

Abstract
The 25-kDa heat shock protein (Hsp25) is associated with various malignancies and is expressed at high levels in biopsies as well as circulating in the serum of breast cancer patients. In this study, we used RNA interference technology to silence the hsp25 gene in 4T1 breast adenocarcinoma cells, known as a poorly immunogenic, highly metastatic cell line. We demonstrate that transfection of 4T1 cells with short interference RNA-Hsp25 dramatically inhibits proliferation as compared with control transfected cells. In addition, we show that 4T1 cells transfected with short interference RNA-Hsp25 abrogates tumor migration potential by a mechanism that is in part due to the repression of matrix metalloproteinase 9 expression and a concomitant upregulation of its antagonist, tissue inhibitor metalloproteinase 1. Taken together, these findings provide a model system for the study of metastatic potential of tumors and are suggestive of an earlier unrecognized role for Hsp25 in tumor migration.
Keywords: Breast, carcinoma, Chaperokine, Heat shock proteins, Metastasis, siRNA
Introduction
The incidence of female breast cancer has continued to rise since 1986 [1]. In the United States, an estimated 215,900 new cases of invasive breast cancer were expected to occur in women in the year 2004. The 5-year relative survival rate for localized breast cancer (cancer that has not spread to lymph nodes or other locations outside the breast) has increased from 72% in the 1940s to 97% today. However, the survival rate is lower (79%) in patients with regional spread, while for women with distant metastases, the rate is 23% [1]. In this ultimate stage of malignant transformation, the tumors become resistant to most therapies, and the disease is almost incurable. Therefore, therapeutic interventions designed to inhibit metastatic spread of cancer cells are critical.
Organs are protected and sustained by several types of tissue barriers like the basement membrane, the extracellular matrix and interstitial connective tissue. For metastasis to be successful, these barriers have to be crossed by tumor cells that have been released from a primary tumor. These are the first steps for the progression of metastasis, involving a proteolytic breakdown. Several enzymes are involved in these processes and studies have shown that metalloproteinases are required to invade tissues, enter the bloodstream and metastasize to other organs [2]. Matrix metalloproteinases (MMP) together with membrane-type MMP and serine proteases are the major enzymes responsible for extracellular matrix degradation [for a review, see 3]. Recent studies have shown that a high tumorigenicity potential in a murine breast cancer model is characterized by a high secretion of MMP together with IL-6 and monocyte chemoattractan protein 1, whereas in cells with a low tumorigenicity potential, the secretion of these factors was significantly lower [4]. The studies done by the group of Saiki [5] further demonstrate the association of MMP-9 with the progression of breast cancer. When mice were given vanillin orally, the number of metastases to the lungs after inoculation of 4T1 cells was significantly reduced and it was demonstrated that vanillin inhibits the production of MMP-9.
The inhibition of apoptosis and the resistance to chemotherapeutic agents have an impact on tumor progression, metastasis and therapy outcome [1]. Small heat shock protein (Hsp) expression is essential to protect differentiating cells against apoptosis [6]. The 25-kDa Hsp (Hsp25) belongs to the family of small Hsp and is the murine homologue of human Hsp27. This protein is ubiquitously expressed and has been implicated in various biological functions. In contrast to large Hsp, Hsp25/27 act through ATP-independent mechanisms, and in vivo, they act in concert with other chaperones by creating a reservoir of misfolded proteins [3]. Activation of the heat shock-responsive element on the hsp25/27 gene results in biological functions associated with cytoprotection from a variety of stressful stimuli. The upregulation of intracellular Hsp25/27 protects cells from tumor necrosis factor-alpha-mediated apoptosis by a mechanism that involves the downregulation of reactive oxygen species [4-6]. Hsp25/Hsp27 has been shown to be associated with estrogen-responsive malignancies and is expressed at high levels in biopsies, as well as circulating in the serum of breast cancer patients [7-9]. Murine breast carcinoma 4T1 is a 6-thioguanine-resistant cell line selected from the 410.4 tumor without mutagen treatment. When injected into BALB/c mice, 4T1 spontaneously produces highly metastatic tumors that can metastasize to the lung, liver, lymph nodes and brain, while the primary tumor is growing in situ [10]. The primary tumor does not have to be removed to induce metastatic growth. The tumor growth and metastatic spread of 4T1 cells in BALB/c mice very closely mimic human breast cancer [11]. In our previous work, we have shown that a percentage of 4T1 cells express Hsp25 on their surface, and this particular phenotype increases their metastatic potential [12] .
Since Hsp25 has been shown to be highly expressed in biopsies of breast cancer patients [13] and has been implicated as a regulator of metastasis [12], we hypothesize that silencing the hsp25 gene in our murine breast cancer model would inhibit cell migration. Recent studies have shown that RNA interference (RNAi) can be used to study the function of different genes and their role on the formation of metastasis in breast cancer [14]. RNAi is a form of post-transcriptional control in which the introduction of a double-stranded RNA (dsRNA) into a cell leads to the homology-dependent degradation of its cognate mRNA [15]. In mammalian cells, short dsRNA (21-23 mer) is specific and is referred as short interference RNA (siRNA). The siRNAs then hybridize into their cognate mRNA which induce specific degradation of the target mRNA [16] .
In this study, we demonstrate that silencing the hsp25 gene of the highly metastatic 4T1 breast adenocarcinoma cell line suppresses proliferation and cell migration. Further, we show for the first time that silencing of the hsp25 gene downregulates MMP-9 and concomitantly upregulates tissue inhibitor of metalloproteinase 1 (TIMP-1), a known MMP-9 antagonist.
