

Abstract
Background
Formalin-fixed paraffin-embedded (FFPE) tissues represent the largest source of archival biological material available for genomic studies of human cancer. Therefore, it is desirable to develop methods that enable whole genome amplification (WGA) using DNA extracted from FFPE tissues. Multiple-strand Displacement Amplification (MDA) is an isothermal method for WGA that uses the large fragment of Bst DNA polymerase. To date, MDA has been feasible only for genomic DNA isolated from fresh or snap-frozen tissue, and yields a representational distortion of less than threefold.
Results
We amplified genomic DNA of five FFPE samples of normal human lung tissue with the large fragment of Bst DNA polymerase. Using quantitative PCR, the copy number of 7 genes was evaluated in both amplified and original DNA samples. Four neuroblastoma xenograft samples derived from cell lines with known N-myc gene copy number were also evaluated, as were 7 samples of non-small cell lung cancer (NSCLC) tumors with known Skp2 gene amplification. In addition, we compared the array comparative genomic hybridization (CGH)-based genome profiles of two NSCLC samples before and after Bst MDA. A median 990-fold amplification of DNA was achieved. The DNA amplification products had a very high molecular weight (> 23 Kb). When the gene content of the amplified samples was compared to that of the original samples, the representational distortion was limited to threefold. Array CGH genome profiles of amplified and non-amplified FFPE DNA were similar.
Conclusion
Large fragment Bst DNA polymerase is suitable for WGA of DNA extracted from FFPE tissues, with an expected maximal representational distortion of threefold. Amplified DNA may be used for the detection of gene copy number changes by quantitative realtime PCR and genome profiling by array CGH.
Background
With growing interest in the genomic characteristics of various human tumors and a steep increase in the availability of genomic tests for both clinical and research purposes, the amount of genomic DNA available from biological samples may limit the practicality of genomic analysis. Having been used for decades, formalin-fixed paraffin-embedded (FFPE) tissues comprise the most common form of human tissue samples archives. Therefore, it is desirable to establish a whole genome amplification (WGA) method specifically for DNA extracted from FFPE tissues. Two main approaches for WGA have been developed: thermocycling protocols and isothermal amplification methods.
Several thermocycling protocols have been used, including the degenerate oligonucleotide primed-polymerase chain reaction (DOP-PCR) [1-4], primer extension preamplification (PEP) [5-7], tagged-PCR (T-PCR) [8], and single cell comparative genomic hybridization (SCOMP), also known as linker adaptor-PCR [9,10]. What common in all these protocols are the PCR principle of temperature-dependent cyclic amplification, and the use of primers with a random sequence to allow for multiple binding sites. They differ in primer design and the sequence of temperature changes. Their amplification magnitude is a few hundredfold and the size of their DNA product ranges from 200–3000 bases. Each technique has its advantages and limitations, varying from incomplete genomic coverage to preferences for certain DNA length (e.g. shorter alleles in DOP-PCR [4]), and inconsistency in the magnitude of amplification and elaborated protocol (SCOMP).
Isothermal amplification methods refer to Hyperbranched Strand Displacement Amplification (HSDA), which is also known as Multiple-strand Displacement Amplification (MDA) [11-13]. MDA is based on two principles [14-16]: (1) the ability of the polymerase to cause strand-displacement, and (2) random initiation points using random primers. The 5' end of each strand is displaced by another upstream strand that is growing in the same direction. Displaced single strands are targeted by new random priming events. As more DNA is generated by strand displacement, an increasing number of random priming events occur, forming a network of hyperbranched DNA structures of high molecular weight. As the reaction proceeds, thousands or even millions of copies of the original DNA are generated. Two enzymes are capable of catalyzing MDA: Φ29 DNA polymerase and the large fragment of Bacillus stearothermophilus (Bst DNA polymerase, large fragment). Previous work has shown that MDA using Bst DNA polymerase on intact DNA (e.g. DNA isolated from fresh or snap-frozen tissue) gives rise to robust amplification with a representational distortion of less than threefold [14-16]. We have investigated the feasibility of MDA on DNA from FFPE tissue using the Bst DNA polymerase, and evaluated the magnitude of representational distortion using quantitative realtime PCR (QPCR) and whole genome tiling array comparative genomic hybridization (CGH) representing complete coverage of the human genome.
Results
The Bst DNA polymerase yielded a median 990-fold (range 613–1618) of DNA amplification (Figure 1). The reaction efficiency for commercial DNA and DNA extracted from snap-frozen samples was comparable and achieved a median amplification of 803-fold (range 613 – 1043), whereas the FFPE derived DNA was amplified slightly more, with a median amplification of 1035-fold (range 839 – 1618). The amplification of the negative control also generated DNA product, which was consistent in amount to the other samples. Amplification of DNA from the human pancreatic ductal epithelium (HPDE) cell line yielded 1422 ± 310- and 1560 ± 144-fold changes without and with DNA shearing, respectively. DNA replication products were of very high molecular weight as they were larger than 23 Kb (Figure 2).
Figure 1.
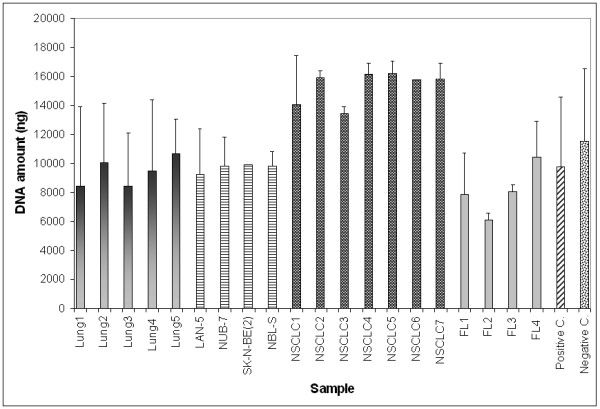
Mean amplification of DNA by Bst polymerase. All reactions started with 10 ng of target DNA. FFPE samples: Lung 1–5, neuroblastoma xenografts (LAN-5, NUB-7, SK-N-BE(2), NBL-S) and NSCLC 1–7. Intact DNA samples: FL (Frozen Lung) 1–4 and Positive C. (Control). Negative C. (Control) contained water in lieu of target DNA. For each sample the mean and SD of 2–6 independent experiments is shown.
Figure 2.
