatory sequences of the translocation partner (Either SIL or TCRD in these examples). SCL exons 1–6 are shown..
A third theme emerges from analysis of the function(s) of the genes located at translocation breakpoints. These genes often encode transcription factors that are known to be important for hematopoietic differentiation, or tyrosine kinases that are important regulators of cell proliferation 1,7. Moreover, it is not uncommon that a leukemia cell will have two translocations; one translocation that generates an aberrant transcription factor, resulting in impaired hematopoietic differentiation, and a second translocation that activates a tyrosine kinase, leading to uncontrolled cellular proliferation. However, only rarely will a leukemic cell have two translocations, each of which impairs differentiation or increases cellular proliferation. These observations have led to the hypothesis that at least two complementary events, one leading to a block in differentiation, and the second leading to hyperproliferation, are required for leukemic transformation 8.
Finally, a number of studies have identified leukemogenic chromosomal translocations, including fusions between IGH and BCL2, BCR and ABL, TEL and AML1, in the peripheral blood of healthy individuals 9. These results were initially quite surprising and controversial, as it had been assumed for decades that these translocations inevitably led to leukemia. In general, these studies have used PCR, often nested, to identify either a fusion mRNA or genomic DNA fusion produced by the translocation. There are important implications of these results. First, they strongly suggest that these oncogenic translocations, some of which have been shown to occur in immature progenitor cells 10, are not sufficient to produce a frank malignancy, but they instead produce a “pre-malignant” clone that requires additional, complementary, events to fully transform the cell. These results demonstrate that detection of an oncogenic translocation is not equivalent to detection of a malignancy, which has important ramifications for clinicians using PCR-based assays to identify small numbers of remaining malignant cells (minimal residual disease, or MRD) in leukemia patients whose disease is in a clinical remission 11. Second, these findings provide a potential explanation for the observation that mice engineered to overexpress an oncogenic fusion protein often do not develop overt leukemia. In these mice, one oncogenic mutation is placed in the mouse germline, but leukemic transformation is not apparent until complementary mutation(s) occur spontaneously as the mouse ages. Although most of these putative spontaneous mutations have not been identified, our lab has recently identified very frequent activating Notch1 mutations that collaborate to induce pre-T LBL in SCL transgenic mice (Y-W. Lin and P.D. Aplan, unpublished data).
Since chromosomal translocations are important events leading to malignant transformation, a better understanding of the causes of these translocations is clearly important in order to understand the general causes of cancer. The discussion which follows will review what is known about host factors that predispose to leukemogenic translocations, and the molecular events that might lead to chromosomal translocation.
Inherited predisposition
A number of inherited syndromes predispose individuals to hematologic malignancy; some of these syndromes are associated with genetic instability and an increased incidence of chromosomal translocations. For instance, patients with ataxia-telangiectasia (AT), which is due to mutation of the ATM gene that plays an important role in the recognition and repair of DNA double strand breaks (DSB), are prone to developing chromosomal translocations that typically involve the T-cell or immunoglobulin antigen receptor loci 12. In addition, mice deficient for the ATM protein develop pre-T lymphoblastic leukemia/lymphoma (pre-T LBL); these tumors also show chromosomal translocations that involve antigen receptor genes 13. Patients with Nijmegen’s breakage syndrome (NBS), which is caused by a mutation in the NBS1 gene that, like ATM, is involved in the repair of DNA DSB, have features similar to patients with AT, including a predisposition to chromosomal translocation and lymphoid malignancies 14. A common mechanism can be implicated in these translocations, namely, illegitimate V(D)J recombination (see below).
Individuals with inherited DNA repair defects, such as Bloom’s syndrome, Fanconi’s anemia, and Li-Fraumeni syndrome are predisposed to a wide spectrum of malignancies, including leukemias and lymphomas 15. Although lymphocytes from patients with Bloom’s syndrome and Fanconi’s anemia are clearly susceptible to chromosomal breakage in vitro, the hematologic malignancies that develop in patients with Bloom’s syndrome and Fanconi’s anemia sometimes, but not always, show chromosomal translocations 16.
Molecular mechanisms
At a most general level, a chromosomal translocation involves a DNA DSB and repair, more specifically, mis-repair. Based largely on the study of translocation breakpoint sequences as well as in vitro and in vivo model systems (Box 3), several specific mechanisms have been proposed to explain the recurrent, balanced chromosomal translocations seen in leukemic cells. These proposed mechanisms include i) illegitimate V(D)J or switch recombination, ii) homologous recombination mediated by repetitive sequences, such as Alu or LINE elements, iii) DNA topoisomerase II subunit exchange, and iv) error prone non-homologous end-joining (NHEJ) following DNA DSB particularly involving regions of the genome that show increased susceptibility to DNA DSB. Several DNA sequence features have been proposed to increase the likelihood of DNA DSB, including purine/pyrimidine repeat regions, scaffold/matrix attachment regions (S/MARs), and preferential DNA topoisomerase II cleavage sites. It is important to note that these proposed mechanisms are not mutually exclusive; for instance, a translocation breakpoint may show evidence of cleavage at a V(D)J heptamer sequence on one side, and cleavage at a purine/pyrimidine repeat at the other side of the breakpoint 17.
BOX 3 Model organisms and systems.