Materials and Methods
Cell Lines and Culture Conditions
Murine breast carcinoma 4T1 cells were a gift from Dr. Christopher Nicchitta, Duke University Medical Center, Durham, N.C., USA. 4T1 cells were maintained in Dulbecco’s modified Eagle media (Invitrogen, Carlsbad, Calif., USA) containing 2 mM L -glutamine and adjusted to contain 1.5 g/l sodium bicarbonate, 4.5 g/l glucose, 10 mM HEPES, 1.0 mM sodium pyruvate and 10% fetal bovine serum at 37 °C in a humidified incubator with 5% CO2 atmosphere. Cells were grown at an exponential growth rate and harvested using 0.1% trypsin-EDTA when cultures are approximately 80% confluent. Cells were passaged only 5-8 times before fresh cells were used.
hsp25 Gene Silencing Using siRNA
hsp25 gene silencing was achieved by using a vector-based system to produce hairpin RNA. The criteria that were used to silence the hsp25 gene have previously been established [16]. To design the siRNA duplexes, a search for the 23-nucleotide motif with AA(N19)TT was performed in which the target sites were selected from a gene sequence beginning at 50-100 nucleotides downstream of the start codon. Hits with approximately 50% G/C content were selected and anti-sense RNA was synthesized as the complement to position 1-21 of the 23-nucleotide motif. Finally, symmetric 3′-overhangs that help to ensure that siRNPs are formed with approximately equal ratios of sense and anti-sense target RNA-cleaving siRNPs. The 21-base pair product, siRNA-Hsp25, was cloned into the pSuper vector (Oligoengine, Seattle, Wash., USA) under the restriction of Bgl II and Hin d III (fig. 1). This construct was then cotransfected with a green fluorescent protein (GFP) plasmid into the 4T1 tumor cell line.
Fig. 1.

Schematic representation of cloning of siRNA-Hsp25. pSuper vector was used to introduce a 64-base oligomer to form the hairpin with a Hsp25-specific sequence. This 21-base was synthesized with defined sequences to provide two restriction sites (Bgl II and Hin d III) and, on transcription, form a hairpin, in order to initiate gene silencing. Eight clones were screened and three positive clones were sequenced. All experiments were performed with one of the matched sequences.
Splicing of the hairpin results in 21-mer-long dsRNA. pSuper vector was used to introduce a 64-base oligomer to form the hairpin with a Hsp25-specific sequence. A chemically synthesized 21-mer nucleotide DNA to silence the hsp25 gene was used and cloned into the pSuper vector-based silencing (Oligoengine). The primers were designed using web-based proprietary software from Oligoengine, and three 21-mer gene sequences were selected, predicting the successful silencing of the hsp25 gene. The sequence used in this study was Hsp25 sequence 5′-AGCCCGAGCTGGGAACCATT-3′ (GenBank Accession No. LO7577). This 21-base was synthesized with defined sequences to provide two restriction sites (Bgl II and Hin d III) and, on transcription, form a hairpin, in order to initiate gene silencing. The total length of the primer is 64 mer including the specific sequences of Hsp25. A complementary oligonucleotide was also synthesized from Oligoengine. The primers were incubated in annealing buffer (100 mM potassium acetate, 30 mM HEPES-KOH, pH 7.4, 2 mM magnesium acetate) at 95 °C for 5 min and incubated overnight at room temperature. After digesting double-stranded oligo and pSuper vector with Hin d III and Bgl II restriction enzymes, primers were annealed and cloned into the pSuper vector. Positive clones were sequenced.
Transfection and Cell Sorting
4T1 cells were transfected with pSuper-Hsp25 and GFPplasmid using the lipid transfection reagent Effectene according to the manufacturer’s instructions (Qiagen, Valencia, Calif., USA). Briefly, 3 × 105 exponentially growing cells were seeded in 60-mm tissue culture plates and a mixture of 1μg GFPplasmid DNA and 1μg pSuper-Hsp25 plasmid in Effectene was added to the cells and incubated for 18 h at 37 °C. After 48 h, cells were harvested and immediately sorted into GFP-positive and -negative subpopulations using a MoFlow cytometer (Dakocytomation, Carpinteria, Calif., USA). Individual cells were gated on the basis of forward scatter (FSC) and orthogonal scatter (SSC). The photomultiplier (PMT) for GFP (FL1-height) was set on a logarithmic scale. Cell debris was excluded by raising the FSC-height PMT threshold. The flow rate was adjusted to × 200 cells/s and at least 105 cells were sorted for each sample group.
Western Blot Analysis
Following various treatment protocols, cells were lyzed using RIPA buffer containing appropriate protease inhibitors, and the protein concentration was determined using the Bradford method (Bio-Rad, Hercules, Calif., USA) with a DU-650 spectrophotometer (Beckman Coulter). Samples were run in a 12% SDS-PAGE gel and transferred onto a nitrocellulose membrane. The membrane was blocked for 1 h at 4 °C with Tween 20-Tris-buffered saline (T-TBS) containing 5% milk, and the membrane was probed with primary antibodies against Hsp72 (StressGen Biotechnologies), 70-kDa heat shock cognate protein (Hsc70; StressGen Biotechnologies), Hsp25 (kind gift of Daniel Ciocca, Mendoza, Argentina), MMP-9 (Chemicon International) and TIMP-1 (Chemicon International). Antibodies against β-actin (Sigma-Aldrich, St. Louis, Mo., USA) were used as loading controls where indicated. Antibodies were diluted in T-TBS containing 5% milk. After 1 h of incubation at room temperature, the membrane was washed in T-TBS three times. Corresponding horseradish peroxidase-conjugated IgG secondary antibodies (Sigma-Aldrich) were added, and the membrane was incubated for 30 min at room temperature. After additional washes, proteins were visualized using enhanced chemiluminescence (Amersham, Little Chalfont, UK).
Formazan Formation Assay
To measure cell proliferation, 4T1 cells were seeded at 1,000 cells/well and allowed to attach for 18 h and treated with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) and an electron coupling reagent (phenazine ethosulfate) for 4 h. The conversion of the tetrazolium salt MTS to a colored formazan by mitochondrial dehydrogenases is a marker of cell viability. The color development was measured spectrophotometrically at 490 nm using a Benchmark plus plate reader from Bio-Rad (Bio-Rad, San Diego, Calif., USA) with Microplate Manager Software for analysis.
DNA Content and Cell Cycle Analysis
Freshly sorted 4T1 cells (105) transfected with GFPplasmid or siRNA-Hsp25 cells were fixed in 80% ethanol and incubated with 10μg/ml propidium iodide plus 50μg/ml RNAse A. Cells were incubated for 1 h at 37 °C to remove interference from RNA. DNA content and cell cycle were performed using flow cytometric analysis. Briefly, samples were analyzed on a FACScan with a CellQuest software program (Becton Dickinson, Mountain View, Calif., USA). Individual nuclei were gated on the basis of FSC and SSC. The PMT for FL2-width/area was set on a linear scale. Cell debris was excluded by raising the FSC-height PMT threshold. The flow rate was adjusted to × 200 cells/s and at least 30,000 nuclei were analyzed for each sample.