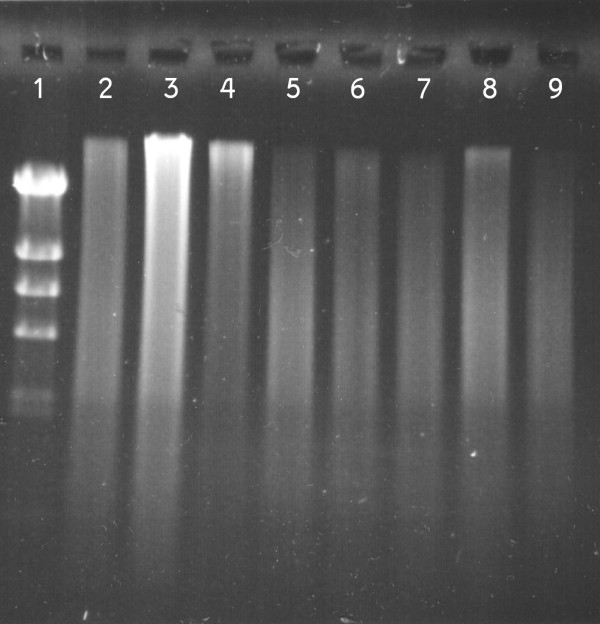
Gel electrophoresis of Bst DNA polymerase amplification products. From left to right: (1) Lambda DNA-Hind III digested ladder; FFPE samples: (2, 3) normal lung 3 & 4; (4, 5) neuroblastoma xenografts LAN-5 & SK-N-BE (2) and (6, 7) NSCLC 3 & 4; (8) Commercial DNA; (9) Negative control. Samples were analyzed in 0.5% agarose gel, stained with SYBR-green II. 10% by volume of the amplification product was used for the gel electrophoresis.
QPCR analysis revealed similar findings in all samples, whether of normal tissue or tumoral nature, carrying known gene copy number abnormalities. All tested genes were found in all FFPE and Bst amplified samples, and their relative gene copy number was within 3-fold range of non-amplified samples (Figure 3 to 5). In normal lung samples, the expected copy number ratio of any given gene to GAPDH was 1. The values shown in Figure 3 are the average ratios of the five samples tested before and after Bst amplification. The average ratios of the non-amplified samples were close to 1 (range 1.08–1.26), while those following amplification were somewhat higher (range 1.20–2.00) but within 3-fold (Figure 3). For neuroblastoma xenografts where the N-myc gene is highly amplified, the representational distortion introduced by Bst amplification was negligible relative to the magnitude of gene amplification (Figure 4). For genes with low amplification levels such as the Skp2 gene in NSCLC (Skp2/PIK3R1 ≤ 6), an increase in gene copy was detected following Bst amplification with a bias of up to 3-fold (Figure 5). It should be noted that in two of the non-amplified samples (NSCLC no. 6 and 7), the Skp2/PIK3R1 ratio was lower than 3, and therefore within the bias range. Nevertheless, it was detectable after Bst amplification.
Figure 3.
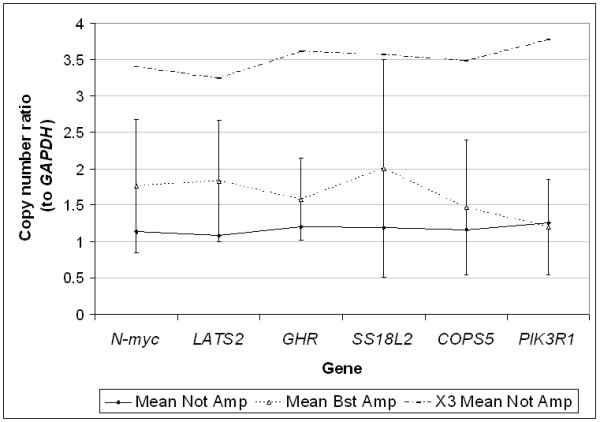
Amplification of genes in formalin-fixed paraffin embedded normal lung tissue. Mean values of relative amount of each gene to GAPDH in five samples before and after Bst polymerase amplification are shown. Error bars for ± 2 SD of mean values of Bst amplified samples are drawn. X3 Mean Not Amp is the calculated expected 3-fold representational distortion range. Gene copy numbers following Bst amplification resembled respective values in non-amplified samples and were within 3-fold change.
Figure 5.
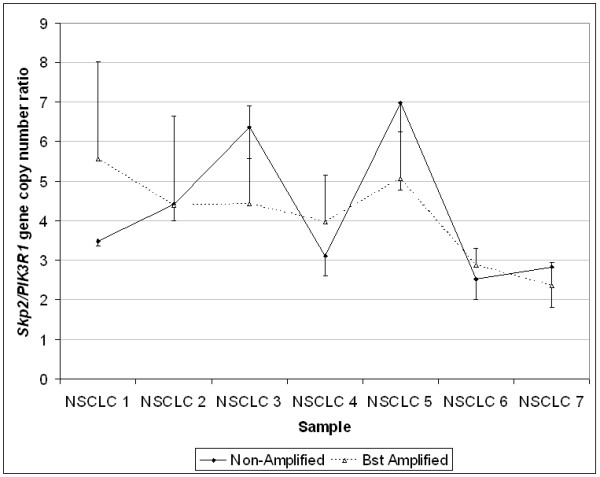
Detection of Skp2 amplification in NSCLC samples following whole genome amplification by Bst DNA polymerase. The ratios of Skp2 to PIK3R1 gene were maintained in Bst amplified vs. non-amplified NSCLC samples. Error bars represent SD.
Figure 4.
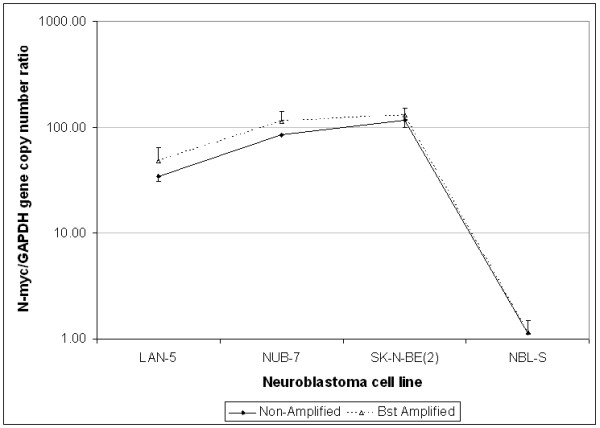
N-myc gene content in Bst amplified vs. non-amplified neuroblastoma xenografts. For neuroblastoma xenografts, where N-myc gene is highly amplified, relative gene content in Bst amplified samples was comparable to the respective values in non-amplified samples and the representational distortion was negligible. Note: NBL-S is a neuroblastoma cell line that lacks N-myc amplification and appropriately the calculated copy numbers were 1.12 ± 0.03 for non-amplified DNA and 1.14 ± 0.35 for Bst amplified DNA. Error bars represent SD.