Numerous insights into the process of malignant transformation have been made through the study of chromosomal translocations in the malignant cells of cancer patients. However, these studies are often descriptive, and, for the most part, study patients with malignant disease, which can be viewed as the end result of one or more mutations. Therefore, investigators have turned to tractable model systems to study the events which might lead or predispose to chromosomal translocation. Several laboratories have used gene targeting experiments to demonstrate that genes important for NHEJ and the repair of DNA DSB help prevent oncogenic chromosomal translocations 50. Other laboratories have used the rare-cutting endonuclease I-SceI to produce DNA DSB in cultured cells. An elegant experimental system has demonstrated that chromosomal translocations can be mediated via HR or SSA in mouse embryonic stem (ES) cells 34,79. In this system, two defective, complementary, neomycin resistance cassettes with recognition sequences for the rare-cutting I-SceI restriction endonuclease are integrated on different chromosomes in ES cells. An I-SceI expression vector is then introduced into the ES cells, and rare translocations between the two defective neomycin resistance cassettes, mediated via HR or SSA, can be selected with the neomycin analogue G418. Of note, two DNA DSB were required to produce HR-mediated translocations in this system; when a single DNA DSB was produced, no chromosomal translocations were detected 79. The budding yeast S. cerivisiae has also been used to study gross chromosomal rearrangements (GCRs), including chromosomal translocations. In these experiments, investigators used a dual selection system to identify yeast cells that had retained one selectable marker, but lost a second selectable marker. They found deletions, inversions, telomere additions, and translocations 80. Some of the translocations showed the hallmarks of HR, whereas others were mediated via NHEJ. In a follow-up series of studies, they demonstrated that yeast mutant for various DNA repair or checkpoint enzymes could have elevated production of GCRs (200x baseline), and double mutants could have markedly elevated production of GCRs (30,000x baseline) 80,81.
Spatial proximity
For any chromosomal translocation to occur, the two chromosomes involved must come in contact with one another. This observation has led to the hypothesis that loci involved in recurrent translocations might be in close proximity to one another in the interphase nucleus Although there has been considerable discussion regarding the optimal design of experiments to investigate this hypothesis, centered principally on how to best measure proximity, and the selection of control or comparison loci, recent studies have produced data that support this hypothesis. Both the BCL6 and MYC loci, which are frequently translocated to the IGH loci in B-cell malignancies, were found to be close to the IGH loci in B-cells 18, and the ABL and BCR loci were found to be close to one another in CD34+ bone marrow cells 19. In addition, the RET and PTC1 loci, involved in a chromosomal inversion in patients with papillary thyroid cancer, were found to be closer to one another in thyroid cells than in lymphocytes 20. The frequency of RET-PTC1 fusions increased after exposure to ionizing radiation, leading to the speculation that proximity of the two loci in the interphase nucleus enhanced radiation-induced recombination of these two loci 21.
Illegitimate V(D)J recombination and class switch recombination
In many cases, chromosomal translocation in lymphoid malignancies seem to be the result of mistakes in normal V(D)J or immunoglobulin class switch recombination (CSR) 22–24 (Figure 3). Typically, the translocations attributed to illegitimate V(D)J recombination juxtapose a proto-oncogene to a locus that codes for an antigen receptor (either an immunogolobulin or a T-cell receptor locus). The proto-oncogene present on the derivative chromosome becomes activated through immunoglobulin or T-cell receptor gene regulatory regions. The assertion that these translocations are the result of illegitimate V(D)J recombinase activity is strengthened by the presence of features typically associated with normal V(D)J recombination, including site-specific DNA cleavage at cryptic heptamer sequences, and non-templated (“N” region) nucleotide addition at the translocation breakpoints 25. There are a number of specific examples of chromosomal translocations where some or all of these features are present (for review see 22).
Figure 3. Chromosomal translocation mediated via V(D)J recombination or class switch recombination (CSR).
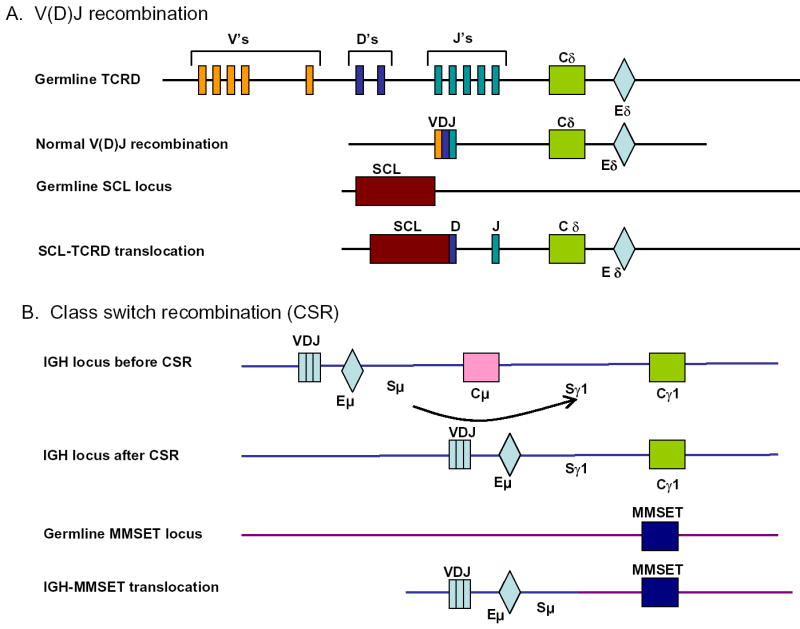
A. In the germline TCRD, numerous V (variable), D (diversity), and J (joining) segments are dispersed over hundreds of kb. Each discrete V, D, or J segment is flanked by a heptamer-nonamer recombination signal sequence. A single C (constant) segment is present, as is a 3′ enhancer (E). Normal V(D)J recombination occurs through reconfiguration of the genomic DNA, with V, D, and J segments becoming spliced together as shown. A cryptic recombination signal sequence within the SCL locus can be recognized by the V(D)J recombinase complex, leading to an SCL-TCRD translocation. A fusion mRNA is formed between SCL (in the 3′untranslated region) and TCRD; SCL expression is now governed by the TCRD enhancer (Eδ). Although TCRD and SCL are shown in this example, translocations involving all antigen receptor loci (except TCRG) have been reported. B. Prior to CSR, IgM is produced as the VDJ segment of the mRNA transcript splices to Cμ. The recombination is mediated via switch (S) regions 5′ of the C regions. The μ and γ subscripts designate immunoglobulin classes. Following CSR, IgG is produced, as the VDJ segment of the mRNA transcript splices to Cγ1. An IGH-MMSET translocation is caused when the switch recombination occurs between a region within the MMSET gene and the Sμ region. MMSET expression is now governed by the immunoglobulin heavy chain enhancer (Eμ). Class switch recombination occurs only at the IGH locus and not at other antigen receptor loci. MMSET is shown as an example, but many other genes have been shown to be fused to the IGH locus via CSR.