Migration Assays
Cell migration was measured using the Matrigel invasion chambers (BD Biocoat Cellware, San Jose, Calif., USA) according to the manufacturer’s instructions. Briefly, conditioned medium was placed in the lower chamber as a chemoattractant. Single-cell suspensions were placed on the upper chamber. Twenty-two hours later, cells that had not penetrated the filter were washed off and the membrane stained with 0.5% crystal violet, mounted on a microscope slide, visualized and photographed. Fifteen different fields were visualized using a light microscope at 10 × magnification.
Immunofluorescence Microscopy
After cell sorting, 4T1 breast adenocarcinoma cells (104) were seeded onto Falcon culture slides (BD Labware, Franklin Lakes, N.J., USA) for 24 h. Slides were then washed to remove non-adherent cells with ice-cold PBS and were simultaneously fixed, permeabilized using the Cytofix/CytopermTM kit (BD Biosciences, San Diego, Calif., USA) according to the manufacturer’s instructions and counter-stained with anti-Hsp25, and isotype-matched Texas red secondary antibody was added and incubated for 30 min in cold and dark. After washing, DAPI was added at a ratio of 1:2,000, incubated for 10 min, and then one drop of mounting media (Vector, Burlingame, Calif., USA) was placed onto the glass slide before the cover slip was sealed with nail polish.
Statistical Analysis
The data were analyzed using a two-tailed t test after applying ANOVA. Differences were considered significant with p < 0.05.
Results
Inhibition of Hsp25 Expression in 4T1 Breast Adenocarcinoma Cells Using siRNA Technology
To determine if we could successfully silence the hsp25 gene in a cell system, 4T1 cells were transfected with siRNA-Hsp25 and sorted into GFP-positive (siRNA-Hsp25) and GFP-negative (GFPplasmid) subpopulations (fig. 2). Immunofluorescent microscopy revealed that sorted 4T1 cells transfected with siRNA-Hsp25 (GFP-positive cells) did not express significant amounts of Hsp25 as compared with 4T1 cells transfected with plasmid (GFP-negative cells). The transfection efficiency was consistently between 22 and 28% (fig. 2).
Fig. 2.

Effect of transfection of siRNA-Hsp25 on 4T1 breast adenocarcinoma cells. 4T1 cells were transfected with GFPplasmid plus siRNA-Hsp25 plasmid as described in ‘Materials and Methods'. After 48 h, cells were sorted using flow cytometry for either GFP-negative or GFP-positive cells. Sorted cells were stained with anti-Hsp25 antibody and a secondary isotype-matched antibody biotinylated with Texas red. Cells were counterstained for nuclei integrity with DAPI (blue fluorescence). Immunofluorescent pictograms show GFP-negative cells express Hsp25 (red fluorescence) and GFP-positive cells are negative for Hsp25. Data are representative of at least three independently performed experiments with similar results.
These results were confirmed by Western blot analysis, which demonstrated that silencing the hsp25 gene in 4T1 cells using siRNA-Hsp25 resulted in significant inhibition of Hsp25 expression (p < 0.05) (fig. 3). However, the expression of the constitutively expressed Hsp70 (Hsc70) was not affected by siRNA-Hsp25 (fig. 3). Hsp25 is a stress-inducible gene that is turned on in response to stress [7]. We demonstrated that at both ambient temperature (37 °C) and in response to non-lethal heat shock treatment (43 °C, 30 min), there was no significant up-regulation in total Hsp25 expression (fig. 4). 4T1 cells transfected with plasmid effectively upregulated Hsp25 expression in response to non-lethal heat shock treatment.
Fig. 3.
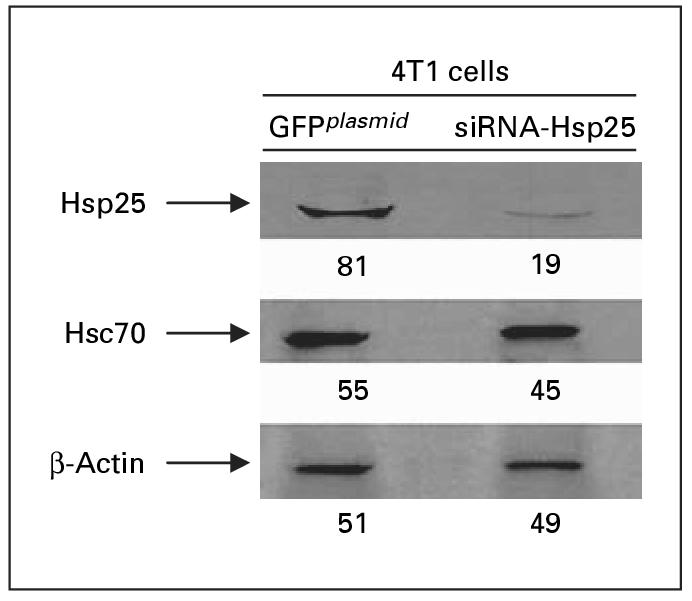
Suppression of Hsp25 expression in 4T1 breast adenocarcinoma cells. Freshly sorted GFPplasmid or siRNA-Hsp25 transfectants were lyzed, and extracted proteins were analyzed by 12% SDS-PAGE and immunoblotted with antibodies specific to Hsp25 or Hsc70 and visualized with a coupled secondary antibody chemiluminescence method. Anti-β-actin antibody was used as loading control on stripped Hsp25 membranes. The intensities of the bands were analyzed by densitometry with a video densitometer (Chemilmager ™ 5500; Alpha Innotech, San Leandro, Calif., USA) using the American Applied Biology software. Figures represent relative density. Results are representative of three independently performed experiments with similar results.
Fig. 4.