In the negative control, the amplification reaction produced substantial amounts of DNA. However, no genes were ever detected by QPCR, indicating that the measured product was the result of a spurious amplification of the primers.
Array CGH genome profiles of the Bst-amplified DNA from two NSCLC tumours were similar to profiles obtained using their respective non-amplified DNA (Figure 6A). Hybridization of Bst-amplified samples against Bst-amplified reference DNA allowed for the identification of genomic changes that were below 3-fold change. For each of the array CGH clones, the ratio of sample to reference signal defines the changes in gene content of a given tumour. This ratio should not change for each clone when an optimal WGA method is used. Figure 6B illustrates that the correlation of such ratios was near ideal (1:1) between the un-amplified and Bst-amplified DNA from NSCLC 8; similar finding was found in NSCLC 9. The four CGH arrays had variable quality; therefore a different number of human bacterial artificial chromosome (hBAC) clones was evaluated for each pair of arrays hybridized to non-amplified and amplified DNA (Table 2). Both NSCLC samples had normal gene content in more than half of the hBAC clones, and detected changes were of low gene-dosage. Analysis using the aCGH-Smooth software showed that 58.3–84.3% of the clones had a matching call (normal gene content/amplification/deletion) following Bst amplification. The percentage of matching clones correlated with the level of genomic changes: the higher the un-amplified sample/reference ratio was (especially > 3), the more likely it was to be detected correctly following Bst amplification. NSCLC 8, which had higher level of genomic gains compared to NSCLC 9 (as reflected by the highest ratio of un-amplified sample/reference of 2.9 vs. 2.1), also exhibited a higher percentage of hBAC clones with matching profiles (84.3% vs. 58.3% respectively). The level of disagreement between the paired arrays was expressed by the number of clones that changed after Bst amplification. Both NSCLC samples had more amplified than deleted clones. Likewise, more amplified than deleted clones were undetected after Bst amplification. However, based on the percentage of originally amplified/deleted clones, gene deletions were more prone to escape detection following Bst MDA (95.83–100%) compared to gene amplifications (67.22–92.31%).
Figure 6.
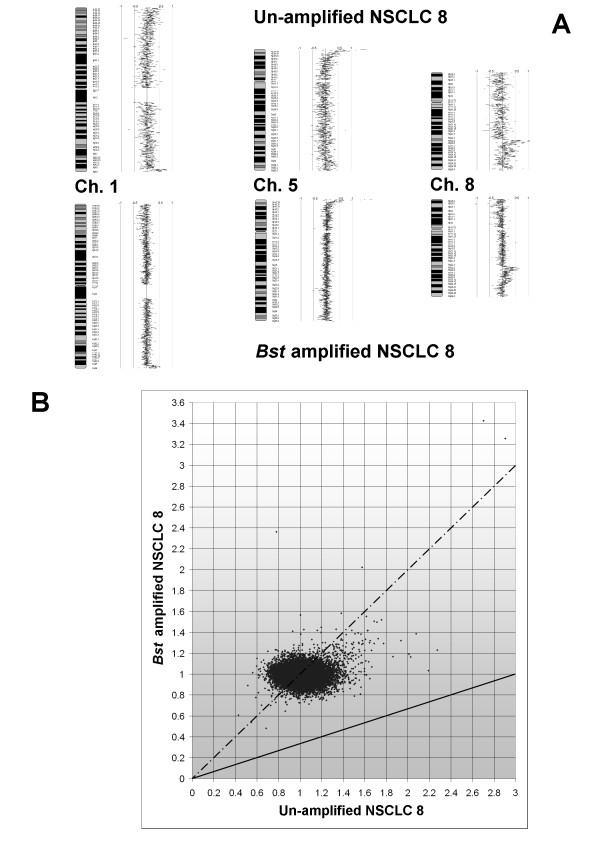
Array CGH of NSCLC before and after Bst amplification. (A) Data for chromosomes 1, 5 & 8 are displayed as a karyotype diagram with values corresponding to log2 ratio of Cy5/Cy3 spot signal (SeeGH v1.6). The genome profile following Bst amplification was similar to the profile of the original sample. Clones with log2 ratio <0.5 at 1p escaped detection following Bst amplification. (B) Scatter diagram comparing ratio of Cy5/Cy3 spot signal of NSCLC 8 before and after Bst amplification. Solid line: expected 3 fold representational distortion. Dashed line: desired (1:1) ratio of ideal WGA devoid of representational distortion. Comparison of the signal ratio for NSCLC 8 before and after Bst amplification shows it is near ideal (1:1) ratio.
Table 2.
Comparison of array CGH genome profiles before and after Bst amplification
| Number of clones | Normal gene content | Gene amplification | Gene deletion | ||
| NSCLC 8 (2.9*) | |||||
| Before Bst MDA | 16277 | 12779 | 3450 | 48 | |
| Comparing after to before MDA | Matching clones | 13725 (84.3%) | 12592 (91.75%) | 1131 (8.24%) | 2 (<0.01%) |
| Clones that changed | 2552 (15.7%) | 187 (7.3%) | 2319 (90.9%) | 46 (1.8%) | |
| % of abnormal clones before MDA | 67.22% | 95.83% | |||
| NSCLC 9 (2.1*) | |||||
| Before Bst MDA | 23942 | 13538 | 9483 | 922 | |
| Comparing after to before MDA | Matching clones | 13960 (58.3%) | 13231 (94.8%) | 729 (5.2%) | 0 (0%) |
| Clones that changed | 9982 (41.7%) | 307 (3.1%) | 8754 (87.7%) | 922 (9.2%) | |
| % of abnormal clones before MDA | 92.31% | 100% | |||
Data from each of the four array CGH experiments (NSCLC 8 & 9 before and after Bst amplification) was normalized and replicate data points with standard deviation of log2 ratio > 0.075 were excluded. Following, data was analyzed with aCGH-Smooth software, which identifies breakpoints and areas of gene amplification and deletion. The numbers presented refer to clones that were evaluable both before and after Bst amplification for each of the tumors. * Highest ratio of un-amplified sample/reference.