In addition, the site-specific recombination event between the SIL and SCL (TAL1) genes is a common feature in leukemic cells from patients with pre-T LBL, and shows all of the aforementioned hallmarks of normal V(D)J recombinase activity. This recombination event replaces the SCL promoter region with that of the SIL promoter, and results in inappropriate expression of SCL, under the control of SIL regulatory elements 26. Since neither SIL nor SCL encode antigen receptor genes, this recombination event demonstrates that oncogenic recombination events caused by illegitimate V(D)J recombinase activity are not restricted to genes encoding antigen receptors.
Translocations caused by mistakes in CSR were initially identified when investigators noted immunoglobulin switch regions joined to the MYC gene in both Burkitt’s lymphoma patients and mouse plasmacytoma cells. More recently, a large number of different translocations involving the immunoglobulin switch region have been cloned in cells from patients with multiple myeloma 24. The mechanism underlying these switch region translocations is thought to involve mistakes in normal CSR. CSR, as well as somatic hypermutation, is mediated, at least in part, by the activation-induced cytidine deaminase (AID) enzyme. AID induces deamination of cytidine residues, leading to dU:dG mismatches. The uracil base is then removed by uracil DNA glycosylase (UNG), creating an abasic site in the DNA strand. This abasic site is then cleaved by an AP endonuclease, resulting in a DNA strand nick. The nicked DNA may then initiate recombination directly, or a second nick within a downstream switch region on the opposite DNA strand would lead to a DNA DSB. However, if the DNA DSB at the two switch regions recombined with a DNA DSB on a non-homologous chromosome, the end result would be a balanced chromosomal translocation rather than an interstitial deletion. For example, the BCL6 gene which is known to be subject to hypermutation in lymphoid cells is also frequently involved in chromosomal translocations into the immunoglobulin switch region 27. Therefore, AID activity can result in hypermutation of the BCL6 and IGH loci or chromosomal translocation involving these same loci. Although an alternate, AID-independent pathway has recently been proposed based on finding recombination involving Igh and Myc genes in Aid deficient mice 28, these results have not been confirmed by others 29.
Homologous recombination and single-strand annealing
Homologous recombination (HR) between repetitive Alu elements is thought to be a common mechanism for recombination in meiotic cells. It seems likely that imperfect exchange during meiosis may be responsible for chromosomal rearrangements associated with non-malignant diseases such as adenosine deaminase deficiency 30. However, there is little convincing evidence for Alu-mediated exchange of chromosomal arms as a cause of acquired chromosomal translocations in malignant disease. Alu elements have been implicated in the etiology of some complex translocations involving BCR and ABL, but these are quite rare 31. The most convincing evidence for Alu-mediated oncogenic rearrangements in leukemic cells involves not a translocation but an intra-chromosomal event that leads to a partial tandem duplication of MLL; in one series, seven of nine patients with a partial tandem duplication had undergone a rearrangement within Alu elements 32. Single-strand annealing (SSA), like HR, is a conservative mode of DNA DSB repair, that takes place via annealing of DNA strands generated after resection at the DNA DSB 33. A recent report using an experimental approach similar to that described in Box 3 demonstrated that chromosomal translocations can be mediated via Alu repeats in mouse embyronic stem cells 34.
Therapy-related AML (t-AML), topoisomerase II poisons, and translocations
Therapy-related AML (t-AML, sometimes called secondary AML) has been noted to develop following treatment for a primary malignancy. In many cases, the treatment for the primary malignancy employed chemotherapeutic agents that targeted DNA topoisomerase (topo) II 35. The topo II poisons (also referred to as topo II inhibitors) most significantly associated with these t-AMLs are the epipodophylotoxins such as etoposide (VP-16) and teniposide (VM-26), although anthracyclines such as daunorubicin and doxorubicin have also been implicated in t-AML 35. The t-AMLs that occur following treatment with topo II poisons often demonstrate balanced chromosomal translocations involving one of a few genes. The most common chromosomal region involved is 11q23 35, although recurrent breakpoints involving 21q22, 17q21, 16p13, and 11p15 have also been reported following therapy with topo II poisons 36–38. These observations have led to the speculation that treatment with topo II poisons can directly induce chromosomal translocations. This speculation is supported by the observation that treatment of peripheral blood lymphocytes in vitro with etoposide can induce chromosomal translocations 39, as well as a preliminary study 40 which suggests that in utero exposure to certain pesticides that are weak topo II poisons might be associated with translocations involving the MLL locus (Box 4).
BOX 4 In utero origin of some oncogenic translocations.