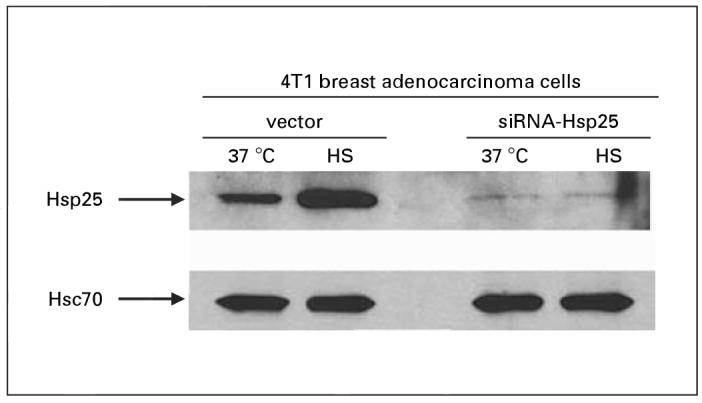
Effect of siRNA-Hsp25 in response to heat stress in 4T1 breast adenocarcinoma cells. Exponentially growing GFPplasmid or siRNA-Hsp25 transfectants were either exposed to non-lethal heat shock (HS) treatment (43 °C, 30 min) and allowed to recover for 18 h or maintained at 37 °C. Cells were then lyzed, and extracted proteins were analyzed by 12% SDS-PAGE and immunoblot with antibodies specific to Hsp25 or Hsc70 and visualized with a coupled secondary antibody chemiluminescence method. Anti-β-actin antibody was used as loading control on stripped Hsp25 membranes. Results are representative of two independently performed experiments with similar results.
Effect of Silencing the hsp25 Gene on 4T1 Breast Adenocarcinoma Cell Proliferation and Cell Cycle Progression
An important characteristic of tumors is their high proliferation rate. We demonstrated that siRNA-Hsp25 significantly inhibited proliferation of 4T1 cells as compared with plasmid control or wild-type 4T1 cells (fig. 5). Since silencing the hsp25 gene results in the inhibition of proliferation, it might be assumed that overexpression of Hsp25 would result in enhanced proliferation. As expected, 4T1 cells transfected with Hsp25Neo resulted in the overexpression of Hsp25 and enhanced proliferation, as compared with vector only (pLXSN) (fig. 5).
Fig. 5.
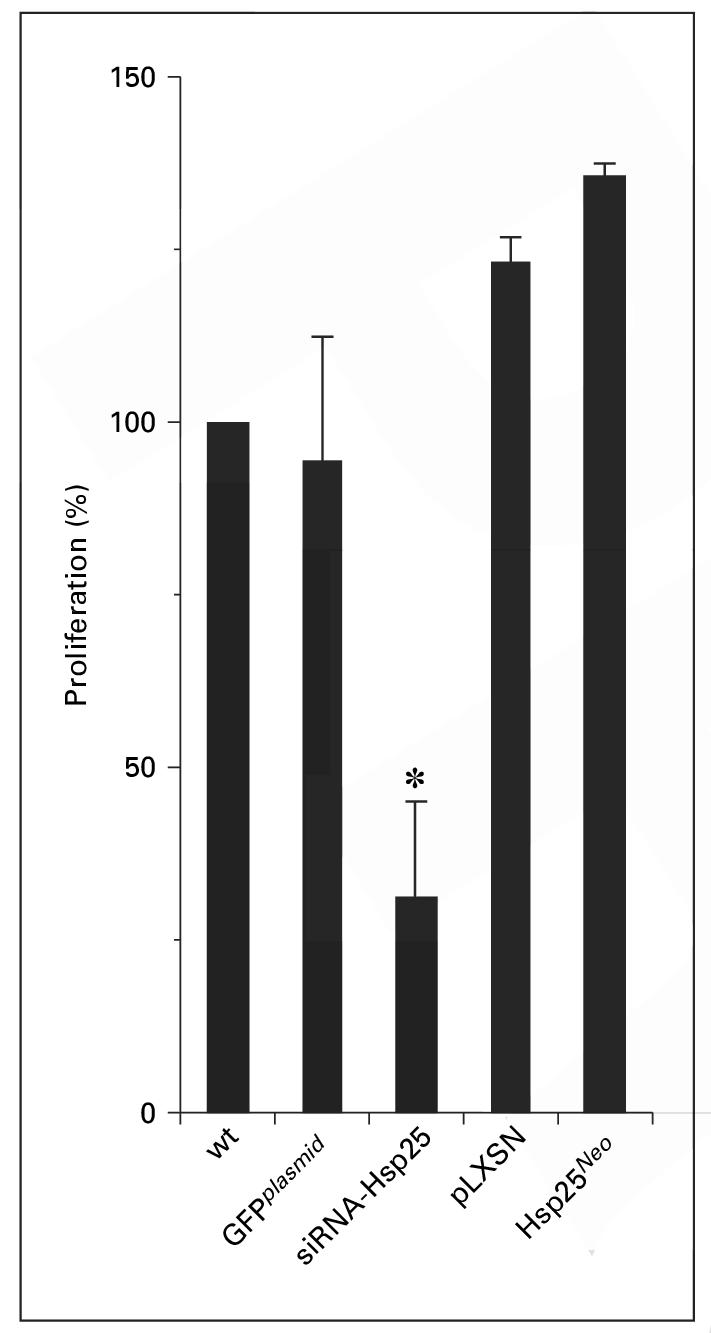
Effect of silencing the hsp25 gene on proliferation of 4T1 breast adenocarcinoma cells. Exponentially growing wild-type 4T1 cells (wt) or 4T1 cells transfected with GFPplasmid, siRNA-Hsp25, pLXSN or Hsp25Neo transfectants were seeded and treated with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium, inner salt (MTS) to determine the proliferation as described in ‘Materials and Methods’. Data are the mean percent proliferation ± SD and represent the sum of three independently performed experiments. * p < 0.05 versus control (Student’s t test).
To examine the specific phase of the cell cycle that silencing the hsp25 gene affects, DNA content of transfected 4T1 cells was measured using flow cytometry. Silencing the hsp25 gene resulted in significant changes in the G0/G1 (57-29%) and G2/M (19-47%) phase, whereas the S phase was not significantly affected (21-22%), as compared with GFPplasmid (fig. 6). 4T1 cells transfected with Hsp25Neo exhibited significant changes in the G0/G1 phase (50-66%) and the S phase (23-2%), whereas the G2/M phase was not significantly affected (22-25%), as compared with pLXSN plasmid (p < 0.05) (fig. 6). Taken together, these results demonstrate that silencing the hsp25 gene in 4T1 cells inhibits proliferation and induces cell cycle arrest at G2/M.
Fig. 6.