In contrast to the successful Bst MDA of FFPE DNA, repeated attempts to amplify FFPE DNA using Φ29 DNA polymerase failed. Although the latter yielded 568 ± 342-fold DNA amplification and the reaction product was visible on a gel, QPCR of genes successfully validated on Bst MDA products consistently failed on Φ29 DNA polymerase WGA products. To rule out the inadequacy of FFPE DNA or of the Φ29 DNA polymerase reaction, we repeated the QPCR reactions on non-amplified FFPE DNA and DNA from frozen tissue before and after Φ29 DNA polymerase amplification and were able to detect all genes.
Discussion
Lage and Dean et al [14,15] reported that MDA demonstrates high-amplification potential and excellent loci representation with less than 3-fold bias. Our study showed for the first time that Bst MDA is feasible and reliable for WGA even on FFPE DNA. We have demonstrated that in three groups of FFPE samples (normal lung tissue, neuroblastoma xenografts and NSCLC), the relative content of different genes was maintained following the amplification. If a bias existed, it was limited to a 3-fold change. We deliberately monitored the content of genes that are located on separate, unrelated regions of the genome to provide a good estimation of the overall amplification process. Array CGH data further supports the adequacy of Bst MDA on FFPE DNA. Hybridization against Bst-amplified reference DNA allows detection of genomic changes that are even below 3-fold change.
In our study, median amplification ranged from 803- to 1035-fold, and was higher for FFPE samples than for intact DNA isolated from snap-frozen tissue or commercial DNA. This is more than the 250-fold reported previously [14]. The discrepancy might be attributed to the method used for quantitation of the template and products (PicoGreen DNA quantitation vs. NanoDrop spectrophotometry). The wide range in amplification yield may be due to variability in DNA quality and tissue fixation. A possible explanation for the higher yield in FFPE compared to intact DNA could be the preferential amplification of shorter DNA fragments by the Bst polymerase [17,18]. However, the partial shearing of intact genomic DNA did not result in a significant change of the amplification yield. Yet, our results contradict previous mathematically-based predictions of a lower yield with sheared DNA [14].
Our QPCR analysis of gene copy content of FFPE samples following Bst amplification demonstrated up to a 3-fold change with respect to the non-amplified samples, fitting earlier reports of up to 3-fold representational bias [14,15]. To compare the bias resulting from various WGA methods is challenging, as the reported characteristics of each method depend on the initial amount of DNA template used, as well as on the application under investigation. The representational bias can be implied from the reported range of efficiency rates for amplification of DNA sequences of several microsatellite loci. When performed in single cells, DOP-PCR efficiency rate ranged from 0–10% and was inferior to PEP-PCR and improved PEP-PCR (I-PEP-PCR) that ranged from 0–20% and 20–50%, respectively [6]. Dean et al [15] specifically compared the representational bias of three WGA methods and reported a 103–106 representational bias with DOP-PCR, 102–104 bias with PEP-PCR, and less than a 3-fold bias with MDA, which remained almost constant between 100- to 100,000-fold amplification.
It is known that MDA by either Φ29 or Bst DNA polymerases gives products even in the absence of DNA template. This is thought to result from spurious amplification of the primers. Lage et al [14] reported that background DNA synthesis was completely suppressed when modified primers with two 5'-nitroindole (universal base) residues were used. We were unable to eliminate primer amplification reactions despite the use of modified primers. However, this spurious primer amplification appears to be tolerable, since tested genes were consistently detected by QPCR in all the Bst-amplified samples but not in the amplified negative control.
The two enzymes used for MDA, Φ29 DNA polymerase and the large fragment of Bacillus stearothermophilus, have distinct qualities. Where as Bst DNA polymerase is devoid of the 3'→5' exonuclease, Φ29 DNA polymerase holds this proofreading activity. Therefore, Φ29 DNA polymerase has a lower error rate, more efficient amplification reactions and appears to be more suitable for additional sequencing studies. However, Bst DNA polymerase seemed to demonstrate greater fidelity for copy number than Φ29 DNA polymerase. One reason is the significantly reduced activity of Φ29 DNA polymerase at the telomeres. In contrast, Bst DNA polymerase can switch templates and is therefore less affected by the proximity of genes to the telomere [14]. Thus, Bst MDA may be a better choice for WGA in array-CGH studies. It was suggested that MDA (by either Φ29 or by large fragment Bst DNA polymerase) is appropriate for array-CGH from the point of view of both sequence fidelity and representation distortion with the following guidelines. First, amplification should be < 1000-fold to keep the representation distortion less than threefold [14-16]; second, only gene copy number changes that are minimally threefold can be reliably detected [14], and finally, array-CGH study design should include amplification of the sample of interest and reference genomic DNA under identical conditions to minimize biases [14]. Based on our results, we also recommend that these guidelines should be adopted whenever Bst MDA is followed by QPCR for gene copy number evaluation. Copy number changes detected following Bst amplification are reliable only if they are higher than the 3-fold representational distortion range. Thus, it is expected that high copy number changes (e.g. N-myc in neuroblastoma xenografts) will be easily detected, since even with 3-fold amplification bias, the change in copy number is conspicuous compared to normal gene content. Although low copy number changes (e.g. Skp2 in NSCLC) are also detectable, the difference in copy number relative to normal gene content may be attenuated with 3-fold representation distortion.
Our array CGH results using FFPE DNA are remarkably similar to results previously obtained on intact DNA. With 1000-cell experiments, Lage et al reported a concordance between the amplified and non-amplified DNA array CGH of 53.6–83.3% [14]. We found concordance of 58.3–84.3% when Bst amplification was applied to 10 ng of DNA starting material. Lage stated that altered loci with relatively high gene-dosage alterations were detected with high reproducibility. Likewise, we observed that gene amplifications were consistently detected, even when smaller than 3-fold, yet the detection sensitivity correlated with the level of genomic changes. We noted that gene deletions were prone to be missed following Bst amplification and similar findings can be seen in the data presented by Lage et al. Array properties, the quality of the amplified DNA, the length of deleted areas and the low level of gene content change in deletions, which is strongly affected by the amplification representation bias introduced by MDA, all contribute to the reduced detection ability of deletions following Bst amplification. Altogether, we found that Bst MDA on FFPE DNA is reliable for following genome profiling by array CGH, in particular for the detection of gene amplification.