Two lines of investigation have clearly demonstrated that some oncogenic translocations occur in utero. Studies of monozygotic twins with leukemia and translocations involving the MLL gene, which are particularly prevalent in infants with leukemia, demonstrated identical MLL breakpoints in the individual twins, indicating that the leukemia had developed in one twin and “metastasized”, through the shared placenta, to the second twin 82. This phenomenon has also been demonstrated for twins with TEL-AML1 fusions 82. Of note, whereas twins with MLL translocations typically present as infants, within the first year of life, twins with TEL-AML1 translocations develop clinical disease later, as old as age 14 82,83. This observation suggests that the MLL fusions may be “more” leukemogenic than TEL-AML1 fusions. It is conceivable that MLL fusions, which can inhibit TP53 function 84 might create a “mutator phenotype” that predisposes patients to developing complementary mutations. Alternatively, MLL fusions may activate a large number of downstream genes and pathways, some blocking differentiation, some activating proliferation, and some blocking apoptosis, in effect copying the impact of several independent mutations. A second line of investigation that has demonstrated the presence of oncogenic fusions in utero stems from the study of newborn screening or “Guthrie” cards 83,85. Shortly after birth, several drops of blood are routinely collected from infants and placed on blotter paper (a “Guthrie” card). These cards have traditionally been used for the detection of inherited diseases, such as sickle cell anemia or phenylketonuria. The cards are often archived, in case a positive screen needs to be verified. When a child develops leukemia with a specific translocation, investigators can now retrieve the Guthrie card and determine, by PCR-based assays, whether the translocation was present at birth. In this manner, MLL-AF4, TEL-AML1, PML-RARA, and CBFB-MYH11 fusions have been detected in clinically healthy newborn infants 83,86. However, a pre-natal origin for childhood leukemia is not universal. For instance, there is no evidence for detection of E2A-PBX1 fusions in infants who later develop leukemia with an E2A-PBX1 fusion83.
The basic molecular mechanism(s) which generate these balanced translocations in patients with t-AML have not been completely defined. DNA topoisomerase II normally functions as a homodimer, and catalyzes a complex reaction consisting of double strand DNA cleavage, strand passage, and DNA religation 41. During this reaction, a short-lived intermediate which consists of topo II monomers covalently bound to the DNA phosphodiester backbone is stabilized by topo II poisons; these intermediates are recognized as damaged DNA, triggering apoptotic cell death. It has been proposed that a topo II “subunit exchange”, in which topo II monomers (subunits) that are covalently bound to DNA exchange partners, might occur, leading to a chromosomal translocation 42,43. DNA recombination events consistent with this hypothesis have been detected in vitro 44 and in tissue culture cells treated with etoposide 42. In addition, the leukemic cells of patients with t-AML have been noted to have precise inter-chromosomal exchanges consistent with such a mechanism, initially in translocations involving NUP98 and TOP143, and more recently with translocations involving MLL and AF9 45 and PML and RARA 46.
Mis-repair of DNA double-strand breaks
It has been estimated that approximately 50 DNA DSB occur during a typical mammalian cell division cycle 47. Since homologous recombination is only available during S and G2/M phases of the cell cycle, NHEJ is the principal mechanism for repair of DNA DSB, especially during G0 and G1 phases of the cell cycle. One could envision an increased frequency of chromosomal aberrations, including translocations, if either NHEJ were impaired or the frequency of DNA DSBs were increased. A detailed review of NHEJ is beyond the scope of this overview, and the reader is referred to recent reviews of NHEJ for additional background 48,49
Several lines of evidence support the hypothesis that impaired NHEJ is associated with increased frequency of chromosomal aberrations, including translocations. Patients with Nijmegen breakage syndrome, which is caused by a mutation in NBS1, a component of the NHEJ repair pathway, have an increased incidence of chromosomal rearrangements and cancer 14. Mice that are deficient in one of several components of the NHEJ pathway, including Ku70, DNA-PKcs, and DNA Ligase IV are prone to developing lymphoma with complex chromosomal translocations, most commonly involving antigen receptor genes and Myc 50. In addition, mice that are deficient for H2ax, a histone H2 variant that is phosphorylated on serine residues in the region of DNA DSBs, also develop chromosomal translocations that involve antigen receptor genes and Myc 51,52. Finally, analysis of both derivative chromosomes involved in a balanced translocation has identified features consistent with repair mediated via an NHEJ pathway at chromosomal translocation breakpoints in patients with leukemia and sarcomas 53,54. These features include short (1–6 nucleotides) stretches of microhomology between the two translocated chromosomes, direct and indirect repeats, short deletions, and insertions at the breakpoints.
Recombination “hot-spots”
Certain DNA sequences are thought to be “recombinogenic”, and prone to DNA DSB. These include purine/pyrimidine repeat regions, scaffold/matrix attachment regions, and preferred topoisomerase II cleavage sites. Several chromosomal translocation breakpoints identified in patients with ALL have been mapped within or near extended (200+ consecutive bp) tracts of alternating purine and pyrimidine residues (Pu/Py tracts) 55,56. In solution, DNA containing Pu/Py tracts can form an unusual left-handed helical structure known as Z-DNA. In vivo, Z-DNA segments are preferentially located at inter-nucleosomal regions, presumably because of the higher energy cost of compacting the more rigid Z-DNA into nucleosomes 57. Thus, it is conceivable that Z-DNA located in inter-nucleosomal regions has an increased susceptibility to DNA DSB compared to histone bound nucleosomal DNA, and is therefore more likely to be involved in DNA recombination events. This speculation is supported by the observations that these types of repeat regions are susceptible to “slippage” during DNA replication.58.
Breakpoints in the MLL, AML1, and AF9 loci are clustered in specific regions within these genes (breakpoint cluster regions or bcr). Several investigators have noted that scaffold or matrix attachment regions (S/MARs) co-localize with these bcrs 59–61. Moreover, these same bcrs have been shown to be preferred cleavage sites during early stages of apoptosis 62,63, when genomic DNA is cleaved to high molecular weight fragments on the order of 50–100 kb 64. This cleavage to high molecular weight DNA fragments precedes the oligonucleosomal fragmentation that is a hallmark of the final stages of apoptosis 64. The co-localization of S/MARs and apoptotic cleavage sites is directed by sequences within the MLL bcr, as episomal vectors containing the MLL bcr also function as S/MARs, and show the same preferential cleavage during apoptosis 60. These observations, that bcrs often map to S/MARs, and show increased susceptibility to apoptotic endonucleases, have led to the provocative hypothesis that aborted apoptosis could lead to chromosomal rearrangements 62,65. This hypothesis suggests that cells might abort an apoptotic program after cleavage at the S/MARs had begun. The cells would then need to repair the cleaved DNA, potentially producing DNA rearrangements during error-prone NHEJ mediated mis-repair. In support of this hypothesis are recent papers suggesting that apoptosis can be reversed in C elegans 66,67, and studies showing production of chromosomal rearrangements involving the MLL and AML1 loci triggered by apoptotic endonucleases 68,69.