Effect of siRNA-Hsp25 on cell cycle distribution. Freshly sorted 4T1 cells (105) transfected with GFPplasmid (a), siRNA-Hsp25 (b), pLXSN (c) or Hsp25Neo (d) were fixed in ethanol and incubated with propidium iodide and RNAse A for 1 h at 37 °C. DNA content and cell cycle were performed using flow cytometric analysis as described in detail in ‘Materials and Methods'. Results are a representative of four independently performed experiments with similar results. Abscissa, fluorescence intensity; ordinate, relative cell number.
Effect of Silencing the hsp25 Gene on 4T1 Breast Adenocarcinoma Cell Migration
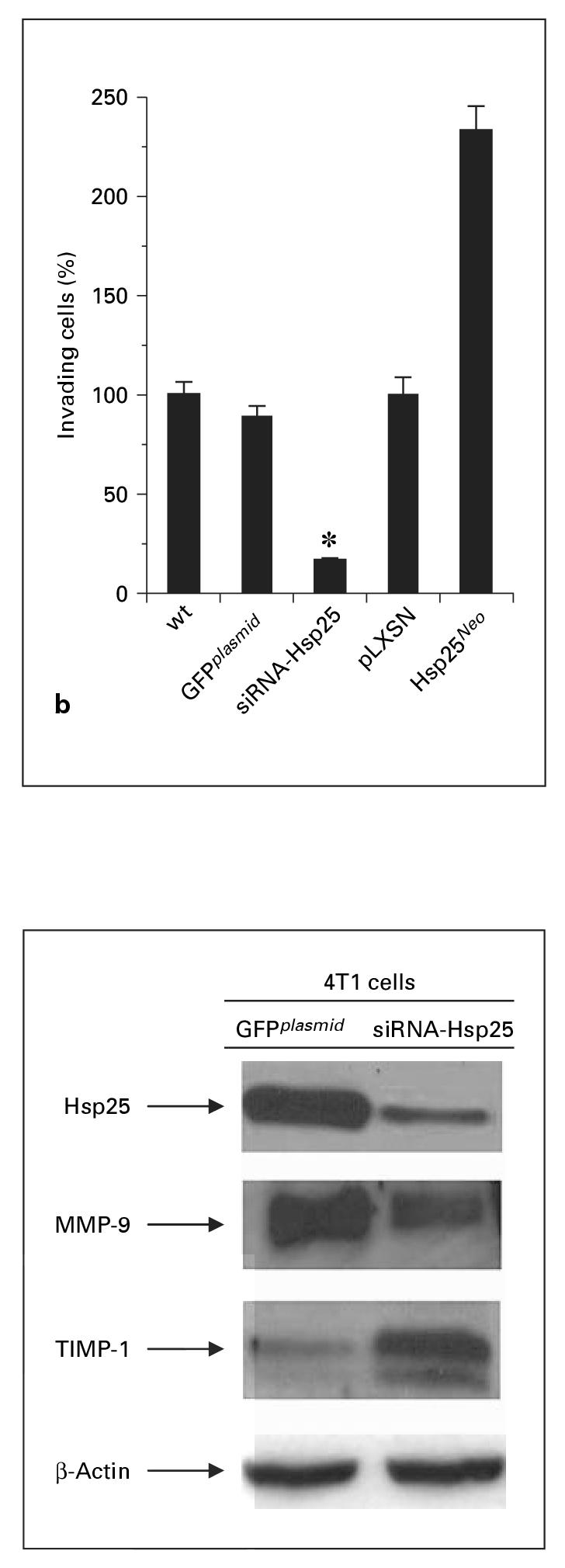
Metastasis of tumors is the primary cause of death for solid tumors [8]. To date, the clinical significance of micrometastasis is incompletely understood for most tumor types [8]. Therefore, inhibition of metastasis is an important step in fighting the disease. We demonstrated that siRNA-Hsp25 treatment of 4T1 cells drastically inhibited migration as judged by the cell invasion assay, as compared with 4T1 cells transfected with GFPplasmid (fig. 7 a, b). In confirmation of the proliferation data, 4T1 cells transfected with Hsp25Neo migrated significantly more than either GFPplasmid or siRNA-Hsp25 (p < 0.05) (fig. 7 a, b). Taken together, these results demonstrate that silencing the hsp25 gene in 4T1 cells eliminates migration.
Fig. 7.

Suppression of migration of 4T1 cells upon transfection with siRNA-Hsp25. a 4T1 (105) cells transfected with siRNA-Hsp25, GFPplasmid or Hsp25Neo were placed on Matrigel invasion chambers (BD Pharmingen). Twenty-two hours later, cells were harvested and stained with 0.5% crystal violet solution and mounted in a microscope slide at 10 × magnification. Data are representative of three independently performed experiments with similar results. b 4T1 (105) cells transfected with siRNA-Hsp25, GFPplasmid or Hsp25Neo were placed on Matrigel invasion chambers (BD Pharmingen). Twenty-two hours later, cells were harvested and stained with 0.5% crystal violet solution and mounted in a microscope slide under 10 × magnification and 15 fields were counted. Bars are the mean percent migration ± SD and represent the sum of three independently performed experiments. * p < 0.05 versus control (Student’s t test).
Effect of Silencing hsp25 Gene on MMP-9 and TIMP-1 Expression
To understand the Hsp25-dependent mechanisms of 4T1 cell migration, we downregulated the Hsp25 protein level in cells by siRNA-mediated gene silencing and determined the MMP-9 protein level. We hypothesized that MMP-9 could be affected, since it has been previously reported as a protein involved in the progression of breast cancer. Western blot analysis demonstrated that silencing Hsp25 downregulates the expression of MMP-9 (fig. 8). Interestingly, the expression of TIMP-1, a known MMP-9 antagonist, was upregulated.
Fig. 8.
Silencing of Hsp25 inhibits the expression of MMP-9 and upregulates TIMP-1 expression. Freshly sorted GFPplasmid or siRNA-Hsp25 transfectants were lyzed. Protein concentration was determined by the Bradford method and wells were equally loaded (5μg) with the extracted proteins as shown by performed β-actin staining on stripped membranes. Proteins were analyzed by 12% SDS-PAGE and immunoblots performed. Anti-Hsp25-, anti-MMP-9- or anti-TIMP-1-specific polyclonal antibodies were used. Visualization of bands was done using enhanced chemiluminescence reagent. Results are representative of two independently performed experiments with similar results.