Prior to 2003, there were very few studies on the use of MDA compared to the widely reported PCR-based WGA [19]. However, there has recently been a growing interest in the method, as reflected by the increasing number of papers published during the past two years. MDA was reported to succeed in a variety of applications, including sequencing [20,21], microsatellite marker analysis [22,23], SNP analysis [24,25], genotyping [26] and array-CGH [14,27]. All these studies used the Φ29 DNA polymerase, and all but one [28] used DNA isolated from fresh or snap-frozen tissue samples. Like others [26], we have failed in our attempts to amplify FFPE samples with the Φ29 polymerase. Although the reaction yielded DNA that was 500-fold greater than the initial amount, no genes could be detected by QPCR and results were consistent with a spurious amplification of the primers. On the other hand, Wang et al [28] reported a successful Φ29 amplification of FFPE-derived DNA after the addition of a preliminary restriction enzyme fragmentation step. The modified protocol was named Restriction and Circularization Aided Rolling Circle Amplification (RCA-RCA). Our work is the first to describe the use of the large fragment of Bst DNA polymerase for WGA of FFPE DNA.
Aside from PCR-based techniques and MDA, a novel approach to WGA is the T7-based linear amplification of DNA (TLAD). TLAD appears free of sequence and length-dependent biases, and is thus applicable to FFPE DNA. However, this technique requires purification following each step and is therefore laborious and vulnerable to sample loss [19,29].
Conclusion
We have shown that the large fragment of Bst DNA polymerase is suitable for WGA of DNA extracted from FFPE tissues, with an expected representational distortion of up to threefold. Amplified DNA may be used for the detection of gene copy number changes by QPCR and genome profiling by array CGH.
The expected application, magnitude of findings and the limits of the method are factors that need to be considered when WGA is chosen. The virtues of Bst DNA polymerase use for WGA should be emphasized: the method is efficient, technically easy, gives high yield and is suitable for FFPE DNA. We believe that in time, Bst DNA amplification will become part of routine molecular laboratory work for research and clinical purposes.
Methods
Tissue materials and genes
The University Health Network Research Ethics Board has approved this study protocol. Tissues were obtained from NSCLC patients who underwent tumour resection at the University Health Network. Snap-frozen tissues were banked within 30 min after resection. Archival paraffin embedded tissues were 4 to10 years old (from 1994–2000); they were fixed in 10% buffered formalin and processed according to routine pathology departmental protocols. DNA was extracted from four snap-frozen normal lung tissue samples and five FFPE normal lung tissues. Commercial human male DNA (Novagen, Madison, WI) served as a positive control, and water in lieu of target DNA served as the negative control. Gene copy number both prior to and following Bst amplification was assayed by QPCR for GAPDH (12p13), N-myc (2p24.1), SS18L2 (3p21), GHR (5p12-13), PIK3R1 (5q13.1), COPS5 (8q13.1) and LATS2 (13q11-12). Four xenografts of neuroblastoma cell lines with known N-myc gene copy number including LAN-5 [30,31], NUB-7 [32,33], SK-N-BE (2) [34,35] and NBL-S [36] were also studied. Seven non-small cell lung cancer (NSCLC) samples with known Skp2 gene amplification [37] were also similarly evaluated. Tumor cells from the xenograft and NSCLC samples were enriched by manual microdissection from sections stained by toluidine blue. To compare the amplification yield in intact and fragmented DNA, we used DNA from three clones of the normal human pancreatic ductal epithelium (HPDE) cell line. DNA was sheared by sonication to strands of less than 4 Kb.
DNA extraction from FFPE tissue
Tissue sections 5 μm thick were de-waxed in toluene with vibration for 10 seconds, followed by 2 ethanol washes (with 100% and 75% ethanol) after brief vortexing. Digestion with 0.5 mg/ml Proteinase K (Roche, Laval, QC, Canada) in digestion buffer (50 mM KCl, 10 mM Tris-HCl (pH 8.3), 0.1 mg/mL gelatin, 0.45% Igepal CA-630 and 0.45% Tween) was carried out over-night, and was inactivated in 100°C for 10 minutes. The samples were purified by Phase Lock Gel (Eppendorf, Westbury, NY) with Phenol:Chloroform:Isoamyl Alcohol 25:24:1, and precipitated in 3 volumes of 0.1 M NaOAc in 95% ethanol overnight at -20°C. DNA was re-suspended in water at 37°C for 1 hour.
Bst DNA polymerase and Φ29 DNA polymerase MDA
DNA was amplified by the large fragment of Bst DNA polymerase according to Lage et al [14]. Briefly, 10 ng of DNA were mixed with 1.5 μL of 10× ThermoPol buffer (New England Biolabs, Ipswich, MA) and primers (random 7-mers with an additional two nitroindole residues at the 5' end and a phosphorothiate linkage at the 3' end) at a concentration of 100 μM in 15 μL. DNA was denaturated at 96°C for 2 minutes, cooled at room temperature for 10 minutes and then placed on ice. The reaction mixture was then brought up to 50 μL with 400 μM dNTPs in 1× ThermoPol buffer, 0.35 units/μL Bst DNA polymerase, large fragment (New England Biolabs, Ipswich, MA) and 4% final concentration of DMSO. T4 gene 32 protein (Amersham Biosciences, Piscataway, NJ) was added to the reaction in final concentration of 30 ng/μL. The reaction was carried out at 50°C for 6 hours and inactivated at 80°C for 15 minutes. Amplified samples were purified using Phase Lock Gel (Eppendorf, Westbury, NY) with Phenol:Chloroform:Isoamyl Alcohol 25:24:1 and precipitated in 3 volumes of 0.1 M NaOAc in 95% ethanol overnight at -20°C. Amplified DNA was re-suspended in water at 37°C for 1 hour and the concentration was measured by NanoDrop (NanoDrop Technologies, Wilmington, DE). 10% by volume of the amplification products were analyzed on 0.5% alkaline agarose gels stained with SYBR-green II (Molecular Probes, Eugene, OR). For Φ29 DNA polymerase, we used the GenomiPhi DNA amplification kit (GE Healthcare, Piscataway, NJ) according to the manufacturer's protocol.