As mentioned above, the precise molecular mechanisms that lead to chromosomal translocations and therapy-related leukemia following therapy with topo II poisons remain poorly understood. Several investigators have noted that the breakpoint junctions in the leukemic cells of patients with t-AML display features that are consistent with repair by NHEJ, such as microhomologies, deletions, and inversions 70,71. In addition, some of these breakpoint junctions were shown to be preferred sites for topo II cleavage in vitro 46, suggesting that mis-repair of topo II induced DNA DSBs by NHEJ could result in chromosomal translocation.
Summary
Chromosomal translocations have been shown to be causal events involved in the development of many forms of malignancy; the clinical utility of these translocations is underscored by their use in devising risk-appropriate treatment strategies. The study of chromosomal translocations has revealed several recurring themes, and yielded important insights into the process of malignant transformation. However, the mechanism(s) which cause these translocations remain elusive. A more thorough understanding of the mechanisms that cause chromosomal translocations will be aided by continuing characterization of translocation breakpoints, and by developing in vitro and in vivo model systems that can generate chromosomal translocations.
BOX Outstanding questions.
Do non-random, recurrent chromosomal translocation breakpoints represent sites within the genome that are “fragile”, and highly susceptible to breakage and religation, or do these breakpoints simply represent sites near growth promoting proto-oncogenes, whose deregulation gives cells a growth advantage?
Why are the balanced chromosomal translocations typically associated with specific hematopoietic cell types?
At what phase of the cell division cycle do chromosomal translocations most often take place?
Why is it that hematologic malignancies are often pseudodiploid (i.e., demonstrate a balanced translocation, but no other numerical or structural abnormalities), whereas epithelial tumors are typically aneuploid, with a large number of structural and numerical abnormalities?
A small minority of patients treated with genotoxic chemotherapy agents develop t-AML with specific chromosomal translocations. Do these patients have subtle DNA repair defects, and/or polymorphisms of enzymes involved in drug metabolism?
Should newborn infants routinely be screened for the presence of oncogenic chromosomal translocations?
Acknowledgments
I would like to thank Drs. W. Michael Kuehl, Ying Wei Lin, Tamas Varga, and Sharon Pine for their critical review of this manuscript. I would also like to thank Dr. Thomas Ried and Hesed Padilla-Nash for the spectral karyotype experiments shown in figure 2. I apologize in advance for authors whose work I was unable to cite due to space limitations.
GLOSSARY BOX
- Actue lymphoblastic leukemia (ALL)
An acute malignancy of lymphocytes, characterized by >25% malignant cells in the bone marrow. There are three major subtypes, B-cell ALL, T-cell ALL (also referred to as pre-T LBL), and “common” ALL. Common ALL is in fact a malignancy of immature B-cells, and is now referred to as B-cell precursor ALL (or pre-B LBL).
- Acute myelogenous leukemia (AML)
An acute malignancy of the hematopoietic system, typified by >25% malignant cells in the bone marrow. There are at least eight major subtypes, traditionally designated M0-M7. These subtypes include acute undifferentiated leukemia, acute promyelocytic leukemia, acute monocytic leukemia, acute erythroblastic leukemia, and acute megakaryocytic leukemia.
- Balanced translocation
A chromosomal translocation that has no substantial net gain or loss of DNA, also referred to as a reciprocal translocation. Note that a translocation that appears to be balanced at a light microscopy (i.e., cytogenetic) level may have losses of small amounts of genomic DNA detected when it is molecularly cloned.
- Class switch recombination (CSR)
A normal feature of B-cell development, this leads to the replacement of one constant (C) region gene segment with another.
- Chromosomal translocation
A chromosomal rearrangement in which part of a chromosome is detached by breakage and subsequently joined to a non-homologous chromosome.
- Chronic myelocytic leukemia (CML)
A chronic malignancy of the hematopoietic system.
- Guthrie cards
named for Dr. Robert Guthrie, these are absorbent strips of paper onto which several drops of peripheral blood (usually obtained by lancing the heel) from newborn infants are placed, and used for disease screening.
- Hematologic malignancy
a form of cancer involving the blood and lymphatic systems, including leukemia, lymphoma, and multiple myeloma.
- Hypermutation
regions of the genome that show a markedly increased frequency in mutation rate, usually in lymphocytes.
- Leukemia
a malignancy of the hematopoietic system
- Lymphoma
a malignancy of lymphocytes
- Non-homologous end joining (NHEJ)
a DNA repair mechanism in which two non-homologous DNA ends are repaired by ligation to one another
- Precursor T-cell leukemia/lymphoma (pre-T LBL)
A malignancy of T-lymphocyte precursors. This designation includes diseases also known at T-cell ALL and T-cell lymphoblastic lymphoma.
- Proto-oncogene
a normal cellular gene whose activation is linked to malignant transformation
- Scaffold/matrix attachment (or associated) region (S/MAR)
Segments of genomic DNA preferentially associated with the proteinaceous material left in the nucleus after high salt extraction.