Discussion
Our experiments indicate that silencing the hsp25 gene has a profound effect on 4T1 breast adenocarcinoma tumors by decreasing cell proliferation and inhibiting cell migration by a mechanism that is dependent on the inhibition of MMP-9, a molecule previously shown to be involved in tumor migration and adhesion. The spread of breast cancer from its primary site to lymph nodes and distant organs continues to be the most important problem in the field of cancer treatment and therapy. Patients who present with metastatic disease at diagnosis or those who develop metastases after successful management of the primary tumor have a poor prognosis. Therefore, a better understanding of the mechanisms that govern tumor metastasis is necessary. In this study, we use RNAi technology to silence the hsp25 gene in 4T1 breast adeno-carcinoma cells. We used the difference in GFP intensity to differentiate between cells that have taken up both plasmids containing siRNA-Hsp25 and those without (fig. 2). The possibility exists that high concentrations of GFP might cause a mild degree of cellular injury that might result in an increase in Hsp25 levels. However, this is not the case using the conditions and concentrations we employed in these experiments. We clearly demonstrate reduced (not elevated) levels of Hsp25 in cells positive for siRNA-Hsp25, which exhibit a high intensity of GFP as compared with siRNA-Hsp25-negative cells which do not express GFP (fig. 3, 4). In addition, when cells were transfected with GFPplasmid alone using the same agent and method and sorted on the basis of similar intensity to the GFP siRNA-Hsp25-positive cells and tested for the expression of Hsp25 or cellular injury, there was no significant increase in Hsp25 expression (as judged by Western blot analysis) or cellular damage (as judged by the lactate dehydrogenase release assay) than cells transfected with plasmid only (GFP negative) (data not shown).
Studies designed to determine the role of Hsp25/27 and Hsp70 during tumor progression [9, 10] and in response to current anti-cancer drugs [11, 12] attribute a modest prognostic value of Hsp27 in breast cancer and have shown it to be implicated in drug resistance [12]. Several studies report that enhanced or maintained expression of Hsp27 is an independent and accurate predictor of poor clinical outcome for individuals with breast [10] or prostate cancer [13]. However, these studies measure total Hsp27 and Hsp70 expression. Studies specifically designed to measure surface bound/expressed and total Hsp25 and Hsp70 on 4T1 tumors suggest that tumor development and metastatic spread favor cells that express high levels of Hsp25 on their plasma surface, while tumors that express Hsp70 on their plasma surface are sensitive to anti-tumor effector cells [14]. These studies indicate that treatment protocols designed to increase the surface expression of Hsp70 on tumors and downregulate Hsp27 expression could inhibit tumor growth and eliminate metastasis [14]. This is a very important distinction because of the dichotomy that has been revealed between intracellular and extracellular expressed Hsp [for a review, see 15]. Enhanced expression of intracellular Hsp70 has clearly been shown to be anti-inflammatory [16], antiapoptotic [17], induce cell cycle arrest [18] and protect cells from stressful stimuli [for a review, see 19]. In contrast, enhanced extracellular expression of Hsp70 either on the surface of tumors or in the extracellular milieu either enhances natural killer cell-mediated lysis [20] or upregulates antigen presenting cell-mediated acute-phase responses [21-24]. Thus, it might be expected that Hsp25 would exhibit a similar differential pattern of activation depending on its cellular location. Contrary to these predictions, tumor development and metastatic potential of 4T1 breast adenocarcinoma cells were demonstrated to favor tumors that overexpress high levels of Hsp25 on their plasma surface, while tumors that express Hsp70 on their plasma surface are sensitive to anti-tumor effector cells [14]. We hypothesized that therapies which can upregulate Hsp70 surface expression and concomitantly suppress Hsp25/27 surface expression on tumors might result in the suppression of tumor growth and abrogate the metastatic potential of the tumors [14] .
Our studies show that the hsp25 gene has an effect on proliferation (fig. 5) and cell cycle arrest (fig. 6) in 4T1 breast adenocarcinoma cells. In agreement with these studies, others have demonstrated an increase in tumor growth by L929 murine fibrosarcoma transfected with plasmids that induces the overexpression of hsp25, when transplanted into nude mice [25]. Lemieux et al. [26] demonstrated that overexpression of hsp27 in the human breast cancer cell line MDA-MB increased cell invasiveness, adhesion and migration in Matrigel assays. Conversely, these authors showed that antisense suppression of hsp27 gene significantly inhibited cell invasiveness [26]. In contrast, studies by Eskenazi et al. [27] did not observe a significant increase in the metastatic capacity of human B16 melanoma cells that overexpress hsp27. In other studies, the overexpression of hsp27 inhibits in vitro proliferation and induces a delay in tumor development and migration when A375 human melanoma cells were transfected with hsp27 [28]. The reason for this discrepancy is unknown. A possible reason could lie in the intrinsic differences between human and murine sequences when overexpressed in a tumor mouse model. Studies looking at the effect of overexpression of Hsp25/27 have shown that it plays an important role in the cycle by a mechanism that involves cycle arrest in G1, S and G2/M [29, 30]. In our studies, we show that 4T1 cells transfected with siRNA-Hsp25 induce suppression of cell cycle arrest at the G2/M phase (fig. 6). We hypothesize that this might account for the anti-proliferative effect of the siRNA on 4T1 cells. However, further studies are required to conclusively answer this question. The cell cycle distribution analyses were performed 48 h after transfection. At this time, we did not observed significant apoptosis in the resting cell population. Nevertheless, this does not negate the possibility that at a later time point significant apoptosis might occur. However, this is beyond the scope of this present article.
Our results do not exclude the possibility that other genes are important in metastases of tumors. Indeed, at the submission of this manuscript, a study using RNAi technology to silence the chemokine receptor CXCR4 in 4T1 cells prevented primary tumor formation in some mice, and surviving mice did not develop macroscopic metastases [31]. In a second study, Liang et al. [32] demonstrate that silencing the chemokine receptor CXCR4, using siRNA, effectively blocks metastasis of the human breast carcinoma cell line MDA-MB-231. In invasion studies, these authors show that each siRNA-CXCR4 constructed (siRNA1 and siRNA2) suppresses migration by approximately 60%. However, a combination of both siRNA1 and siRNA2 blocks migration by 90% [32]. In our studies, we demonstrate that a single siRNA-Hsp25 effectively blocks migration by > 90% (fig. 7). In vivo studies are currently underway to test the efficacy of the siRNA-Hsp25 in a tumor animal model [Bausero et al., in preparation].