Gene copy number evaluation based on quantitative realtime PCR
QPCR was performed using the SYBR Green technique in a Mx3000P® QPCR System (Stratagene, Cedar Creek, TX). Primers were designed using Primer Express software v1.5 [38] (Applied Biosystems, Foster City, CA) and tested for their specificity by alignment using the BLASTN program, followed by dissociation curve and primer efficiency tests. The target amplicons were 61-144 bp in intron 1 of each gene. The sequences of the genomic primers used for subsequent QPCR assays are listed in Table 1. Five ng of non-amplified target DNA was used in each QPCR reaction, compared to 50 ng of Bst-amplified DNA. Gene copy number was normalized to that of GAPDH, and pooled DNA from five FFPE normal lung samples was used as a calibrator. Relative gene copy number was calculated using the formula: 2-ΔΔCt [38,39]. In NSCLC samples, Skp2 was normalized against PIK3R1 (instead of GAPDH) since PIK3R1 was previously reported to show no amplification in NSCLC, unlike GAPDH [37,40].
Table 1.
Primers sequences
| Gene | Amplicon | Forward primer | Reverse primer |
| GAPD | 125 bp | 5'-GGTAAGGAGATGCTGCATTCG-3' | 5'-CGCCCAATACGACCAAATCTAA-3' |
| NMYC | 111 bp | 5'-CGCAAAAGCCACCTCTCATTA-3' | 5'-TCCAGCAGATGCCACATAAGG-3' |
| SS18L2 | 127 bp | 5'GTAGGGATGAGGTCTCCCTTTGT-3' | 5'-GAAATGCGGAGCTGGTGTG-3' |
| GHR | 120 bp | 5'-GACTGGCCACTTAGCTGTCTTTG-3' | 5'-GGAGTCCTTTGAGTAGCAGCAACT-3' |
| PIK3R1 | 144 bp | 5'-TCATTTGTGGGATGACTTAGATTTG-3' | 5'-AAAGTTGACAGTCCTGAATATTTTTAATATATAAA-3' |
| COPS5 | 61 bp | 5'-TCGACATGCACCTTGTTTGG-3' | 5'-TGAAAACAGCTGCAATCCCC-3' |
| LATS2 | 101 bp | 5'-GAGTCAGGGAACCTGGCTTTAA-3' | 5'-ATATGACTCTTCGGCAGCTGC-3' |
| SKP2 | 102 bp | 5'-GGGTACCATCTGGCACGATT-3' | 5'-GATACTGCTATTCTGAAAGTCTTTTTCTTC-3' |
Array CGH of non-amplified and Bst amplified samples
Paired un-amplified and Bst-amplified DNA from two NSCLC tumours were studied and compared for genomic profile changes using whole genome array CGH. We used the "27 K" high-density hBAC Sub Megabase Resolution Tiling (SMRT) set array CGH (BCCRC, Vancouver, BC), which contains two replicates for each clone and has a resolution of 70–80 Kb. The experiments were performed as previously described [41,42]. Briefly, 400 ng of both sample and reference male genomic DNA (Novagen, Mississauga, ON, Canada) were labeled with Cyanine-5 and Cyanine-3 dCTPs (PerkinElmer, Woodbridge, ON, Canada), respectively. Bst-amplified samples were hybridized against Bst-amplified reference DNA. All MDA DNA was sheared by sonication into 3–4 Kb prior to labeling. Following hybridization, arrays images were captured by the charge-coupled device (CCD) scanner system (PerkinElmer, Wellesley, MA, USA) and analyzed with SoftWoRx Tracker (Applied Precision, Issaquah, WA, USA). Data were normalized using a three-step normalization framework [43]; replicate data points with a log2 ratio that exceeded a standard deviation of 0.075 were excluded. Data were analyzed with SeeGH v1.6 [44] and aCGH-Smooth [45] software with the Lambda and breakpoint per chromosome settings set to 6.75 and 100, respectively. This analysis defines chromosomal breakpoints and identifies chromosomal sections with abnormal gene content, namely areas of gene amplification or deletion. As the chromosomal location of each hBAC clone is known, the breakpoint information can be presented at the clone level where each clone can be defined as having either amplification, normal content or deletion. Each of the arrays was independently analyzed and evaluable clones before and after Bst amplification were compared. The concordance between paired arrays was indicated by the number and percentage of clones with matching profiles, while discordance was shown as the number and percentage of clones that changed after Bst amplification (Table 2). The concordance between paired arrays was also demonstrated by the correlation of the sample to reference signal ratio between un-amplified and Bst-amplified DNA (Figure 6B).
Abbreviations
CGH – comparative genomic hybridization, FFPE – formalin-fixed paraffin-embedded, hBAC – human bacterial artificial chromosome, MDA – multiple-strand displacement amplification, NSCLC – non-small cell lung cancer, PCR – polymerase chain reaction, QPCR – quantitative realtime PCR, WGA – whole genome amplification.
Authors' contributions
SAR carried out the molecular genetic studies, performed the statistical analysis, contributed to the conception of the study and its design and drafted the manuscript. CQZ participated in the molecular genetic studies and contributed to the statistical analysis. BPC participated in statistical analysis. NL and SKW participated in molecular genetic studies. WLL contributed to the conception and design of the study. MST conceived of the study, participated in its design and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work was supported by the Canadian Cancer Society and National Cancer Institute of Canada (NCIC) grant #015184. Dr. Aviel-Ronen is a fellow of the Canadian Institutes of Health Research
(CIHR) Training Program for Clinician Scientists in Molecular Oncologic Pathology (STP-53912) and a recipient of the Ontario Cancer Institute Knudson Research Fellowship and NCIC Terry Fox Foundation Clinical Research Fellowship. Dr. Tsao is the M. Qasim Choksi Chair in Lung Cancer Translational Research.
The authors thank Dr. Herman Yeger for his generosity in providing the paraffin blocks of neuroblastoma xenograft tumors, Dr. Roya Navab for her help with the gel electrophoresis, Ms. Suzanne K. Lau for her comments and Dr. Jeremy Squire for advice.