- V(D)J recombination
the process by which discontiguous V (variable), D (diversity), and J(joining) segments become joined in lymphocytes, leading to a rearrangement of the germline DNA in these cells
References
- 1.Rabbitts TH. Chromosomal translocation master genes, mouse models and experimental therapeutics. Oncogene. 2001;20 (40):5763–5777. doi: 10.1038/sj.onc.1204597. [DOI] [PubMed] [Google Scholar]
- 2.Mitelman F, et al. Prevalence estimates of recurrent balanced cytogenetic aberrations and gene fusions in unselected patients with neoplastic disorders. Genes Chromosomes Cancer. 2005;43 (4):350–366. doi: 10.1002/gcc.20212. [DOI] [PubMed] [Google Scholar]
- 3.Chou WC, Dang CV. Acute promyelocytic leukemia: recent advances in therapy and molecular basis of response to arsenic therapies. Curr Opin Hematol. 2005;12 (1):1–6. doi: 10.1097/01.moh.0000148552.93303.45. [DOI] [PubMed] [Google Scholar]
- 4.Hunger SP. Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood. 1996;87 (4):1211–1224. [PubMed] [Google Scholar]
- 5.Rubnitz JE, et al. 11q23 rearrangements in acute leukemia. Leukemia. 1996;10 (1):74–82. [PubMed] [Google Scholar]
- 6.Begley CG, Green AR. The SCL gene: from case report to critical hematopoietic regulator. Blood. 1999;93 (9):2760–2770. [PubMed] [Google Scholar]
- 7.Rowley JD. Chromosome translocations: dangerous liaisons revisited. Nat Rev Cancer. 2001;1 (3):245–250. doi: 10.1038/35106108. [DOI] [PubMed] [Google Scholar]
- 8.Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002;1 (5):417–420. doi: 10.1016/s1535-6108(02)00081-8. [DOI] [PubMed] [Google Scholar]
- 9.Janz S, et al. Lymphoma- and leukemia-associated chromosomal translocations in healthy individuals. Genes Chromosomes Cancer. 2003;36 (3):211–223. doi: 10.1002/gcc.10178. [DOI] [PubMed] [Google Scholar]
- 10.Mori H, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. 2002;99 (12):8242–8247. doi: 10.1073/pnas.112218799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izraeli S, Waldman D. Minimal residual disease in childhood acute lymphoblastic leukemia: current status and challenges. Acta Haematol. 2004;112 (1–2):34–39. doi: 10.1159/000077558. [DOI] [PubMed] [Google Scholar]
- 12.Rotman G, Shiloh Y. ATM: from gene to function. Human Molecular Genetics. 1998;7 (10):1555–1563. doi: 10.1093/hmg/7.10.1555. [DOI] [PubMed] [Google Scholar]
- 13.Liyanage M, et al. Multicolour spectral karyotyping of mouse chromosomes. Nature Genetics. 1996;14 (3):312–315. doi: 10.1038/ng1196-312. [DOI] [PubMed] [Google Scholar]
- 14.Digweed M, Sperling K. Nijmegen breakage syndrome: clinical manifestation of defective response to DNA double-strand breaks. DNA Repair (Amst) 2004;3 (8–9):1207–1217. doi: 10.1016/j.dnarep.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Horwitz M. The genetics of familial leukemia. Leukemia. 1997;11 (8):1347–1359. doi: 10.1038/sj.leu.2400707. [DOI] [PubMed] [Google Scholar]
- 16.Maarek O, et al. Faconi anemia and bone marrow clonal chromosome abnormalities. Leukemia. 1996;10 (11):1700–1704. [PubMed] [Google Scholar]
- 17.Boehm T, et al. Alternating purine-pyrimidine tracts may promote chromosomal translocations seen in a variety of human lymphoid tumours. EMBO Journal. 1989;8 (9):2621–2631. doi: 10.1002/j.1460-2075.1989.tb08402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roix JJ, et al. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat Genet. 2003;34 (3):287–291. doi: 10.1038/ng1177. [DOI] [PubMed] [Google Scholar]
- 19.Neves H, et al. The nuclear topography of ABL, BCR, PML, and RARalpha genes: evidence for gene proximity in specific phases of the cell cycle and stages of hematopoietic differentiation. Blood. 1999;93 (4):1197–1207. [PubMed] [Google Scholar]
- 20.Nikiforova MN, et al. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290 (5489):138–141. doi: 10.1126/science.290.5489.138. [DOI] [PubMed] [Google Scholar]
- 21.Caudill CM, et al. Dose-dependent generation of RET/PTC in human thyroid cells after in vitro exposure to gamma-radiation: a model of carcinogenic chromosomal rearrangement induced by ionizing radiation. J Clin Endocrinol Metab. 2005;90 (4):2364–2369. doi: 10.1210/jc.2004-1811. [DOI] [PubMed] [Google Scholar]
- 22.Tycko B, Sklar J. Chromosomal translocations in lymphoid neoplasia: a reappraisal of the recombinase model. Cancer Cells. 1990;2 (1):1–8. [PubMed] [Google Scholar]
- 23.Chatterji M, et al. New concepts in the regulation of an ancient reaction: transposition by RAG1/RAG2. Immunol Rev. 2004;200:261–271. doi: 10.1111/j.0105-2896.2004.00167.x. [DOI] [PubMed] [Google Scholar]
- 24.Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene. 2001;20 (40):5611–5622. doi: 10.1038/sj.onc.1204641. [DOI] [PubMed] [Google Scholar]
- 25.Grawunder U, et al. Antigen receptor gene rearrangement. Current Opinion in Immunology. 1998;10 (2):172–180. doi: 10.1016/s0952-7915(98)80246-x. [DOI] [PubMed] [Google Scholar]
- 26.Aplan PD, et al. Disruption of the human SCL locus by "illegitimate" V-(D)-J recombinase activity. Science. 1990;250 (4986):1426–1429. doi: 10.1126/science.2255914. [DOI] [PubMed] [Google Scholar]
- 27.Pasqualucci L, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412 (6844):341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 28.Unniraman S, et al. Identification of an AID-independent pathway for chromosomal translocations between the Igh switch region and Myc. Nat Immunol. 2004;5 (11):1117–1123. doi: 10.1038/ni1127. [DOI] [PubMed] [Google Scholar]