Our studies show that downregulation of Hsp25 affects the expression of both MMP-9 and TIMP-1. Here, we describe for the first time an association between Hsp25 repression and the downregulation of MMP-9. MMP-9 has been previously demonstrated to be involved in the migration of tumor cells and their metastatic potential and is influenced by cell adhesion molecules including cadherins [for a review, see 3]. In the present study, we demonstrate that Hsp25 repression downregulates MMP-9 expression and concomitantly upregulates its antagonist TIMP-1, suggesting an additional mechanism by which Hsp25 increases the metastatic potential of 4T1 cells [14]. The MMP-9 antagonist TIMP-1 is able to activate Ras involving the Tyk/mitogen-activated protein kinase pathway [33]. A specific blockade of the extracellular signal-regulated kinase pathway suppresses the expression of MMP-9 and markedly inhibits the invasiveness of tumor cells [34]. Our results show that TIMP-1 is actually induced when Hsp25 is silenced, suggesting a complex regulation of these proteolytic enzymes by mechanisms that may involve Hsp25.
In summary, our experiments indicate that silencing the hsp25 gene has a profound effect on tumor proliferation and migration. This implies that therapies targeted at repression of the hsp25/27 gene may be beneficial to patients with metastatic disease or those who develop metastases after successful management of the primary tumor.
Table 1.
Effect of siRNA-Hsp25 on cell cycle distribution
| 4T1 transfection1 | Cells in respective phases of cell cycle2, % |
|||
|---|---|---|---|---|
| Sub-G0/G1 | G0/G1 | S | G2/M | |
| None | 2.4±3 | 63.5±5 | 17.1±2 | 17.8±2 |
| GFPplasmid | 1.0±1 | 58.5±4 | 20.6±2 | 13.2±1 |
| siRNA-Hsp25 | 2.8±2 | 29.6±3 | 21.6±1 | 47.2±2 |
| pLXSN | 3.4±2 | 50.3±4 | 22.8±2 | 21.6±2 |
| Hsp25Neo | 5.6±2 | 56.5±5 | 20.6±2 | 19.3±2 |
Results are expressed as mean ± SD.
Freshly sorted 4T1 cells (105) were transfected with GFPplasmid, siRNA-Hsp25, pLXSN or Hsp25Neo and were fixed in ethanol and incubated with propidium iodide and RNAse A for 1 h at 37 ° C as described in detail in ‘Materials and Methods’.
Cell cycle analysis was performed using flow cytometric analysis as described in detail in ‘Materials and Methods’. Results are the percentage of cells in respective phases of cell cycle ± SD and are the sum of two to three independently performed experiments with similar results.
Acknowledgements
The authors thank Dr. Susana Fiorentino (INSERM U462, Saint-Louis Hospital, Paris, France) for helpful discussions. This work was supported in part by the National Institute of Health Grant RO1CA91889, the Joint Center for Radiation Therapy Foundation Grant, Harvard Medical School, institutional support from the Department of Medicine, Boston University School of Medicine, Scott & White Clinic, and an Endowment from the Cain Foundation (to A.A.).
References
- 1.Weir HK, Thun MJ, Hankey BF, Ries LA, Howe HL, Wingo PA, Jemal A, Ward E, Anderson RN, Edwards BK. Annual report to the nation on the status of cancer, 1975-2000, featuring the uses of surveillance data for cancer prevention and control. J Natl Cancer Inst. 2003;95:1276–1299. doi: 10.1093/jnci/djg040. [DOI] [PubMed] [Google Scholar]
- 2.Kim J, Yu W, Kovalski K, Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998;94:353–362. doi: 10.1016/s0092-8674(00)81478-6. [DOI] [PubMed] [Google Scholar]
- 3.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 4.Neumark E, Sagi-Assif O, Shalmon B, Ben-Baruch A, Witz IP. Progression of mouse mammary tumors: MCP-1-TNFalpha cross-regulatory pathway and clonal expression of proma lignancy and antimalignancy factors. Int J Cancer. 2003;106:879–886. doi: 10.1002/ijc.11337. [DOI] [PubMed] [Google Scholar]
- 5.Lirdprapamongkol K, Sakurai H, Kawasaki N, Choo MK, Saitoh Y, Aozuka Y, Singhirunnusorn P, Ruchirawat S, Svasti J, Saiki I. Vanillin suppresses in vitro invasion and in vivo metastasis of mouse breast cancer cells. Eur J Pharm Sci. 2005;25:57–65. doi: 10.1016/j.ejps.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 6.Soldes OS, Kuick RD, Thompson IA, 2nd, Hughes SJ, Orringer MB, Iannettoni MD, Hanash SM, Beer DG. Differential expression of Hsp27 in normal oesophagus, Barrett’s metaplasia and oesophageal adenocarcinomas. Br J Cancer. 1999;79:595–603. doi: 10.1038/sj.bjc.6690094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jantschitsch C, Trautinger F. Heat shock and UV-B-induced DNA damage and mutagenesis in skin. Photochem Photobiol Sci. 2003;2:899–903. doi: 10.1039/b301253k. [DOI] [PubMed] [Google Scholar]
- 8.Wieder R. Insurgent micrometastases: sleeper cells and harboring the enemy. J Surg Oncol. 2005;89:207–210. doi: 10.1002/jso.20199. [DOI] [PubMed] [Google Scholar]
- 9.Oesterreich S, Hilsenbeck SG, Ciocca DR, All-red DC, Clark GM, Chamness GC, Osborne CK, Fuqua SA. The small heat shock protein HSP27 is not an independent prognostic marker in axillary lymph node-negative breast cancer patients. Clin Cancer Res. 1996;2:1199–1206. [PubMed] [Google Scholar]
- 10.Ciocca DR, Vargas-Roig LM. Hsp27 as a prognostic and predictive factor in cancer. Prog Mol Subcell Biol. 2002;28:205–218. doi: 10.1007/978-3-642-56348-5_11. [DOI] [PubMed] [Google Scholar]
- 11.Ciocca DR, Green S, Elledge RM, Clark GM, Pugh R, Ravdin P, Lew D, Martino S, Osborne CK. Heat shock proteins hsp27 and hsp70: lack of correlation with response to tamoxifen and clinical course of disease in estrogen receptor-positive metastatic breast cancer (a Southwest Oncology Group Study) Clin Cancer Res. 1998;4:1263–1266. [PubMed] [Google Scholar]