Contributor Information
Sarit Aviel-Ronen, Email: saronen@uhnres.utoronto.ca.
Chang Qi Zhu, Email: czhu@uhnres.utoronto.ca.
Bradley P Coe, Email: bcoe@bccrc.ca.
Ni Liu, Email: niliu@uhnres.utoronto.ca.
Spencer K Watson, Email: swatson@bccrc.ca.
Wan L Lam, Email: wanlam@bccrc.ca.
Ming Sound Tsao, Email: ming.tsao@uhn.on.ca.
References
- Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A. Degenerate oligonucleotide-primed PCR: General amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718–725. doi: 10.1016/0888-7543(92)90147-K. [DOI] [PubMed] [Google Scholar]
- Cheung VG, Nelson SF. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA. Proc Natl Acad Sci USA. 1996;93:14676–14679. doi: 10.1073/pnas.93.25.14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, Okita K, Shiraishi K, Kusano N, Furuya T, Oga A, Kawauchi S, Kondoh S, Sasaki K. Detection of genetic alterations in pancreatic cancers by comparative genomic hybridization coupled with tissue microdissection and degenerate oligonucleotide primed polymerase chain reaction. Oncology. 2002;62:251–258. doi: 10.1159/000059573. [DOI] [PubMed] [Google Scholar]
- Grant SF, Steinlicht S, Nentwich U, Kern R, Burwinkel B, Tolle R. SNP genotyping on a genome-wide amplified DOP-PCR template. Nucleic Acids Res. 2002;30:e125. doi: 10.1093/nar/gnf125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N. Whole genome amplification from a single cell: Implications for genetic analysis. Proc Natl Acad Sci USA. 1992;89:5847–5851. doi: 10.1073/pnas.89.13.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietmaier W, Hartmann A, Wallinger S, Heinmoller E, Kerner T, Endl E, Jauch KW, Hofstadter F, Ruschoff J. Multiple mutation analyses in single tumor cells with improved whole genome amplification. Am J Pathol. 1999;154:83–95. doi: 10.1016/S0002-9440(10)65254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang VW, Bell DA, Berkowitz RS, Mok SC. Whole genome amplification and high-throughput allelotyping identified five distinct deletion regions on chromosomes 5 and 6 in microdissected early-stage ovarian tumors. Cancer Res. 2001;61:4169–4174. [PubMed] [Google Scholar]
- Grothues D, Cantor CR, Smith CL. PCR amplification of megabase DNA with tagged random primers (T-PCR) Nucleic Acids Res. 1993;21:1321–1322. doi: 10.1093/nar/21.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CA, Schmidt-Kittler O, Schardt JA, Pantel K, Speicher MR, Riethmuller G. Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells. Proc Natl Acad Sci USA. 1999;96:4494–4499. doi: 10.1073/pnas.96.8.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklein NH, Erbersdobler A, Schmidt-Kittler O, Diebold J, Schardt JA, Izbicki JR, Klein CA. SCOMP is superior to degenerated oligonucleotide primed-polymerase chain reaction for global amplification of minute amounts of DNA from microdissected archival tissue samples. Am J Pathol. 2002;161:43–51. doi: 10.1016/S0002-9440(10)64155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat Genet. 1998;19:225–232. doi: 10.1038/898. [DOI] [PubMed] [Google Scholar]
- Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11:1095–1099. doi: 10.1101/gr.180501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizardi PM. Multiple Displacement Amplification. Connecticut, United States; 2000. [Google Scholar]
- Lage JM, Leamon JH, Pejovic T, Hamann S, Lacey M, Dillon D, Segraves R, Vossbrinck B, Gonzalez A, Pinkel D, Albertson DG, Costa J, Lizardi PM. Whole genome analysis of genetic alterations in small DNA samples using hyperbranched strand displacement amplification and array-CGH. Genome Res. 2003;13:294–307. doi: 10.1101/gr.377203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward P, Sun Z, Zong Q, Du Y, Du J, Driscoll M, Song W, Kingsmore SF, Egholm M, Lasken RS. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook MS, Delach SM, Deyneko G, Worlock A, Wolfe JL. Whole genome amplification of DNA from laser capture-microdissected tissue for high-throughput single nucleotide polymorphism and short tandem repeat genotyping. Am J Pathol. 2004;164:23–33. doi: 10.1016/S0002-9440(10)63092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M, Sedmak D, Jewell S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol. 2002;161:1961–1971. doi: 10.1016/S0002-9440(10)64472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siwoski A, Ishkanian A, Garnis C, Zhang L, Rosin M, Lam WL. An efficient method for the assessment of DNA quality of archival microdissected specimens. Mod Pathol. 2002;15:889–892. doi: 10.1097/01.MP.0000024288.63070.4F. [DOI] [PubMed] [Google Scholar]
- Hughes S, Arneson N, Done S, Squire J. The use of whole genome amplification in the study of human disease. Prog Biophys Mol Biol. 2005;88:173–189. doi: 10.1016/j.pbiomolbio.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Monstein HJ, Olsson C, Nilsson I, Grahn N, Benoni C, Ahrne S. Multiple displacement amplification of DNA from human colon and rectum biopsies: Bacterial profiling and identification of helicobacter pylori-DNA by means of 16S rDNA-based TTGE and pyrosequencing analysis. J Microbiol Methods. 2005;63:239–247. doi: 10.1016/j.mimet.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Mai M, Hoyer JD, McClure RF. Use of multiple displacement amplification to amplify genomic DNA before sequencing of the alpha and beta haemoglobin genes. J Clin Pathol. 2004;57:637–640. doi: 10.1136/jcp.2003.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono S, Faruqi AF, Dean FB, Du Y, Sun Z, Wu X, Du J, Kingsmore SF, Egholm M, Lasken RS. Unbiased whole-genome amplification directly from clinical samples. Genome Res. 2003;13:954–964. doi: 10.1101/gr.816903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook JF, Stabley D, Sol-Church K. Exploring whole genome amplification as a DNA recovery tool for molecular genetic studies. J Biomol Tech. 2005;16:125–133. [PMC free article] [PubMed] [Google Scholar]