- 29.Ramiro AR, et al. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118 (4):431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Markert ML. Molecular basis of adenosine deaminase deficiency. Immunodeficiency. 1994;5 (2):141–157. [PubMed] [Google Scholar]
- 31.Zhang JG, et al. Characterization of genomic BCR-ABL breakpoints in chronic myeloid leukaemia by PCR. British Journal of Haematology. 1995;90 (1):138–146. doi: 10.1111/j.1365-2141.1995.tb03392.x. [DOI] [PubMed] [Google Scholar]
- 32.Strout MP, et al. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95 (5):2390–2395. doi: 10.1073/pnas.95.5.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elliott B, Jasin M. Double-strand breaks and translocations in cancer. Cell Mol Life Sci. 2002;59 (2):373–385. doi: 10.1007/s00018-002-8429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elliott B, et al. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17 (6):885–894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 35.Smith MA, et al. The secondary leukemias: challenges and research directions. Journal of the National Cancer Institute. 1996;88 (7):407–418. doi: 10.1093/jnci/88.7.407. [DOI] [PubMed] [Google Scholar]
- 36.Olney HJ, et al. Unique balanced chromosome abnormalities in treatment-related myelodysplastic syndromes and acute myeloid leukemia: report from an international workshop. Genes Chromosomes Cancer. 2002;33 (4):413–423. doi: 10.1002/gcc.10045. [DOI] [PubMed] [Google Scholar]
- 37.Block AW, et al. Rare recurring balanced chromosome abnormalities in therapy-related myelodysplastic syndromes and acute leukemia: report from an international workshop. Genes Chromosomes Cancer. 2002;33 (4):401–412. doi: 10.1002/gcc.10044. [DOI] [PubMed] [Google Scholar]
- 38.Slovak ML, et al. 21q22 balanced chromosome aberrations in therapy-related hematopoietic disorders: report from an international workshop. Genes Chromosomes Cancer. 2002;33 (4):379–394. doi: 10.1002/gcc.10042. [DOI] [PubMed] [Google Scholar]
- 39.Maraschin J, et al. Chromosome aberrations induced by etoposide (VP-16) are not random. International Journal of Cancer. 1990;46 (5):808–812. doi: 10.1002/ijc.2910460511. [DOI] [PubMed] [Google Scholar]
- 40.Alexander FE, et al. Transplacental chemical exposure and risk of infant leukemia with MLL gene fusion. Cancer Res. 2001;61 (6):2542–2546. [PubMed] [Google Scholar]
- 41.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3 (6):430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 42.Zhou RH, et al. A precise interchromosomal reciprocal exchange between hot spots for cleavable complex formation by topoisomerase II in amsacrine-treated Chinese hamster ovary cells. Cancer Research. 1997;57 (21):4699–4702. [PubMed] [Google Scholar]
- 43.Ahuja HG, et al. Potential role for DNA topoisomerase II poisons in the generation of t(11;20)(p15;q11) translocations. Genes Chromosomes Cancer. 2000;29 (2):96–105. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1013>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 44.Bae Ys, et al. Illegitimate recombination mediated by calf thymus DNA topoisomerase II in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1988;85 (7):2076–2080. doi: 10.1073/pnas.85.7.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitmarsh RJ, et al. Reciprocal DNA topoisomerase II cleavage events at 5′-TATTA-3′ sequences in MLL and AF-9 create homologous single-stranded overhangs that anneal to form der(11) and der(9) genomic breakpoint junctions in treatment-related AML without further processing. Oncogene. 2003;22 (52):8448–8459. doi: 10.1038/sj.onc.1207052. [DOI] [PubMed] [Google Scholar]
- 46.Mistry AR, et al. DNA topoisomerase II in therapy-related acute promyelocytic leukemia. N Engl J Med. 2005;352 (15):1529–1538. doi: 10.1056/NEJMoa042715. [DOI] [PubMed] [Google Scholar]
- 47.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A. 2003;100 (22):12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lieber MR, et al. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4 (9):712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 49.Rassool FV. DNA double strand breaks (DSB) and non-homologous end joining (NHEJ) pathways in human leukemia. Cancer Lett. 2003;193 (1):1–9. doi: 10.1016/s0304-3835(02)00692-4. [DOI] [PubMed] [Google Scholar]
- 50.Mills KD, et al. The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev. 2003;194:77–95. doi: 10.1034/j.1600-065x.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 51.Bassing CH, et al. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114 (3):359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- 52.Celeste A, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114 (3):371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zucman-Rossi J, et al. Chromosome translocation based on illegitimate recombination in human tumors. Proc Natl Acad Sci U S A. 1998;95 (20):11786–11791. doi: 10.1073/pnas.95.20.11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reichel M, et al. Fine structure of translocation breakpoints in leukemic blasts with chromosomal translocation t(4;11): the DNA damage-repair model of translocation. Oncogene. 1998;17 (23):3035–3044. doi: 10.1038/sj.onc.1202229. [DOI] [PubMed] [Google Scholar]
- 55.Aplan PD, et al. Disruption of the SCL gene by a t(1;3) translocation in a patient with T cell acute lymphoblastic leukemia. Journal of Experimental Medicine. 1992;176 (5):1303–1310. doi: 10.1084/jem.176.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thandla SP, et al. ETV6-AML1 translocation breakpoints cluster near a purine/pyrimidine repeat region in the ETV6 gene. Blood. 1999;93 (1):293–299. [PubMed] [Google Scholar]