- 12.Ciocca DR, Rozados VR, Cuello Carrion FD, Gervasoni SI, Matar P, Scharovsky OG. Hsp25 and Hsp70 in rodent tumors treated with doxo-rubicin and lovastatin. Cell Stress Chaperones. 2003;8:26–36. doi: 10.1379/1466-1268(2003)8<26:hahirt>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cornford PA, Dodson AR, Parsons KF, Desmond AD, Woolfenden A, Fordham M, Neoptolemos JP, Ke Y, Foster CS. Heat shock protein expression independently predicts clinical outcome in prostate cancer. Cancer Res. 2000;60:7099–7105. [PubMed] [Google Scholar]
- 14.Bausero MA, Page DT, Osinaga E, Asea A. Surface expression of Hsp25 and Hsp72 differentially regulates tumor growth and metastasis. Tumor Biol. 2004;25:243–251. doi: 10.1159/000081387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asea A. Exogenous Hsp70: principles and application of the chaperokine activity of Hsp70; In: Henderson B, Pockley AG, editors. The Extracellular Biology of Molecular Chaperones. Cambridge University Press; London: 2005. [Google Scholar]
- 16.Asea A, Ara G, Teicher BA, Stevenson MA, Calderwood SK. Effects of the flavonoid drug quercetin on the response of human prostate tumours to hyperthermia in vitro and in vivo. Int J Hyperthermia. 2001;17:347–356. doi: 10.1080/02656730110053146. [DOI] [PubMed] [Google Scholar]
- 17.Jaattela M, Wissing D, Kokholm K, Kallunki T, Egeblad M. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 1998;17:6124–6134. doi: 10.1093/emboj/17.21.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calderwood SK, Asea A. Targeting HSP70-induced thermotolerance for design of thermal sensitizers. Int J Hyperthermia. 2002;18:597–608. doi: 10.1080/0265673021000019666. [DOI] [PubMed] [Google Scholar]
- 19.Jaattela M. Programmed cell death: many ways for cells to die decently. Ann Med. 2002;34:480–488. doi: 10.1080/078538902321012423. [DOI] [PubMed] [Google Scholar]
- 20.Farkas B, Hantschel M, Magyarlaki M, Becker B, Scherer K, Landthaler M, Pfister K, Gehr-mann M, Gross C, Mackensen A, Multhoff G. Heat shock protein 70 membrane expression and melanoma-associated marker phenotype in primary and metastatic melanoma. Melanoma Res. 2003;13:147–152. doi: 10.1097/00008390-200304000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 22.Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calder-wood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 23.Asea A, Kabingu E, Stevenson MA, Calder-wood SK. HSP70 peptide-bearing and peptidenegative preparations function as chaperokines. Cell Stress Chaperones. 2000;5:425–431. doi: 10.1379/1466-1268(2000)005<0425:hpbapn>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basu S, Suto R, Binder RJ. Srivastava PK: Heat shock proteins as novel mediators of cytokine secretion by macrophages. Cell Stress Chaperones. 1998;3:11–16. [Google Scholar]
- 25.Blackburn RV, Galoforo SS, Berns CM, Armour EP, McEachern D, Corry PM, Lee YJ. Comparison of tumor growth between hsp25- and hsp27-transfected murine L929 cells in nude mice. Int J Cancer. 1997;72:871–877. doi: 10.1002/(sici)1097-0215(19970904)72:5<871::aid-ijc26>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 26.Lemieux P, Oesterreich S, Lawrence JA, Steeg PS, Hilsenbeck SG, Harvey JM, Fuqua SA. The small heat shock protein hsp27 increases invasiveness but decreases motility of breast cancer cells. Invasion Metastasis. 1997;17:113–123. [PubMed] [Google Scholar]
- 27.Eskenazi AE, Powers J, Pinkas J, Oesterreich S, Fuqua SA, Frantz CN. Induction of heat shock protein 27 by hydroxyurea and its relationship to experimental metastasis. Clin Exp Metastasis. 1998;16:283–290. doi: 10.1023/a:1006553127695. [DOI] [PubMed] [Google Scholar]
- 28.Aldrian S, Trautinger F, Frohlich I, Berger W, Micksche M, Kindas-Mugge I. Overexpression of Hsp27 affects the metastatic phenotype of human melanoma cells in vitro. Cell Stress Chaperones. 2002;7:177–185. doi: 10.1379/1466-1268(2002)007<0177:oohatm>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SH, Lee YS, Osawa Y, Hachiya M, Akashi M. Hsp25 regulates the expression of p21(Waf1/Cip1/Sdi1) through multiple mechanisms. J Biochem (Tokyo) 2002;131:869–875. doi: 10.1093/oxfordjournals.jbchem.a003177. [DOI] [PubMed] [Google Scholar]
- 30.Lee YJ, Cho HN, Jeoung DI, Soh JW, Cho CK, Bae S, Chung HY, Lee SJ, Lee YS. HSP25 overexpression attenuates oxidative stress-induced apoptosis: roles of ERK1/2 signaling and manganese superoxide dismutase. Free Radic Biol Med. 2004;36:429–444. doi: 10.1016/j.freeradbiomed.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64:8604–8612. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 32.Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005;65:967–971. [PMC free article] [PubMed] [Google Scholar]
- 33.Wang T, Yamashita K, Iwata K, Hayakawa T. Both tissue inhibitors of metalloproteinases-1 (TIMP-1) and TIMP-2 activate Ras but through different pathways. Biochem Biophys Res Commun. 2002;296:201–205. doi: 10.1016/s0006-291x(02)00741-6. [DOI] [PubMed] [Google Scholar]
- 34.Tanimura S, Asato K, Fujishiro SH, Kohno M. Specific blockade of the ERK pathway inhibits the invasiveness of tumor cells: down-regulation of matrix metalloproteinase-3/-9/-14 and CD44. Biochem Biophys Res Commun. 2003;304:801–806. doi: 10.1016/s0006-291x(03)00670-3. [DOI] [PubMed] [Google Scholar]