- Tzvetkov MV, Becker C, Kulle B, Nurnberg P, Brockmoller J, Wojnowski L. Genome-wide single-nucleotide polymorphism arrays demonstrate high fidelity of multiple displacement-based whole-genome amplification. Electrophoresis. 2005;26:710–715. doi: 10.1002/elps.200410121. [DOI] [PubMed] [Google Scholar]
- Pask R, Rance HE, Barratt BJ, Nutland S, Smyth DJ, Sebastian M, Twells RC, Smith A, Lam AC, Smink LJ, Walker NM, Todd JA. Investigating the utility of combining phi29 whole genome amplification and highly multiplexed single nucleotide polymorphism BeadArray genotyping. BMC Biotechnol. 2004;4:15. doi: 10.1186/1472-6750-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoholm MI, Hoffmann G, Lindgren S, Dillner J, Carlson J. Comparison of archival plasma and formalin-fixed paraffin-embedded tissue for genotyping in hepatocellular carcinoma. Cancer Epidemiol Biomarkers Prev. 2005;14:251–255. [PubMed] [Google Scholar]
- Hughes S, Lim G, Beheshti B, Bayani J, Marrano P, Huang A, Squire JA. Use of whole genome amplification and comparative genomic hybridisation to detect chromosomal copy number alterations in cell line material and tumour tissue. Cytogenet Genome Res. 2004;105:18–24. doi: 10.1159/000078004. [DOI] [PubMed] [Google Scholar]
- Wang G, Maher E, Brennan C, Chin L, Leo C, Kaur M, Zhu P, Rook M, Wolfe JL, Makrigiorgos GM. DNA amplification method tolerant to sample degradation. Genome Res. 2004;14:2357–2366. doi: 10.1101/gr.2813404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Schreiber SL, Bernstein BE. Development and validation of a T7 based linear amplification for genomic DNA. BMC Genomics. 2003;4:19. doi: 10.1186/1471-2164-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger RC, Danon YL, Rayner SA, Hoover F. Definition of a thy-1 determinant on human neuroblastoma, glioma, sarcoma, and teratoma cells with a monoclonal antibody. J Immunol. 1982;128:983–989. [PubMed] [Google Scholar]
- Hamano S, Ohira M, Isogai E, Nakada K, Nakagawara A. Identification of novel human neuronal leucine-rich repeat (hNLRR) family genes and inverse association of expression of Nbla10449/hNLRR-1 and Nbla10677/hNLRR-3 with the prognosis of primary neuroblastomas. Int J Oncol. 2004;24:1457–1466. [PubMed] [Google Scholar]
- Dimitroulakos J, Squire J, Pawlin G, Yeger H. NUB-7: A stable I-type human neuroblastoma cell line inducible along N- and S-type cell lineages. Cell Growth Differ. 1994;5:373–384. [PubMed] [Google Scholar]
- Torkin R, Lavoie JF, Kaplan DR, Yeger H. Induction of caspase-dependent, p53-mediated apoptosis by apigenin in human neuroblastoma. Mol Cancer Ther. 2005;4:1–11. [PubMed] [Google Scholar]
- Squire JA, Thorner PS, Weitzman S, Maggi JD, Dirks P, Doyle J, Hale M, Godbout R. Co-amplification of MYCN and a DEAD box gene (DDX1) in primary neuroblastoma. Oncogene. 1995;10:1417–1422. [PubMed] [Google Scholar]
- Smith AG, Popov N, Imreh M, Axelson H, Henriksson M. Expression and DNA-binding activity of MYCN/Max and Mnt/Max during induced differentiation of human neuroblastoma cells. J Cell Biochem. 2004;92:1282–1295. doi: 10.1002/jcb.20121. [DOI] [PubMed] [Google Scholar]
- Cohn SL, Salwen H, Quasney MW, Ikegaki N, Cowan JM, Herst CV, Kennett RH, Rosen ST, DiGiuseppe JA, Brodeur GM. Prolonged N-myc protein half-life in a neuroblastoma cell line lacking N-myc amplification. Oncogene. 1990;5:1821–1827. [PubMed] [Google Scholar]
- Zhu CQ, Blackhall FH, Pintilie M, Iyengar P, Liu N, Ho J, Chomiak T, Lau D, Winton T, Shepherd FA, Tsao MS. Skp2 gene copy number aberrations are common in non-small cell lung carcinoma, and its overexpression in tumors with ras mutation is a poor prognostic marker. Clin Cancer Res. 2004;10:1984–1991. doi: 10.1158/1078-0432.CCR-03-0470. [DOI] [PubMed] [Google Scholar]
- PE Applied Biosystems . User Bulletin #2: Relative Quantitation of Gene Expression. Foster City, CA: Applied Biosystems; 1997. [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Massion PP, Kuo WL, Stokoe D, Olshen AB, Treseler PA, Chin K, Chen C, Polikoff D, Jain AN, Pinkel D, Albertson DG, Jablons DM, Gray JW. Genomic copy number analysis of non-small cell lung cancer using array comparative genomic hybridization: Implications of the phosphatidylinositol 3-kinase pathway. Cancer Res. 2002;62:3636–3640. [PubMed] [Google Scholar]
- Ishkanian AS, Malloff CA, Watson SK, DeLeeuw RJ, Chi B, Coe BP, Snijders A, Albertson DG, Pinkel D, Marra MA, Ling V, MacAulay C, Lam WL. A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet. 2004;36:299–303. doi: 10.1038/ng1307. [DOI] [PubMed] [Google Scholar]
- Coe BP, Lockwood WW, Girard L, Chari R, Macaulay C, Lam S, Gazdar AF, Minna JD, Lam WL. Differential disruption of cell cycle pathways in small cell and non-small cell lung cancer. Br J Cancer. 2006 doi: 10.1038/sj.bjc.6603167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khojasteh M, Lam WL, Ward RK, MacAulay C. A stepwise framework for the normalization of array CGH data. BMC Bioinformatics. 2005;6:274. doi: 10.1186/1471-2105-6-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi B, DeLeeuw RJ, Coe BP, MacAulay C, Lam WL. SeeGH – a software tool for visualization of whole genome array comparative genomic hybridization data. BMC Bioinformatics. 2004;5:13. doi: 10.1186/1471-2105-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong K, Marchiori E, Meijer G, Vaart AV, Ylstra B. Breakpoint identification and smoothing of array comparative genomic hybridization data. Bioinformatics. 2004;20:3636–3637. doi: 10.1093/bioinformatics/bth355. [DOI] [PubMed] [Google Scholar]