- 57.Garner MM, Felsenfeld G. Effect of Z-DNA on nucleosome placement. Journal of Molecular Biology. 1987;196 (3):581–590. doi: 10.1016/0022-2836(87)90034-9. [DOI] [PubMed] [Google Scholar]
- 58.Samadashwily GM, et al. Trinucleotide repeats affect DNA replication in vivo. Nature Genetics. 1997;17 (3):298–304. doi: 10.1038/ng1197-298. [DOI] [PubMed] [Google Scholar]
- 59.Strissel PL, et al. DNA structural properties of AF9 are similar to MLL and could act as recombination hot spots resulting in MLL/AF9 translocations and leukemogenesis. Hum Mol Genet. 2000;9 (11):1671–1679. doi: 10.1093/hmg/9.11.1671. [DOI] [PubMed] [Google Scholar]
- 60.Stanulla M, et al. Mechanisms of MLL gene rearrangement: site-specific DNA cleavage within the breakpoint cluster region is independent of chromosomal context. Hum Mol Genet. 2001;10 (22):2481–2491. doi: 10.1093/hmg/10.22.2481. [DOI] [PubMed] [Google Scholar]
- 61.Stanulla M, et al. Topoisomerase II inhibitors induce DNA double-strand breaks at a specific site within the AML1 locus. Leukemia. 1997;11 (4):490–496. doi: 10.1038/sj.leu.2400632. [DOI] [PubMed] [Google Scholar]
- 62.Stanulla M, et al. DNA cleavage within the MLL breakpoint cluster region is a specific event which occurs as part of higher-order chromatin fragmentation during the initial stages of apoptosis. Molecular & Cellular Biology. 1997;17 (7):4070–4079. doi: 10.1128/mcb.17.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ploski JE, Aplan PD. Characterization of DNA fragmentation events caused by genotoxic and non-genotoxic agents. Mutat Res. 2001;473 (2):169–180. doi: 10.1016/s0027-5107(00)00147-0. [DOI] [PubMed] [Google Scholar]
- 64.Oberhammer F, et al. Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO Journal. 1993;12 (9):3679–3684. doi: 10.1002/j.1460-2075.1993.tb06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khodarev NN, et al. Abortive apoptosis as an initiator of chromosomal translocations. Med Hypotheses. 1999;52 (5):373–376. doi: 10.1054/mehy.1997.0672. [DOI] [PubMed] [Google Scholar]
- 66.Reddien PW, et al. Phagocytosis promotes programmed cell death in C. elegans. Nature. 2001;412 (6843):198–202. doi: 10.1038/35084096. [DOI] [PubMed] [Google Scholar]
- 67.Hoeppner DJ, et al. Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature. 2001;412 (6843):202–206. doi: 10.1038/35084103. [DOI] [PubMed] [Google Scholar]
- 68.Eguchi-Ishimae M, et al. Breakage and fusion of the TEL (ETV6) gene in immature B lymphocytes induced by apoptogenic signals. Blood. 2001;97 (3):737–743. doi: 10.1182/blood.v97.3.737. [DOI] [PubMed] [Google Scholar]
- 69.Betti CJ, et al. Apoptotic stimuli initiate MLL-AF9 translocations that are transcribed in cells capable of division. Cancer Res. 2003;63 (6):1377–1381. [PubMed] [Google Scholar]
- 70.Langer T, et al. Analysis of t(9;11) chromosomal breakpoint sequences in childhood acute leukemia: almost identical MLL breakpoints in therapy-related AML after treatment without etoposides. Genes Chromosomes Cancer. 2003;36 (4):393–401. doi: 10.1002/gcc.10167. [DOI] [PubMed] [Google Scholar]
- 71.Atlas M, et al. Cloning and sequence analysis of four t(9;11) therapy-related leukemia breakpoints. Leukemia. 1998;12 (12):1895–1902. doi: 10.1038/sj.leu.2401223. [DOI] [PubMed] [Google Scholar]
- 72.Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer. 2003;3 (9):685–694. doi: 10.1038/nrc1168. [DOI] [PubMed] [Google Scholar]
- 73.Tonon G, et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet. 2003;33 (2):208–213. doi: 10.1038/ng1083. [DOI] [PubMed] [Google Scholar]
- 74.Jhiang SM. The RET proto-oncogene in human cancers. Oncogene. 2000;19 (49):5590–5597. doi: 10.1038/sj.onc.1203857. [DOI] [PubMed] [Google Scholar]
- 75.Tognon C, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2 (5):367–376. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 76.Mitelman F, et al. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet. 2004;36 (4):331–334. doi: 10.1038/ng1335. [DOI] [PubMed] [Google Scholar]
- 77.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344 (14):1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 78.Bloomfield CD, et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998;58 (18):4173–4179. [PubMed] [Google Scholar]
- 79.Richardson C, Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000;405 (6787):697–700. doi: 10.1038/35015097. [DOI] [PubMed] [Google Scholar]
- 80.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23 (1):81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 81.Myung K, Kolodner RD. Suppression of genome instability by redundant S-phase checkpoint pathways in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2002;99 (7):4500–4507. doi: 10.1073/pnas.062702199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Greaves MF, et al. Leukemia in twins: lessons in natural history. Blood. 2003;102 (7):2321–2333. doi: 10.1182/blood-2002-12-3817. [DOI] [PubMed] [Google Scholar]
- 83.Greaves M. Pre-natal origins of childhood leukemia. Rev Clin Exp Hematol. 2003;7 (3):233–245. [PubMed] [Google Scholar]
- 84.Wiederschain D, et al. Multiple MLL fusion proteins suppress p53-mediated response to DNA damage. J Biol Chem. 2005 doi: 10.1074/jbc.M412237200. [DOI] [PubMed] [Google Scholar]
- 85.Gale KB, et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci U S A. 1997;94 (25):13950–13954. doi: 10.1073/pnas.94.25.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McHale CM, et al. Prenatal origin of childhood acute myeloid leukemias harboring chromosomal rearrangements t(15;17) and inv(16) Blood. 2003;101 (11):4640–4641. doi: 10.1182/blood-2003-01-0313. [DOI] [PubMed] [Google Scholar]